
ABSTRACT
Head and neck squamous cell carcinomas (HNSCC) are well suited for cancer vaccination strategies. In addition to tumor-associated antigens (TAAs) and endogenous retrovirus (ERV) encoded proteins, HNSCCs have a relatively high tumor mutational burden encoding potential neoepitopes. Peptide vaccine candidates are prioritized by predicted high-affinity to major histocompatibility complex (MHC) class I with MHC-II affinity largely not being considered. Herein, we extend previous studies to evaluate therapeutic vaccination in the mouse oral cancer (MOC) 22 model. Two distinct MOC22 derived SLPs were tested – a TSA-oriented mutant intercellular adhesion molecule 1 (mICAM1) and p15E, an ERV encoded antigen. In silico prediction revealed mICAM1 SLP bore strong MHC-I and MHC-II epitopes sharing a mutant residue with vaccination significantly increasing both antigen-specific IFN-γ producing CD4+ and CD8+ T cells. By contrast, p15E SLP had a predicted high-affinity MHC-I epitope but lacked an MHC-II epitope and vaccination induced antigen-specific CD8+ but not CD4+ T cell responses. Therapeutic mICAM1 vaccination attenuated tumor growth effectively with mICAM1-specific T cells displaying durable IFN-γ production compared with p15E SLP. Furthermore, mICAM1 SLPs carrying weakened MHC-II binding epitopes were unable to control tumor growth. These data underscore the potential value of therapeutic targeting of HNSCC epitopes and highlight the importance of studying distinct antigen classes in this setting. Moreover, they raise the possibility that, at least in part, CD4+ T cell help is critical for cancer vaccination in this preclinical HNSCC model and suggest in silico prediction approaches prioritize overlapping MHC-I and MHC-II epitopes to generate potent cancer vaccines.
Introduction
The ultimate purpose of cancer vaccination, encompassing peptide, DNA, or RNA formats, is to improve clinical outcomes by enhancing adaptive immunity, including antigen-specific CD4+/ CD8+ T cells.Citation1 This antigenic stimulation is a crucial part of the cancer immunity cycle that eventually contributes to T cell trafficking and differentiation of antigen-specific T cells.Citation2,Citation3 Sources of antigens include tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs) including overexpressed proteins and endogenous retrovirus (ERV) encoded proteins. As CD8+ T cells are the major immune cell population that eradicate cancer cells, many studies have focused on the predicted affinity for major histocompatibility complex (MHC) class I to select candidate antigens for vaccine treatment.Citation4 Intriguingly, clinically responsive cases often display an increase of activated antigen-specific CD4+ T cells,Citation4 which is consistent with induction of CD4+ T cell-specific responses as a critical component of cellular immunity against cancer.Citation5,Citation6 Schreiber and colleagues showed a non-overlapping role for an integrated MHC-I and II response at the tumor site in the endogenous and checkpoint inhibitor treatment setting.Citation6 Further understanding of the interplay of MHC-I and II neoepitopes and CD4+ T cells in the cancer vaccination setting is required.
Head and neck squamous cell carcinoma (HNSCC) is the predominant cancer type in the head and neck region.Citation7,Citation8 Two phase III studies (Checkmate 141 and Keynote 048) led to approval of anti-PD-1 in recurrent and/or metastatic HNSCC treatment, underscoring the efficacy of immunotherapy.Citation9,Citation10 However, only 15–20% of patients benefit from these treatments, emphasizing the need for new immunotherapeutic methods. In addition, HNSCCs do not harbor identified frequent oncogenic driver mutationsCitation11 and exhibit a relatively high tumor mutational burdenCitation12,Citation13 and immune infiltration,Citation14 highlighting this tumor type as perhaps better suited for cancer vaccination therapy than oncogene targeting strategies. Previously, we developed mouse oral cancer (MOC) cell lines with similar characteristics to human HNSCC.Citation15 One of these, MOC22, showed high immunogenicity and represents a suitable model for evaluating HNSCC vaccine therapy strategies. Using an immunogenomic approach,Citation16 we previously selected TSAs as candidate neoepitopes and identified mutant intercellular adhesion molecule 1 (mICAM1)-derived MHC-I epitope reactivity in MOC22 tumor-infiltrating lymphocytes (TILs). Notably, a synthetic long peptide (SLP) with mICAM1 neoepitope conferred anti-cancer immunity in the preventive setting,Citation16 underscoring the efficacy of neoantigen cancer vaccination treatment for HNSCC.
SLPs containing MHC-I epitopes are an established vaccine format that can be efficiently processed by dendritic cells (DCs) and stimulate CD4+/CD8+ T cells.Citation17,Citation18 In this study, to extend our previous findings, we completed an analysis of two distinct MOC22-derived SLPs. The mICAM1 SLP had predicted high-affinity MHC-I and MHC-II epitopes. By contrast, the p15E SLP, derived from a p15E ERV transcript and often immunodominant in C57BL/6 derived tumors,Citation19,Citation20 only had predicted high-affinity MHC-I binding. Notably, although both SLPs demonstrated preventive anti-cancer efficacy, only the mICAM1 SLP showed a significant MOC22 tumor control activity in the therapeutic vaccine setting. Depletion assays using anti-CD40L antibody with mICAM1 SLP vaccination setting significantly attenuated anti-cancer effects, consistent with a crucial role of CD4+ T cells in therapeutic cancer vaccination. Furthermore, altered mICAM1 SLPs with predicted reduced MHC-II binding showed impaired tumor eradication capacity following therapeutic vaccination. These data in the MOC22 HNSCC model highlight considerations in vaccine candidate selection, the essential role of antigen-specific CD4+ T cell help for therapeutic cancer vaccination and define a strategy of selecting overlapping MHC-I and II neoepitopes for cancer vaccines.
Materials and methods
Cell culture and mice
MOC22 was generated and cultured as describedCitation16,Citation21 with MOC medium composed of IMDM modified (HyClone #SH30228.02), Ham’s F10-F12 nutrient mixture (HyClone #SH30026.01) with 5% heat-inactivated FBS (Sigma #F2442), 100 U/ml penicillin-streptomycin (Lonza #17-603E), 5 ng/ml EGF (Millipore #E5160), 400 ng/ml hydrocortisone (Sigma #H0135), and 5 μg/ml insulin (Sigma #I0516). Cell lines were tested for mycoplasma every 6 months. Female wild-type C57BL/6 mice (Taconic Biosciences, 6–8 weeks age) were housed in pathogen-free animal facility. All animal studies were conducted in compliance with regulations of the institutional animal care and use committee (IACUC) of Dana-Farber Cancer Institute.
Cancer cell inoculation and tumor size examination
MOC22 cells were cultured with 150 cm2 flasks with MOC medium. When harvesting, flasks were washed 2 times with 10 mL of PBS with 1 mM EDTA, then treated with 4 mL of 1 × 0.25% EDTA Trypsin (Gibco #25200-056) for 6–8 minutes at 37°C. After quickly neutralizing with 20 mL of MOC medium, MOC22 cells were harvested and washed 2 times with PBS, then resuspended in 100 μl PBS with Basement membrane extract (Thermo #343200501, 30% final) and were inoculated into mice flanks subcutaneously (150 ul/ flank in final). Mice flanks were shaved at least 3 d before the cell inoculation to prevent inflammation. 1 × 106 cells/flank were inoculated for prophylactic vaccination study according to the previous report.Citation16 More cells (5 × 106 cells/flank) were inoculated for therapeutic vaccination to more easily distinguish the effect of each vaccination. Inoculated cell numbers are shown in associated figure legends. All cells were at least 85% viable for in vivo experiments. Tumor volumes were determined 2–3 times per week using this formula: V = Length × Width × Height × π/6.Citation22 The human endpoint was set at 1000 mm3.
Peptide synthesis and vaccination
Peptides (Peptide 2.0) were dissolved in DMSO at 10 mM and stored at −20°C. For vaccination, 50 μg of each SLP and 100 μg of poly(I:C) (Invivogen #vac-pic) were mixed with 100 μl of sterile 0.9% NaCl physiological saline and injected subcutaneously at the tail base. Vaccine injection regions were shaved at least 3 d before the cell inoculation to prevent inflammation. The mICAM1 (TVYNFSAL (wild type ICAM1: TVYNFSAP)) and p15E (KSPWFTTL) MHC-I epitope peptides were used for ELISA and CD8+ ELISPOT analysis. The original mICAM1 SLP (DQILETQRTLTVYNFSALVLTLSQLEVS), altered-1 mICAM1 SLP (DQILETQRTLTVYNFSALVFFLSQLEVS), altered-2 mICAM1 SLP (DQILETQRTLTVYNFSALVDDLSQLEVS) SLP and p15E SLP (GLFNKSPWFTTLISTIMGPLIILLLILL) were used for CD4+ ELISPOT analysis and vaccination. Underlined sequences represent altered amino acids.
Tissue harvesting
MOC22 tumor processing: Flesh-harvested tumors were minced with razors and added to digestion buffer (D-PBS (Gibco #14190-136) with 100 U/ml penicillin-streptomycin, 100 × NEAA (LONZA #13-114E), 20 mM of HEPES (Gibco #15630-080), 100 × Glutamax (Gibco #35050-061) and 0.05 mM of 2-Mercaptoethanol (Sigma #M3148). Then, 1 mg of Liberase TL (Roche, #05401020001) was added, and the mixture incubated for 10 minutes at 37°C. Afterward, 2 mL of washing buffer (digestion buffer with 5% of FBS) was added to neutralize, and tumors were thoroughly minced and passed through 70 um of filter. Single-cell suspensions were analyzed using flow cytometry. Splenocyte processing: to obtain single-cell suspensions of fresh splenocytes, C57BL/6 mouse spleens were gently disaggregated with microscope glass slides in 3 mL of RPMI1640 (Gibco #11875-093). After centrifugation at 500 × g, 5 min 4°C, supernatant was removed. Then, 3 mL of red blood cell lysis buffer (Sigma #R7757) was added, and cells incubated for 3 min at RT. 15 mL of MOC medium was added to neutralize. After splenocytes were passed through 40 um mesh filters and resuspended with 20 mL of 1 mM EDTA PBS, they were counted and used for experiments. Draining lymph nodes (DLNs) processing: For harvesting DLNs, mice inguinal lymph nodes were disaggregated with microscope glass slides in 3 mL of RPMI1640 and passed through 40 um mesh filters. After spinning down and counting, cells were used for each study.
Tumor-infiltrating lymphocyte (TIL) culture
Freshly harvested tumor tissues were cut into 1–2mm pieces and individually placed into single wells of a 24-well dish in 1 mL of mouse T cell medium (RPMI1640, 10% FBS, 20 mM HEPES, 1 mM sodium pyruvate (Sigma #8636), 0.05 mM 2-mercaptoethanol, 2 mM L-glutamine (Gibco #25030-081), 100 ug/ml streptomycin, 100 U/ml penicillin and 100 IU/mL of human IL-2 (Roche TECIN #Ro 23–6019)). After 48 h of culture, TILs were harvested by passing through a 40 um filter, and dead cell removal was performed (Miltenyi #130-090-101). TILs were used for ELISPOT and flow cytometry experiments.
Mouse IFN-γ enzyme-linked immunospot (ELISPOT) assays
ELISPOT was performed using mouse IFN-γ single-color ELISPOT kit (CTL). Fresh splenocytes, untouched CD8+/CD4+ cells from splenocytes or cultured TILs were incubated at 37 degrees with 1 uM of each peptide for 24 h. When cancer cells were used as targets, MOC22 was stimulated with 1000 U/mL of recombinant mouse IFN-γ (Peprotech #315-05) for 48 h prior to use. PMA (Santa Cruz biotechnology #SC-3576, final concentration 2 μg/mL) and ionomycin (Santa Cruz biotechnology #SC-3592, final concentration 0.1 μg/mL) were used for positive control. Untouched CD4+/CD8+ T cells were isolated using CD4+/ CD8+ T cell isolation kits (Miltenyi #130-104-454, #130-104-075) with MS columns (Miltenyi #130-042-201) according to manufacturer instructions. Cell numbers used in each experiment are shown in associated figures, and naïve splenocytes from wild-type (WT) C57BL/6 female mice were used as antigen-presenting cells (APCs). The number of ELISPOT positive spots were counted using an Immunospot analyzer (CTL).
Mouse IFN-γ enzyme-linked immunosorbent (ELISA) assays
ELISA assays were performed with mouse IFN-γ quantikine ELISA kit (Bio-Techne #MIF00) according to manufacturer’s instructions. Freshly harvested mouse splenocytes (1 × 106 cells/well in 96 well plate) were mixed with 65 uL of T cell medium (without IL-2) added with 1 uM of mICAM1 (TVYNFSAL) or p15E (KSPWFTTL) MHC-I epitope peptides. When cancer cells were used as targets, IFN-γ stimulated MOC22 cells (1 × 105 cells/well in 96 well plate) were used. PMA (2 μg/mL) and ionomycin (0.1 μg/mL) were used for positive control. After 24-h culture at 37°C, cells were centrifuged in 2,000 × g for 5 min at 4°C and 50 ul of supernatant was used for ELISA assay. ELISA results were read at 450 nm and 570 nm using i-control 1.10 software and TECAN infinite M200 PRO. Using a standard curve, the IFN-γ concentration of samples were calculated. Technical duplicates were performed for each sample and averaged for analysis.
MHC-II epitope prediction
Peptide affinities for MHC-II were predicted with NetMHCII2.3 software.Citation23 Briefly, mICAM1 and p15E SLPs sequences were submitted to NetMHCII2.3, and affinity levels between 15 mer candidates from each SLP and H-2-IAb were analyzed. Epitopes of %Rank <2 defined strong binding candidates, and 2 ≤ %Rank ≤10 defined weak binding candidates.
Monoclonal antibody-based depletion assays
For CD8+/CD4+ depletion, monoclonal antibodies control rat IgG2A (250 ug, BioXCell Clone#2A3), anti-CD4 antibody (250 ug, BioXCELL Clone#GK1.5), or anti-CD8 antibody (250 ug, BioXCell Clone#YTS169.4)) were injected intraperitoneally on days −1, 6, 13, 20, 27 in the mICAM1 vaccinated setting. For CD40L blocking in vivo, control rat IgG2A (250 ug) or anti-CD40L antibody (250 ug, BioXCELL Clone#MR-1) was intraperitoneally injected on days −1, 6, 13 in the mICAM1 therapeutic vaccine setting and analyzed. Each treatment schedule is shown in associated figures.
Flow cytometry analysis
Single-cell suspension was generated (1 × 106 cells in PBS (100 ul)) from MOC22 tumors, spleens or DLNs of mice. Then, cells were first stained with Zombie aqua (Biolegend #423102, 1:500, 15 min at room temperature (RT) in PBS). After washing with 1 mL of PBS, cells were resuspended in FACS buffer (0.5% FBS and 1 mM of EDTA (Invitrogen #AM9260G) in PBS). Treatment with Fc-block (BD Pharmingen #553142, 1:100, 15 min at 4°C) was performed, then appropriate cell surface fluorophore-conjugated anti-mouse antibodies (CD45.2; clone 104, CD11c; clone N418, CD80; clone 16–10A1, CD86; clone GL-1, CD3e; clone 17A2, CD8a; clone 53–6.7, CD4; RM4-5, CD137; 17B5) were added at 1:200, incubated for 20 min at 4°C. Washing with 1 mL of FACS buffer was performed, and cells were analyzed by flow cytometry (Miltenyi MACSQuantX). FlowJo v10.6.2 (BD) software was used to analyze.
Tetramer staining
MHC-I specific mICAM1 (from the NIH Tetramer Core Facility) and p15E (MBL #TS-M507-1) tetramers were used to detect antigen-specific CD8+ TILs. After staining with Zombie Aqua, cultured TILs were treated with Fc-block, then tetramers (mICAM1, 1:100 and p15E, 3:100) were added for 30 min at 4°C. Subsequent fluorophore-conjugated anti-mouse antibodies staining (CD45.2; clone 104, CD8; clone KT15) was performed at 1:200 and analyzed. Compensation was performed using beads (Invitrogen #01-2222-42 for CD45.2/ CD8, and Invitrogen #A10346 for Zombie Aqua).
Statistical analysis
Before analysis, the Shapiro–Wilk normality test was performed. For samples with normal distributions, parametric tests (student t-test, one-way ANOVA) were performed. When the test of normality was rejected, non-parametric tests (Mann–Whitney U-test and Kruskal–Wallis test) were performed. The statistical method and sample numbers are shown in associated figure legends. A box-and-whisker plot was used to show distribution median and lower and upper quartiles. For survival analyses, log-rank (Mantel-Cox) test was performed. Statistical analyses were carried out using GraphPad Prism 8 software (GraphPad) with *p < .05, **p < .01, ***p < .001, ****p < .0001 and ns = not significant.
Results
mICAM1 SLP can activate both antigen-specific CD4+ and CD8+ T cells
We previously defined mICAM1 as a novel H2-Kb restricted neoantigen and successfully used a mICAM1 SLP in preventive vaccination against MOC22.Citation16 To extend these studies on the mICAM1 SLP and assess additional MOC22 vaccine targets, we compared endogenous T cell responses against two different antigens in progressively growing MOC22 tumors. We used a TSA-derived mICAM1 MHC-I epitope (TVYNFSAL) and a TAA-derived p15E MHC-I epitope (KSPWFTTL). Both peptides showed strong predicted affinity to MHC-I (mICAM1 = 2.45 nM and p15E = 10.48 nM in NetMHC4.0).Citation24,Citation25 ELISPOT and MHC-I tetramer staining of MOC22 TILs validated the existence of antigen-specific CD8+ T cells to both antigens () and (b), Supplementary Figure S1a). Notably, more p15E-specific IFN-γ producing CD8+ T cells were infiltrated in MOC22 relative to mICAM1-specific CD8+ T cells, suggesting the potential immunodominance of p15E-specific CD8+ T cells in MOC22 TILs () and (b)).
Figure 1. The mICAM1 and p15E SLPs show comparable reactivity against MOC22 tumor. (a) IFN-γ ELISPOT data for 5 × 104 MOC22 CD8+ TILs (day 14) cultured with mICAM1 (TVYNFSAL) or p15E (KSPWFTTL) short peptides. N = 3 for each. Quantification shows the number of positive spots in each group. Unstimulated MOC22 TILs were used for the negative control (NC,) and PMA/ionomycin stimulated MOC22 TILs were used for the positive control (PC). Mann–Whitney U test was performed to analyze. (b) Representative flow cytometry plots of mICAM1 and p15E MHC-I tetramer staining against MOC22 TILs (day 14). The gated areas show percentage of tetramer-positive cells in CD45+CD8+ TILs. FMO: fluorescence minus one. (c) MHC-II specific epitope prediction for mICAM1 and p15E SLPs. Predictions were performed using NetMHCII 2.3 software. Pos: position, SB: strong binding candidate, WB: weak binding candidate. Underlined sequences represent predicted MHC-I epitopes. (d) Schematic of vaccine schedule. Each vaccine was performed on days −7, −5, and splenocytes were harvested on day 0. IFN-γ stimulated (100 U/mL, 48 h) 1 × 105 MOC22 were mixed with (e) 5 × 105 (ELISPOT) or (f) 1 × 106 (ELISA) splenocytes per well from vaccinated WT mice and cultured for 24 h. VAC: vaccination. (E) IFN-γ ELISPOT data (n = 5 mice for each group). Quantification shows the number of positive spots in each group. one-way ANOVA was performed for multiple analyses. (F) IFN-γ ELISA assay data (n = 5 mice for each group). Kruskal–Wallis test was performed for multiple analyses
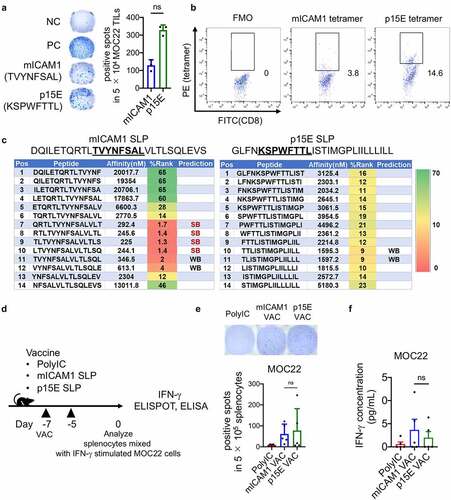
Next, to analyze the effect of MHC-II epitopes for anti-tumor response, we generated 28-mer mICAM1 and p15E SLPs ()). We used the same mICAM1 SLP sequence as in our previous report.Citation16 Predicted affinity levels for MHC-II H-2-IAb showed mICAM1 SLP to have four strong and two weak MHC-II predicted neoepitope candidates incorporating the mutated residue, while p15E SLP had two weak MHC-II candidates ()).
Differential loading and presentation of these two antigens on Class I may impact anti-tumor immune responses. To assess the presentation of mICAM1 and p15E epitopes on MOC22 tumor cells, we first generated antigen-specific T cells by separately vaccinating WT mice with each SLP with poly(I:C) adjuvant. Splenocytes from these mice were then used as a source for antigen-specific T cells. IFN-γ production following mixed culturing of MOC22 with splenocytes from either mICAM1 or p15E vaccinated mice was measured using ELISPOT assays and ELISAs ()). IFN-γ secretion was similar for the two peptide vaccines () and €). These data confirm the presence of cell surface mICAM1- and p15E-Class I complexes and suggest that the expression levels may be comparable.
Antigen-specific CD8+ T cells activation is sufficient for preventive vaccination efficacy against MOC22
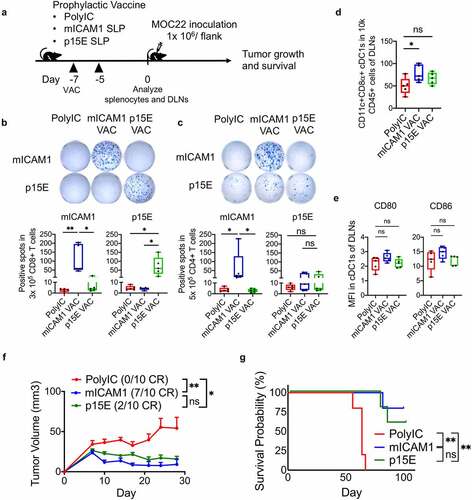
To examine the immunogenicity of these SLPs, subcutaneous vaccination was performed in WT mice with a prophylactic vaccine protocol (vaccinated on days −7 and −5) ()). IFN-γ ELISA assay (day 0) showed the increase of IFN-γ after each SLP vaccination (Supplementary Figure 2(a)). IFN-γ ELISPOT assay demonstrated a significant increase of IFN-γ producing antigen-specific CD8+ T cells in both vaccinated groups () and Supplementary Figure S2b), showing that both mICAM1 and p15E SLPs are immunogenic. Notably, mICAM1 SLP significantly induced IFN-γ producing antigen-specific CD4+ T cells () and Supplementary Figure S2c), confirming that mICAM1 SLP activated both CD8+ and CD4+ T cells while p15E SLP activated CD8+ T cells only. To examine the impact of each SLP vaccine on dendritic cells (DCs), we examined conventional type 1 dendritic cells (cDC1s) and co-stimulatory factors (CD80 and CD86) after prophylactic vaccination. Notably, the mICAM1 SLP vaccine induced a significant increase of cDC1s in DLNs () and Supplementary Figure S2d), suggesting the efficient promotion of antigen-presentation via cDC1s. The CD80 and CD86 co-stimulatory factors in cDC1s were not statistically significantly increased in the mICAM1 SLP vaccinated condition ()). Next, MOC22 cells were inoculated on day 0 to analyze the efficacy of mICAM1 and p15E SLP prophylactic vaccination ()). The mICAM1 SLP vaccinated group significantly suppressed MOC22 tumor formation, and 7/10 tumors were completely rejected ()) with significantly prolonged survival ()), which was consistent with previous findings.Citation16 Notably, the p15E SLP also showed significant suppression of tumor growth and prolonged survival () and (g)), but only 2/10 tumors were completely rejected ()). In addition, re-challenge experiments of MOC22 in the mice with completely rejected tumors were performed. All of 7/7 mice of mICAM1 vaccinated mice and 2/2 mice of p15E SLP vaccinated mice completely rejected MOC22 tumor re-challenge (data not shown), indicating the formation of an immune memory response in mice that initially rejected MOC22. These data suggest that activating either p15E or mICAM1 antigen-specific CD8+ T cells is sufficient to suppress MOC22 tumor growth in the prophylactic vaccine setting.
Figure 2. The mICAM1 and p15E SLPs both show preventive anti-cancer effects against MOC22. (a) Schematic of prophylactic vaccination schedule. Each vaccine was performed on days −7, −5, and 1 × 106 of MOC22 cells were inoculated on day 0. VAC: vaccination, DLNs: draining lymph nodes. (b-c) IFN-γ ELISPOT analysis of (b) purified 3 × 105 CD8+ and (c) purified 5 × 105 CD4+ T cells per well from vaccinated WT splenocytes (n = 5 mice for each group). Quantification shows the number of spots in each group. Kruskal–Wallis test was performed for analyses. Short peptides (mICAM1:TVYNFSAL or p15E:KSPWFTTL) were used for stimulating (B) CD8+ T cells, SLPs (mICAM1:DQILETQRTLTVYNFSALVLTLSQLEVS or p15E: GLFNKSPWFTTLISTIMGPLIILLLILL) were used for stimulating (C) CD4+ T cells. The underlined sequences represent MHC-I epitopes of SLPs. (d) Flow cytometry analysis for CD45+CD11c+CD8α+ cDC1s in draining lymph nodes (DLNs) (n = 5 mice for each group). One-way ANOVA test was performed to analyze. (e) Mean fluorescent intensity (MFI) of CD80 and CD86 of cDC1s in DLNs from vaccinated mice (n = 5 mice for each group). Kruskal–Wallis tests were used for multiple analyses. (f) Tumor volumes in prophylactic poly(I:C) (control) and mICAM1 or p15E SLPs vaccinated groups (n = 10 tumors for each group). Data was analyzed with one-way ANOVA. CR: complete response. (g) Kaplan–Meier survival analysis of each prophylactic vaccinated group (n = 5 mice per group)
Therapeutic mICAM1 but not p15E vaccination of MOC22 bearing mice activates antigen-specific CD4+/CD8+ T cells and prolongs survival
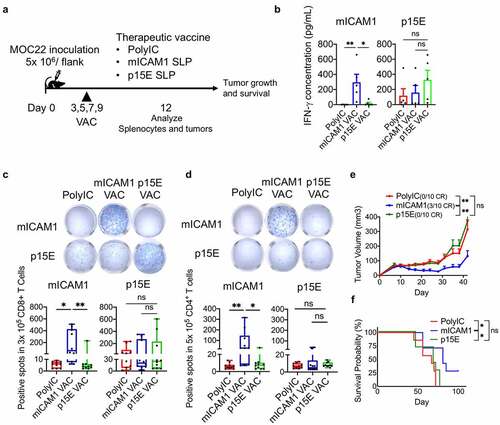
Next, we extended our analysis to therapeutic vaccination in established MOC22 tumor-bearing mice ()). ELISA (day 12) confirmed both SLPs promoted IFN-γ secretion from antigen-specific T cells ()). As expected, mICAM1 SLP significantly activated CD4+ and CD8+ T cells in IFN-γ ELISPOT assay performed on day 12 () and (d)). By contrast, p15E SLP therapeutic vaccination was unable to induce CD4+ T cell activation in tumor-bearing mice ()), while CD8+ T cell activation was observed ()). Notably, in the therapeutic vaccine setting, mICAM1 SLP displayed significant tumor growth control relative to p15E SLP ()). Three of 10 tumors were completely rejected with mICAM1 vaccination, while none of 10 tumors were rejected with p15E vaccine or poly(I:C) control group, respectively ()). In addition, the mICAM1 vaccinated group significantly prolonged overall survival ()), highlighting the efficacy of mICAM1 SLP vaccination. We hypothesized that these results are related to a supportive role of the mICAM1 Class II epitope and CD4+ T cell help being critical to reject MOC22 in the therapeutic vaccination setting.
Figure 3. The mICAM1 SLP vaccination showed more significant therapeutic vaccination impact on MOC22 tumors than p15E SLP vaccine. (a) Schematic of therapeutic vaccination protocol. Each vaccine was performed on days 3,5,7,9, and 5 × 106 of MOC22 were inoculated on day 0. VAC: vaccination. (b) IFN-γ ELISA assay for splenocytes harvested on day 12 using therapeutic vaccine protocol stimulated with mICAM1 (TVYNFSAL) and p15E (KSPWFTTL) short peptides for 24 h before analysis (n = 5 mice per group). Kruskal–Wallis tests were performed. (c-d) IFN-γ ELISPOT assays for purified (c) 3 × 105 CD8+ or (d) 5 × 105 CD4+ T cells per well from splenocytes of vaccinated MOC22 tumor-bearing mice. T cells were stimulated with each peptide (for CD8+ T cells; mICAM1:TVYNFSAL or p15E:KSPWFTTL, for CD4+ T cells; mICAM1:DQILETQRTLTVYNFSALVLTLSQLEVS or p15E: GLFNKSPWFTTLISTIMGPLIILLLILL). The Underlined sequences represent MHC-I epitopes in SLPs. N = 10 mice per group were used in two independent experiments. Quantification shows the ELISPOT positive numbers of each group. Kruskal–Wallis tests were performed for multiple analyses. (e) Tumor volumes in poly(I:C) (control) and mICAM1 or p15E SLPs vaccinated mice (n = 10 tumors for each group). Data were analyzed with one-way ANOVA. CR: complete response. (f) Kaplan–Meier survival analysis of each therapeutic vaccinated group (n = 7 mice per group)
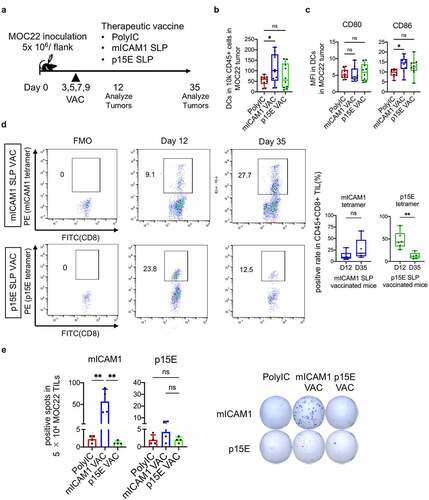
Therapeutic mICAM1 SLP vaccination suppresses tumor growth with sustained CD8+ T cell IFN-γ production
To connect the activation of antigen-specific CD4+ T cells in the MOC22 anti-tumor mICAM1 vaccine response, we next evaluated the MOC22 tumor microenvironment (TME) using the therapeutic vaccination protocol ()). Both DCs and CD86 expression on DCs in MOC22 tumors were significantly increased in the mICAM1 SLP vaccinated group on day 12 () and (c), Supplementary Figure S3a). We did not see a significant increase of CD8+ T cells or the T cell activation marker 4–1BB in the mICAM1 vaccinated condition (Supplementary Figure S3b and d). Notably, tetramer staining indicated the existence of antigen-specific T cells for both p15E and mICAM1 in MOC22 TILs in later stages of tumor growth (day 35). Mutant ICAM1 tetramer-positive cells averaged 26.9% of MOC22 CD8+ TILs and p15E tetramer-positive cells averaged 12.59% of MOC22 CD8+ TILs ()). However, mICAM1-specific TILs exhibited sustained IFN-γ production while p15E-specific TILs did not ()). In addition, mICAM1 tetramer-positive CD8+ TILs were durably maintained on day 35, while p15E tetramer-positive CD8+ TILs were significantly decreased ()). Although there are several explanations for why the p15E SLP was not as active therapeutically, these data indicate that mICAM1-specific T cells in mICAM1 SLP vaccinated mice have a long-lasting anti-cancer effect in MOC22 tumor that likely contributes to tumor control.
Figure 4. The mICAM1 SLP vaccination showed robust anti-cancer efficacy through prolonged IFN-γ production. (a) Schematic of therapeutic vaccination. MOC22 Tumors were analyzed on day 12 and 35. VAC: vaccination. (b) Flow cytometry analysis for CD45+CD11c+DCs in MOC22 tumors on day 12 (n = 10 tumors for each group). One-way ANOVA test was performed for analysis. (c) Mean fluorescent intensity (MFI) for CD80/ CD86 of DCs in MOC22 tumors on day 12 (n = 10 tumors for each group). One-way ANOVA tests were performed for analysis. (d) The mICAM1 and p15E MHC-I tetramer-positive rate in vaccinated CD45+CD8+ MOC22 TILs on day 12 (n = 6 for each group) and day 35 (n = 5 for each group) from therapeutic vaccinated mice and representative flow cytometry plots. Data were analyzed with Mann–Whitney U test. (e) ELISPOT assay of MOC22 TILs from therapeutic vaccinated mice on day 35. TILs were collected from 5 mice for each group, then divided into 5 × 104 each well (n = 4 for each group) and stimulated with mICAM1 (TVYNFSAL) and p15E (KSPWFTTL) short peptides
CD8+ and CD4+ T cells synergistically contribute to immunotherapy efficacy in MOC22
To examine the contribution of CD4+ T cells for anti-tumor immunity in the therapeutic vaccine setting, monoclonal antibody-based depletion was performed concurrently with mICAM1 vaccination ()). Depleting CD4+ T cells with vaccination restored tumor growth, but these data were not significant ()). Notably, depletion of both CD4+ and CD8+ T cells showed accelerated tumor growth ()), suggesting CD4+ and CD8+ T cells work synergistically in mICAM1 vaccinated mice.
Figure 5. CD4+ and CD8+ T cells are both required for therapeutic MOC22 control. (a) A schematic illustration of CD4 or CD8 depletion in mICAM1 SLP vaccinated mice. Depletion antibodies were injected on days −1,6,13,20,27 and vaccines were performed on days 3,5,7,9. 5 × 106 of MOC22 were inoculated on day 0. VAC: vaccination. (b) Average MOC22 tumor volumes in IgG plus poly(I:C), IgG plus mICAM1, anti-CD4 plus mICAM1, anti-CD8 plus mICAM1 and anti-CD4/CD8 plus mICAM1 conditions (n = 10 tumors per group). Data were analyzed with one-way ANOVA. (c) A schematic illustration of CD40L depletion in mICAM1 SLP vaccinated mice. Depletion antibodies were injected on days −1,6,13 and vaccines were performed on days 3,5,7,9. 5 × 106 of MOC22 were inoculated on day 0. VAC: vaccination. (d) Average MOC22 tumor volumes in IgG plus poly(I:C), IgG plus mICAM1, anti-CD40L plus poly(I:C), anti-CD40L plus mICAM1 (n = 10 tumors per group). Data were analyzed with one-way ANOVA. CR: complete response. (e) ELISPOT assay of day 35 MOC22 TILs from IgG plus poly(I:C), anti-CD40L plus poly(I:C), IgG plus mICAM1, anti-CD40L plus mICAM1 treated mice. TILs were collected from 5 mice for each group, then divided into 5 × 104 each well. TILs were stimulated with mICAM1 (TVYNFSAL) peptide
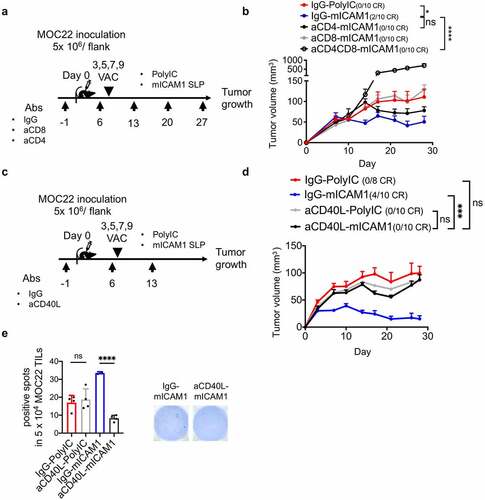
As a more robust phenotype with CD4+ depletion was not seen, we reasoned that depletion of regulatory T cells (Tregs) in addition to helper T cells might have been a limitation. To further investigate this CD4+ cellular contribution, we next examined whether blocking CD40L signaling along with the mICAM1 SLP vaccine would highlight the impact of CD4+ T cells (especially type 1 helper T cells) ()).Citation5,Citation26 Strikingly, CD40L depletion significantly recovered MOC22 tumor volume in the mICAM1 vaccinated condition, underscoring the critical role of CD4+ T cells ()). TIL harvested from tumors in these individual conditions showed significantly decreased ELISPOT activity only in the mICAM1 SLP vaccinated with CD40L blocking condition ()). These data confirm CD4+ T cell importance for mICAM SLP therapeutic vaccination.
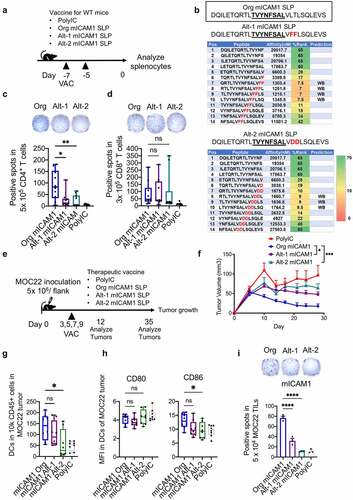
The mICAM1-specific MHC-II epitope is indispensable for anti-cancer immunity in MOC22
Finally, to dissect the contribution of the Class II epitope in mICAM1 SLP, we mutated the predicted MHC-II motif in mICAM1 SLP with an altered mICAM1 SLP sequence and importantly preserved the MHC-I motif ()). The altered mICAM1 SLPs (Alt-1 mICAM1 SLP and Alt-2 mICAM1 SLP) lost predicted MHC-II strong binding epitopes ()). We then analyzed CD4+/ CD8+ T cell responses after vaccination of each SLP in wild-type mice ()). Notably, antigen-specific CD4+ T cell responses were significantly disrupted in Alt-1 or Alt-2 mICAM1 SLPs while antigen-specific CD8+ responses were preserved ()). The MHC-II epitope disruption failed to attenuate tumor progression in therapeutic vaccinations of Alt-1 or Alt-2 mICAM1 SLPs, highlighting the critical importance of antigen-specific CD4+ T cells to induce robust anti-cancer immunity ( and (f)). The DC infiltration and CD86 co-stimulatory factor expression in MOC22 tumors were impaired in the Alt-2 mICAM1 SLP vaccinated group compared to Org mICAM1 vaccinated group ()). Furthermore, long-lasting IFN-γ production capacity was disrupted in Alt-1 or Alt-2 mICAM1 SLPs vaccinated TILs on day 35 ()), consistent with a role for mICAM1 antigen-specific CD4+ T cells in supporting an efficacious, more durable anti-cancer CD8+ T cell response.
Figure 6. The mICAM1 specific MHC-II epitopes are indispensable for anti-cancer immunity in MOC22. (a) A schematic schedule of vaccination protocol for WT mice. VAC: vaccination. Org: original, Alt: altered. (b) Altered mICAM1 SLPs sequence and predicted affinity level with MHC-II using NetMHCII 2.3 software. Amino acids in red were altered. The underlined sequence represents MHC-I epitope. (c-d) ELISPOT quantification showing the number of IFN-γ positive spots of mICAM1-specific CD4+/CD8+ T cells from splenocytes of each vaccinated mouse (n = 10 mice for each group from two independent experiments). One-way ANOVA was used for analysis. (c) Purified 5 × 105 CD4+ T cells per well were stimulated with Org mICAM1 SLP (DQILETQRTLTVYNFSALVLTLSQLEVS). (d) Purified 3 × 105 CD8+ T cells per well were stimulated with mICAM1 short peptide (TVYNFSAL). (e) Schematic of therapeutic vaccination schedule with Org mICAM1, Alt-1 mICAM1, and Alt-2 mICAM1 SLPs. 5 × 106 of MOC22 were injected on day 0. VAC: vaccination. (f) Average tumor volumes in poly(I:C) control, Org mICAM1, Alt-1 mICAM1, and Alt-2 mICAM1 SLPs vaccinated MOC22 tumor-bearing mice, respectively (n = 8 tumors poly(I:C), n = 10 tumors in other groups). Tumor volumes were analyzed with one-way ANOVA. (g) The number of CD45+CD11c+ DCs in MOC22 tumors on day 12 (n = 8 tumors for each group). (h) The mean fluorescent intensity (MFI) of CD80/CD86 in DCs in MOC22 tumors on day 12 (n = 8 tumors for each group). (i) Representative ELISPOT image of MOC22 TILs from vaccinated mice on day 35 and quantitation (Triplicate assay from mixed TILs in 5 mice for each group). TILs were stimulated with mICAM1 (TVYNFSAL) peptide. One-way ANOVA was performed to analyze
Discussion
The robustness of personalized cancer vaccination therapy is greatly influenced by antigen selection.Citation27,Citation28 Use of SLPs that includes predicted high-affinity MHC-I epitopes is an established format for cancer vaccination.Citation18 In addition to TSAs from somatically mutated proteins, TAAs from over-expressed proteins or from ERVs constitute additional targets. As the MOC22 model contained both a defined mICAM1 neoantigen and an ERV p15E derived antigen, we aimed to define vaccine responses to both these types of antigens. In addition, we were able to examine the integration of CD4+ T cell help in therapeutic vaccination of the MOC22 HNSCC model. Several recent studies have highlighted the essential function of antigen-specific CD4+ T cells to support priming, memory T cell formation, and maintenance of cytotoxic CD8+ T cells.Citation5,Citation6 We evaluated the mICAM1 and p15E high-affinity MHC-I antigens and found that they were immunogenic and conferred prophylactic tumor control on MOC22, highlighting the central role of antigen-specific CD8+ T cells for cancer vaccination. Notably, mICAM1 SLP vaccine more efficaciously repressed MOC22 tumor growth than p15E SLP in prophylactic and therapeutic settings. In contrast to the p15E SLP, mICAM1 SLP contained predicted strong MHC-II epitopes and was able to increase IFN-γ producing mICAM1-specific CD4+ T cells in vivo, which supported mICAM1-specific CD8+ T cells to exhibit durable IFN-γ production in MOC22 tumors. These results are consistent with the stabilizing influence of CD4+ T cell help in functional enhancement of the mICAM1 antigen-specific CD8+ T cells. These findings are also consistent with recent findings regarding the critical role of CD4+ T cell help in therapeutic cancer immunity.Citation6,Citation18,Citation29,Citation30 Considering that conventional type 1 dendritic cells (cDC1s) process SLPs and present epitopes to activate both CD8+ and CD4+ T cells,Citation5,Citation31 vaccine platforms that encode both MHC-I and MHC-II epitopes represent an ideal method to induce durable anti-cancer immunity via effective activation of both cell types. In particular, our findings emphasize the integration of predicted high-affinity and mutant-derived MHC-I and MHC-II epitopes within the same SLP as an efficient method to induce potent cellular immunity. However, as in silico prediction of MHC-II strong epitope candidates is more complex than MHC-I prediction because of the open structure of MHC-II binding groove,Citation6,Citation30 improved methods to predict MHC-II epitopes are needed.Citation32
There are two major limitations of this study. First, mICAM1 and p15E are different kinds of antigens, which may provoke different anti-tumor effects. We clearly showed mICAM1 SLP to have strong MHC-I and II epitopes while p15E SLP has only strong MHC-I epitopes. ELISPOT and ELISA with mICAM1- and p15E-antigen specific T cells mixed with MOC22 showed that both these antigen specific T cells produced similar IFN-γ levels, suggesting antigen expression/presentation of these two antigens on MOC22 are comparable. Limitations include that we are not currently able to phenotype antigen-specific cells for these two targets directly in tumors. In addition, the lack of therapeutic efficacy of the p15E SLP may be related to the different antigen source. As the p15E antigen is a TAA antigen, it may be subject to central or peripheral tolerance.Citation33 However, therapeutic responses to p15E were seen with B16 melanoma where CD40 ligand pretreated, p15E-pulsed DCs inhibited established lung metastases.Citation34 A recent clinical trial where an RNA vaccine incorporating four distinct TAA combined with heterologous MHC-II epitopes induced robust and durable anti-cancer immunity in melanoma patients.Citation35 These data suggest the presence of MHC-II epitopes may overcome central/peripheral tolerance. The MOC22 model may allow testing of approaches to increase p15E TAA-specific responses.
To further strengthen the importance of mICAM1 antigen-specific CD4+ T cells, we performed CD4+ T cell depletion and partially restores tumor growth but these data were not significant. One likely explanation for these results is that our approach simultaneously depleted CD4+ helper and regulatory T cells (Tregs). To directly address the CD4+ T cell and DC interaction, we performed additional experiments with a monoclonal antibody blocking CD40L in mICAM1 therapeutic vaccinated condition.Citation5,Citation26 Strikingly, CD40L blockade significantly recovered MOC22 tumor volume, and significantly decreased mICAM1-specific IFN-γ producing T cells in the MOC22 tumor. These data underscore the critical function of CD4+ T cells (mainly type 1 helper T cells) to augment mICAM-1 specific responses. In addition, we synthesized the altered mICAM1 SLPs with reduced predicted MHC-II binding to directly elucidate the importance of mICAM1 antigen-specific CD4+ T cells. The altered mICAM1 SLPs lost the capacity to suppress MOC22 tumor growth, confirming the crucial function of antigen-specific CD4+ T cells, especially for durable anti-cancer immunity.
The second limitation of our study is that although mICAM1 SLP plus poly(I:C) adjuvant could largely protect against MOC22 tumor formation in the prophylactic setting, only 3/10 tumors were rejected in the therapeutic setting. These results demonstrated that an optimized SLP with overlapping MHC-I and MHC-II neoantigens plus poly(I:C) was still insufficient to completely eradicate tumors. Note, we did not combine our vaccines with anti-PD1 blockade as MOC22 is rejected with checkpoint blockade alone.Citation16 Evaluating other methods, including vaccination using multiple overlapping MHC-I and MHC-II neoepitopes SLPs, selecting different adjuvants, or combination therapies such as checkpoint inhibitors to augment vaccine effects will be important for translating preclinical findings to clinical implementation.Citation4,Citation6,Citation29,Citation30,Citation36,Citation37
This MOC22 model may be used to dissect the role of tumor-specific versus heterologous MHC-II epitopes and CD4+ T cell help. CD4+ type 1 helper T cells (Th1s) produce cytokines such as IFN-γ, IL-2, and IL-21,Citation38,Citation39 all of which support cytotoxic CD8+ T cells. In support of enhanced T cell anti-tumor activity via heterologous CD4+ T cell stimulation, one study showed that a heterogenous CD4 neoantigen RNA vaccine reinforced activation of radiation-induced gp70 TAA-specific CD8+ T cells,Citation40 and an independent study demonstrated tetanus toxin-derived universal helper epitope improved therapeutic responses against MHC-I neoepitopes.Citation26 Together, these results raise the important point that heterologous MHC-II epitopes can activate CD4+ T cell help to complement tumor antigen-specific CD8+ T cell responses. Thus, evaluating whether endogenous overlapping or heterologous MHC-II epitopes are similar or different in their contribution to cancer vaccination will be important.
In conclusion, we extended our previous findings on MOC22 SLP vaccination by identifying that MHC-I and MHC-II overlapping neoepitopes induce robust anti-cancer impact in the MOC22 cancer vaccination model. These data highlight that prioritizing overlapping MHC-I and MHC-II epitopes in therapeutic cancer vaccines may represent an important approach.
Author Contribution
HS designed and performed all experiments, analyzed data, and wrote the manuscript. NX performed experiments and edited the manuscript. SS, FC, and OI performed experiments. JW supported experiments. LZ and PZ provided technical and experimental suggestions. AM provided technical and experimental suggestions, and supervised statistical analysis. RU supervised study design and drafted the document. All authors read and approved the final manuscript.
Availability of data and material
All relevant data is included in the manuscript.
Abbreviations
Supplemental Material
Download ()Acknowledgments
We are grateful to J.Baginska and S.Mariano in the Center for Immune-Oncology (CIO) of DFCI for assistance with ELISPOT analysis.
Disclosure statement
RU serves on a Merck scientific advisory board. The MOC models developed by RU have been filed with the Washington University Office of Technology Management and are currently licensed for distribution by Kerafast.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–13.
- Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221.
- Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635–647.
- Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574(7780):696–701.
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
- Chaturvedi AK, Anderson WF, Lortet-Tieulent J, Curado MP, Ferlay J, Franceschi S, Rosenberg PS, Bray F, Gillison ML. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol. 2013;31:4550–4559.
- Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867.
- Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, De Castro G Jr., Psyrri A, Basté N, Neupane P, Bratland Å, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394(10212):1915–1928.
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582.
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339.
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–2608.
- Mandal R, Şenbabaoğlu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, Lee KW, Ganly I, Hakimi AA, Chan TA, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1(17):e89829.
- Campbell KM, Lin T, Zolkind P, Barnell EK, Skidmore ZL, Winkler AE, Law JH, Mardis ER, Wartman LD, Adkins DR, et al. Oral cavity squamous cell carcinoma xenografts retain complex genotypes and intertumor molecular heterogeneity. Cell Rep. 2018;24(8):2167–2178.
- Zolkind P, Przybylski D, Marjanovic N, Nguyen L, Lin T, Johanns T, Alexandrov A, Zhou L, Allen CT, Miceli AP, et al. Cancer immunogenomic approach to neoantigen discovery in a checkpoint blockade responsive murine model of oral cavity squamous cell carcinoma. Oncotarget. 2018;9(3):4109–4119.
- Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW, Silva AL, Jiskoot W, Van Hall T, Van Veelen PA, et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur J Immunol. 2013;43(10):2554–2565.
- Melief CJ, van Hall T, Arens R, Ossendorp F, Van Der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015;125:3401–3412.
- Friedman J, Moore EC, Zolkind P, Robbins Y, Clavijo PE, Sun L, Greene S, Morisada MV, Mydlarz WK, Schmitt N, et al. Neoadjuvant PD-1 immune checkpoint blockade reverses functional immunodominance among tumor antigen-specific T cells. Clin Cancer Res. 2020;26(3):679–689.
- Ye X, Waite JC, Dhanik A, Gupta N, Zhong M, Adler C, Malahias E, Ni M, Wei Y, Gurer C, et al. Endogenous retroviral proteins provide an immunodominant but not requisite antigen in a murine immunotherapy tumor model. Oncoimmunology. 2020;9(1):1758602.
- Judd NP, Allen CT, Winkler AE, Uppaluri R. Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol Head Neck Surg. 2012;147:493–500.
- Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154.
- Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154:394–406.
- Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017.
- Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–517.
- Swartz AM, Congdon KL, Nair SK, Li QJ, Herndon JE 2nd, Suryadevara CM, Riccione KA, Archer GE, Norberg PK, Sanchez-Perez LA, et al. A conjoined universal helper epitope can unveil antitumor effects of a neoantigen vaccine targeting an MHC class I-restricted neoepitope. NPJ Vaccines. 2021;6(1):12.
- Jou J, Harrington KJ, Zocca MB, Ehrnrooth E, Cohen EEW. The changing landscape of therapeutic cancer vaccines-novel platforms and neoantigen identification. Clin Cancer Res. 2021;27:689–703.
- Lam H, McNeil LK, Starobinets H, DeVault VL, Cohen RB, Twardowski P, Johnson ML, Gillison ML, Stein MN, Vaishampayan UN, et al. An empirical antigen selection method identifies neoantigens that either elicit broad antitumor T-cell responses or drive tumor growth. Cancer Discov. 2021;11(3):696–713.
- Dey S, Sutanto-Ward E, Kopp KL, DuHadaway J, Mondal A, Ghaban D, Lecoq I, Zocca MB, Merlo LMF, Mandik-Nayak L, et al. Peptide vaccination directed against IDO1-expressing immune cells elicits CD8(+) and CD4(+) T-cell-mediated antitumor immunity and enhanced anti-PD1 responses. J Immunother Cancer. 2020;8:2.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696.
- Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson JTT, Bagadia P, Liu T, Briseño CG, et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature. 2020;584(7822):624–629.
- Abelin JG, Harjanto D, Malloy M, Suri P, Colson T, Goulding SP, Creech AL, Serrano LR, Nasir G, Nasrullah Y, et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. 2019;51(4):766–779.e717.
- Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135–146.
- Kershaw MH, Hsu C, Mondesire W, Parker LL, Wang G, Overwijk WW, Lapointe R, Yang JC, Wang RF, Restifo NP, et al. Immunization against endogenous retroviral tumor-associated antigens. Cancer Res. 2001;61(21):7920–7924.
- Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, Maurus D, Schwarck-Kokarakis D, Kuhn AN, Omokoko T, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585(7823):107–112.
- Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67–73.
- Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, Lin JJ, et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell. 2020;183(2):347–362.e324.
- Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021;28:5–17.
- Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, Cui W. CD4(+) T cell help is required for the formation of a cytolytic CD8(+) T cell subset that protects against chronic infection and cancer. Immunity. 2019;51:1028–1042.e1024.
- Salomon N, Vascotto F, Selmi A, Vormehr M, Quinkhardt J, Bukur T, Schrörs B, Löewer M, Diken M, Türeci Ö, et al. A liposomal RNA vaccine inducing neoantigen-specific CD4(+) T cells augments the antitumor activity of local radiotherapy in mice. Oncoimmunology. 2020;9(1):1771925.