
ABSTRACT
Heat shock protein 70 (Hsp70), a protein chaperone, is known to promote cell survival and tumor progression. However, its role in the tumor microenvironment (TME) is largely unknown. We specifically evaluated Hsp70 in the TME by implanting tumors in wild-type (WT) controls or Hsp70−/- animals, thus creating a TME with or without Hsp70. Loss of Hsp70 led to significantly smaller tumors; there were no differences in stromal markers, but interestingly, depletion of CD8 + T-cells abrogated this tumor suppressive effect, indicating that loss of Hsp70 in the TME affects tumor growth through the immune cells. Compared to WT, adoptive transfer of Hsp70−/- splenocytes exhibited greater antitumor activity in immunodeficient NSG and Rag 1−/- mice. Hsp70−/- dendritic cells showed increased expression of MHCII and TNF-α both in vitro and in vivo. These results suggest that the absence of Hsp70 in the TME inhibits tumors through increased dendritic cell activation. Hsp70 inhibition in DCs may emerge as a novel therapeutic strategy against pancreatic cancer.
Introduction
Patients with pancreatic ductal adenocarcinoma (PDAC) show resistance to conventional as well as novel therapies contributing to dismal outcomes. For instance, while recent advances in immunotherapy have improved prognosis of aggressive malignancies like advanced melanoma and lung cancer, studies looking at the effectiveness of these approaches in PDAC have been largely disappointing.Citation1 One of the reasons for the poor response of pancreatic cancer to various therapies is believed to be its complex tumor microenvironment (TME). Pancreatic cancer has an intricate TME composed of a variety of cell types, including cancer associated fibroblasts (CAFs), immune cells and endothelial cells, among others.Citation2 Several studies have highlighted the role of various cellular and acellular components of TME in promoting cancer progression and metastases. For instance, CAFs have been shown to stimulate the growth and progression of pancreatic cancer cells.Citation3 Even the noncellular ECM, which is largely laid down by CAFs, has been shown to inhibit delivery of chemotherapeutic agents in pancreatic cancer.Citation4 It appears that there is an ever-evolving cross talk between the cancer cells and TME, where the cancer cells modulate the surrounding tumor microenvironment to acquiesce to its increased metabolic and immunodefensive need.Citation5 We have previously shown that several molecules and pathways in the TME are responsible for mediating resistance to chemotherapy.Citation6 In addition, studies from our group as well as other laboratories have shown that TME dysregulates anticancer immunity to enable cancer growth.Citation7–10 A better understanding of the pathways in TME remodulation that induce resistance to various therapies will help us develop novel therapeutic strategies to overcome it, improving outcomes of patients suffering from pancreatic cancer.
Heat shock proteins are part of an evolutionarily conserved cellular machinery, which are geared toward protecting cells and tissues from various stresses, including thermal distress.Citation11 Heat Shock Protein 70, or Hsp70, is a member of heat shock protein family, which is ubiquitously expressed in a variety of cell types.Citation12 We have previously demonstrated that Hsp70 is overexpressed in pancreatic cancer cells and that it plays a prosurvival and antiapoptotic role in pancreatic cancer epithelial cells. However, the role of Hsp70 in the TME is unknown.Citation13 In the current study, we have investigated the role of Hsp70 in TME in the progression of cancer. Our results suggest that selective genetic deletion of Hsp70 in the TME significantly attenuates tumor growth. Our results also suggest that this effect is due to the deletion of Hsp70 in immune cellsand not due to depletion of Hsp70 in CAFs. Using a combination of in vitro and in vivo approaches, we demonstrate that lack of Hsp70 in dendritic cells energizes the antigen presentation machinery, which, in turn, leads to the development of a robust anticancer immune response. These findings pave the way for a more complete understanding in modulating and designing effective therapeutic approaches that can complement immunotherapy and dendritic cell vaccination against pancreatic cancer.
Methods
All animal experiments were authorized and overseen by the Institutional Animal Care and Use Committee (IACUC) in accordance with approved protocol.
In vitro methods
Primary cell isolation
Tumor-bearing KPC ((B6.LSL-KrasGCitation12D and p53R127H Pdx-cre) or PKT mice ((Ptf1acre/+; LSL-KrasGCitation12D/+;Tgfbr2flox/flox) were euthanized at 3–6 months for KPC tumors and 3 weeks for PKT tumors. These tumors were mechanically dissociated followed by enzymatic digestion using collagenase IV.Citation9,[]Citation9,[][[ Noncancerous epithelial cells were then magnetically depleted using the following antibodies (CD326þ, CD90 and CD45) at passages 4 to 6. Pancreatic stellate cells were isolated using the method published by Apte et al.Citation14Citation15 KPCs and PKT cells were grown in DMEM: F12 media with 1% Penicillin-Streptomycin and 10% Fetal Bovine Serum. Pancreatic stellate cells were grown in Iscove’s Modified Dulbecco’s Medium with 15% fetal bovine serum and 1% penicillin streptomycin.
Histology, immunofluorescence and Western blot
For histological examination, pancreatic tumor specimens were fixed in formalin, dehydrated in ethanol, embedded in paraffin and stained with hematoxylin and eosin (H&E). For immunofluorescence and immunohistological analysis, glass slides containing paraffinized tissue sections were rehydrated and antigen retrieval was performed with a citrate buffer and probed against respective antibodies. Immunohistological slides were counterstained with hematoxylin and 3,3′-diaminobenzidine (DAB) was used as primary chromogen for the antibodies. Details of the antibodies used are given in Supplementary Table 1. All images were acquired with Leica DFC 7000 T (Leica Microsystems).
For immunoblotting, tumors were lysed in ice cold RIPA buffer with phosphatase and protease inhibitor (Catalog number 78440; Thermo Fisher Scientific). Bicinchoninic acid assay (BCA assay, Catalog number 23222; Thermo Fisher Scientific) was used to quantify the protein concentration according to the manufacturer’s protocol. Gradient (4–20%) SDS PAGE gels (Catalog number 456–109, BioRad) were used for separation followed by transfer to a PVDF membrane (0.2uM) under wet conditions while utilizing a BioRad Mini Trans blot cell (Catalog number 1703930; BioRad). Primary antibodies used are detailed in Supplementary Table 1. Manufacturer’s protocol was used for blocking PVDF membranes prior to incubation with primary antibodies. After incubation, the protein antibody complex of interest was detected using secondary antibody conjugated to HRP (Catalog number 7074S and 7077S; Cell Signaling Inc). Chemiluminescence was detected using Pierce ECL enhanced chemiluminesence reagent (Catalog number 32106, Thermo Fisher Scientific) in myECL Imager (PI62236) using the manufacturer’s protocol. ImageJ (National Institute of Health) was used for densitometric analysis.
Flow cytometry
Flow cytometry was performed using standard methods. Briefly, single cell suspension of tumor cells was prepared after harvest of tumor tissue as described in the primary cell isolation section. The tumor cells were then washed with flow cytometry staining buffer composed of phosphate buffered saline (PBS) with 1% Bovine Serum Albumin (Catalog Number Catalog #: A9418-100 G, Sigma Aldrich) and 3 mM EDTA. The antibodies used are provided in Supplementary Table 1. Intracellular staining was performed using eBioscience Intra-cellular Fixation/Permeabilization Buffer Set (Catalog number 88–8824, Thermo Fisher Scientific). Flow cytometric acquisition was performed on the BD LSR-II and Beckman Coulter CytoFLEX machines and analysis was performed using FlowJo software version 7.6.5 (FlowJo LLC, Oregon, USA).
Cytotoxicity assays
For cytotoxicity assays, two approaches were used, LDH cytotoxicity and phase microscopy. An LDH cytotoxicity assay kit (Catalog number 88953; Thermo Scientific) was used as per manufacturer’s instructions. KPC cancer cells were plated on 96-well clear bottom plates as target cells. At around 60% confluency (24 hours), effector cells were introduced in 5% fetal bovine serum and 5 ng/ml IL-2 (Catalog number 212–12; Peprotech Inc) in RPMI-1640 medium. Effector CD8 + T cells at a 5:1 ratio were used against KPC target cells. The LDH level was measured in cell supernatant at 24 hours after effector cell addition using manufacturer suggested spectrophotometric settings.
For phase microscopy, similar experimental design was used. KPC cancer cells (3000) were plated as target cells in clear bottomed 96-well plates. Effector cells (splenocytes) were harvested from KPC orthotopic tumor-bearing mice at 2 weeks. These effector cells were added to KPC cells at a 1: 25 target:effector cell ratio when KPC cells reached 60% confluency. Incucyte live cell analysis software (Essen Biosciences) was used to track cell proliferation for real-time live cell imaging. Cell proliferation was assessed over 21 hours of incubation of effector cells with target cells.
CD8 T cell MACS sorting and Real‐Time PCR (qPCR)
CD8+ T cells were MACS sorted from splenocytes of WT and Hsp70−/- mice using CD8 + T Cell Isolation Kit, Order no. 130–104-075 (Miltenyi Biotec, Germany) as per the manufacturer’s protocol and their RNA was isolated using RNeasy® Micro kit (Qiagen, Germany). The RNA was reverse transcribed into cDNA with a SuperScript™ IV Reverse Transcriptase kit (Invitrogen, USA) as per the protocol. These samples from WT and knockout mice were profiled for the expression of IFNγ, IL-2, TNFα, GZMB, GZMK, and PFN via RT-PCR employing the standard SYBR Green method and StepOnePlus™ Real-Time PCR System (Applied Biosystems™, USA). The expressions of the target genes were normalized to endogenous control, GAPDH. For data analysis, the result files were extracted in XLSX format and cycle threshold (Ct) values were averaged out. The mean Ct values of target genes were normalized to the mean Ct values of endogenous control for corresponding samples, which gives ΔCt value. ΔΔCt was calculated as Test ΔCt – average of Control ΔCt and the fold change was calculated using the formula 2−ΔΔCT. In this way, the differential expression for a specific gene is calculated as compared to the control. Details of primers used can be found in supplementary table 2.
Dendritic cell isolation
Dendritic cells from WT or HSP70−/- mice were derived from splenocytes from tumor-naïve mice. Splenocytes were mechanically digested and strained through a 70 µm cell strainer. Dendritic cells were then magnetically separated using magnetic microbeads (Catalog No: 130–100-875 Pan Dendritic cell isolation kit; Miltenyi Biotec). Isolated dendritic cells were grown in RPMI-1640 medium supplemented with 2% FBS, 1% Penicillin-Streptomycin and 20 ng/ml GM-CSF and IL-4. DCs were grown at a density of 300,000 cells/well in a 12-well plate. KPC cancer cells were used as tumor lysates after four freeze thaw cycles in a transmembrane insert for tumor lysate costimulation experiment. After 72 hours, expression of MHC II and TNFα was evaluated using flow cytometry.
In vivo methods
Splenocyte adoptive transfer
NSG mice (NOD-scid IL2Rgammanull) had 3 million WT or Hsp70−/- splenocytes injected intravenously into the tail vein from either WT or Hsp70−/- mice (Day −1). These animals were then tumor challenged with 3000 KPC cancer cells orthotopically into the pancreas (Day 0). Subsequently, animals were then again given respective WT or Hsp70−/- splenocytes on day 14 and euthanized on day 28. Tumor weights were measured at endpoint. In a similar experiment, Rag1 KO mice were intravenously injected with 1 × 106 WT or Hsp70−/- splenocytes (Day −3) and tumors were induced with orthotropic injection of 3000 KPC cancer cells (Day 0). Tumor weight was measured at endpoint (Day 28) and metastatic sites were enumerated.
Dendritic cell adoptive transfer
Orthotopic KPC tumor-bearing WT and Hsp70−/- mice were euthanized at endpoint and splenic dendritic cells were isolated using methods described above and cultured for 72 hours. WT mice were implanted with 3000 KPC cells orthotopically. On day 7 postimplantation, mice were randomized to receive intraperitoneal injections of 1 × 106 WT or Hsp70−/- dendritic cells dissolved in 200uL of PBS. Tumor weights were measured at endpoint.
CD8+ cell and macrophage depletion
For CD8+ depletion, KPC cancer cells were injected orthotopically into WT or Hsp70−/- mice as described above. Following this, mice were randomized and injected intraperitoneally with neutralizing monoclonal antibodies directed against CD8 (clone YTS169.4; BioXcell, West Lebanon, NH) or its corresponding isotype (clone: LTF2; BioXcell) for 4 weeks at a dose of 20ug/mouse biweekly. Mice were sacrificed after 28 days of tumor challenge and the efficiency of CD8 depletion was evaluated in spleen by flow cytometry.
Macrophage depletion was achieved according to methods previously described using liposomal clodronate.15
Orthotopic KPC tumor-bearing WT or HSP070−/- mice were injected intraperitoneally twice per week with clodronate liposomes weight (C-005; Liposma) at 1.4 mg/20 g body or with an equivalent volume of PBS liposomes (P-005; Liposma). Tumor weights were measured at endpoint (day 28) and macrophage depletion was confirmed using flow cytometry for F4/80 in splenocytes.
Pancreatic stellate cell coinjection with KPC cancer cells
Pancreatic stellate cells (PSCs) were isolated from WT and Hsp70 −/- mice as described previously by our group and others.Citation9,Citation14 After isolation, WT or Hsp70−/- PSCs were coinjected with KPC cancer cells into the pancreas of WT mice to simulate an environment where only the PSCs are with or without Hsp70. Mice were euthanized at day 25 and tumor weight was taken as endpoint.
Mice and animal models
Animal experiments were authorized and overseen by the Institutional Animal Care and Use Committee (IACUC) in accordance with approved protocols. Hsp70−/- mice were acquired from Mutant Mouse Resource and Research Center (MMRC) with the National Institute of Health. These mice lack 12 kb deletion of both Hspa1a and Hspa1b (https://www.mmrrc.org/catalog/sds.php?mmrrc_id=372). Confirmation of the absence of Hsp70 expression in cells from these mice was made via qPCR (data not shown). For in vivo experiments, WT (Hsp70+/+) and Hsp70−/- mice were generated from littermate control mice after breeding Hsp70± mice with Hsp70± mice, which, in turn, were generated by breeding Hsp70−/- mice with C57BL/6 J (Hsp70+/+) mice (Supplementary Figure 1A). Immunodeficient Rag1 KO (Rag1tmCitation1Mom) and NSG (NOD-scid IL2Rgammanull) mice were obtained from the Jackson Laboratories (Bar Harbor, ME). KPC mice, the genetic model for spontaneous pancreatic cancer expressing mutant KRAS and p53, were generated by crossing LSR-KrasGCitation12D/p and LSR Trp53R172H/P with Pdx-Cre mice. Age- (4–9 weeks) and sex-matched mice were used in these experiments. The orthotopic tumor model was generated after administering intrapancreatic injections of tumor cells (dissolved in 10 µl of Matrigel) and animals were sacrificed at indicated time points. The survival fraction was determined using Kaplan-Meier survival curves after implantation of 3000 KPC cells in the pancreas. The surgical implantation model was performed using methods previously published by our group.Citation16 PKT (Ptf1acre/+; LSL-KrasGCitation12D/+;Tgfbr2flox/flox) mice were a gift from Dr. Nipun Merchant (University of Miami). Tumor-bearing mice were euthanized and single cell suspension was prepared from these mice as described in the protocol for KPC mice. PKT cells were suspended in Matrigel and 10,000 cells were injected in 10 ul volume into the pancreas. MC-38 murine colon adenocarcinoma cells were a gift from Dr. Eli Gilboa at the University of Miami. MC-38 cells (100,000) were injected subcutaneously into flanks of C57BL/6 J mice. The tumor volume was measured at endpoint at 30 days. B-16-F10 (1000,000) melanoma cells were injected subcutaneous into the flanks of WT or Hsp70-/- mice and tumor volumes were measured after 16 days.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM) for all experiments, except when indicated otherwise in the figure legend. Respective figures have their sample size (n) included in the figure legend and for in vivo experiment, each data point represents one single animal. Kaplan-Meier curves were used for survival model analysis with the log rank p value for statistical significance. The nonparametric Mann-Whitney test using GraphPad Prism 6 (GraphPad Software, La Jolla, CA) was used for statistical analysis for in vivo experiments. For dendritic cell incubation with tumor lysates, paired student t-test analysis was used. Individual figures also have p-values for each experiment included in their legends. p-values <0.05 were considered statistically significant.
Results and discussion
Hsp70 in the TME modulates cancer growth independent of stromal elements
To evaluate the role of Hsp70 in the TME, we implanted KPC ((B6.LSL-KrasGCitation12D and p53R127H Pdx-cre) pancreatic cancer cells into the pancreas of Hsp70−/- or their WT littermate control mice (Supplementary Figure 1A). This simulated a model where the cancer cells themselves had Hsp70 intact, but the surrounding tumor microenvironment (immune cells, stromal cells, etc.) either had endogenous (WT mice) or was null for (Hsp70−/-) heat shock protein 70 (Supplementary Figure 1B). In the absence of Hsp70 in the TME, we saw reduced KPC tumor growth (). In a separate experiment, this decrease in tumor growth translated to improved survival in mice lacking Hsp70 in their TME (Median survival Hsp70−/- vs WT: 60 vs 49 days, log rank p value<.001) (). To rule out model and cell line-specific effects, we performed similar experiments using alternate models and cell lines. In a surgical implantation model derived from tumor-bearing KPC mice,Citation16 Hsp70−/- mice had much smaller tumors (Supplementary Figure s1C). To exclude cancer type-specific effects, we confirmed these results using the colon cancer cell line MC-38. Subcutaneous implantation of MC-38 colon cancer cells in Hsp70−/- mice resulted in a similar remarkable decrease in tumor growth, when compared to WT mice (). Similarly, when B16-F10 cells, a highly aggressive malignant melanoma cell line, were subcutaneously implanted in Hsp70−/- mice (), significantly decreased tumor growth was observed when compared with WT mice. In a more aggressive model of syngeneic pancreatic cancer, orthotopic implantation of PKT cancer cells also resulted in smaller tumors, but the findings did not reach statistical significance (Supplementary figure 1D).
Figure 1. Hsp70 in the TME modulates tumor growth and metastasis in multiple animal models independent of the stromal compartment
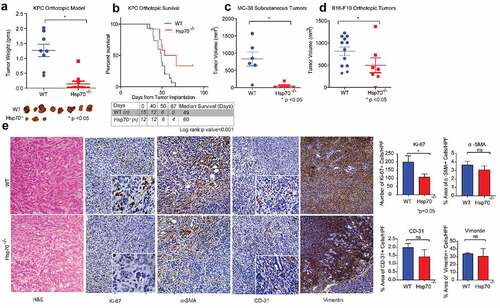
Since our studies demonstrated that Hsp70 in TME supports tumor growth, we next focused on identifying the mechanism. For this, TME was broadly divided into the cancer-associated fibroblast (CAF) compartment and the immune compartment. We first focused on the stromal component of the TME. While Hsp70−/- mice had smaller tumors and lower mitotic index as seen with lower Ki-67 staining, there was no difference in morphology of tumors as well as the expression of fibroblast activation markers (α-SMA and vimentin) or in angiogenesis, as measured by CD-31 expression in tumor sections, between WT and Hsp70−/- tumors. (). These findings were confirmed with immunoblotting where expression of α-SMA and CD-31 was not significantly different between tumors in WT and Hsp70−/- mice (Supplementary Figure 1E).
We further evaluated the role of Hsp70 in CAFs using an experimental model simulating specific absence of HS70 in the pancreatic stellate cells (PSCs), the major source of CAFs in pancreatic cancer (Supplementary Figure s1F). In this experiment, PSCs were harvested from Hsp70−/- or WT mice and coinjected with KPC cancer cells into WT mice. This modeled a TME with specific absence of Hsp70 in the stromal cells of mice that were injected with Hsp70−/- PSCs. KPC cancer cells injected without PSCs were used as control without any external stromal cell coinjection. At endpoint, mice lacking stromal specific Hsp70 showed no difference in tumor growth compared to control mice. Taken together, this points to a low possibility of Hsp70 in CAFs modulating tumor growth in PDAC TME.
Hsp70 in immune cells blunts the antitumor response
To evaluate the mechanism by which Hsp70 modulates tumor growth in the TME, we next focused on the other major component of the TME, the immune cells. We first specifically evaluated the effect of immune cells lacking Hsp70 on tumor growth by comparing the ability of in vivo primed WT and Hsp70−/- splenocytes (obtained from tumor bearing WT or Hsp70−/- mice) to kill cancer cells in vitro. For this, we coincubated splenocytes with cancer cells in vitro and cell proliferation was measured by phase microscopy. As seen, splenocytes deficient in Hsp70 affected extensive cancer cell death (Supplementary Figure 2A). Next, we specifically evaluated the effect of intracellular Hsp70 on the ability of CD8+ cells to kill cancer cells. For this, we isolated CD8+ cells from KPC orthotopic tumors growing in WT and Hsp70−/- mice and performed T cell cytotoxicity assay. As seen in (Schematic depiction in supplementary Figure 2B), CD8+ cells obtained from tumors in Hsp70−/- mice exerted greater cytotoxicity against cancer cells compared to those obtained from tumor in WT mice. To further confirm the role of cytotoxic T cell response in decreased tumor growth observed when Hsp70 is lacking in the TME, we used CD8+ T cell depletion approach. For this, KPC cells were implanted into WT and Hsp70−/- mice and antibody-mediated CD8+ cell depletion was performed. We found that in the absence of these effector immune cells, tumor growth reduction was not seen in Hsp70−/- mice, confirming that a functioning anticancer immune system is required for Hsp70 in the TME to modulate tumor growth (). Confirmation of CD8 depletion was performed using flow cytometry (Supplementary figure s2C). We also evaluated the cytokine expression in CD8+ T cells using RT-PCR to gauge their functional phenotype. Although there was no difference in the expression of Ifnγ, TNFα and IL-2 between CD8+ T cells isolated from tumor bearing WT or Hsp70−/- mice, intriguingly, expression of cytotoxic markers like Granzymes B and K was significantly upregulated in CD8+ T cells from Hsp70−/- mice (Supplementary figure 2D). It must be noted that not only these CD8+ cells lack Hsp70 but also these cells were in the presence of antigen presentation machinery, which lacked Hsp70. This suggests that Hsp70 affects the immune response and subsequently the cytotoxic activity of CD8+ cells.
Figure 2. Hsp70 in the immune cells exerts an immunomodulatory effect
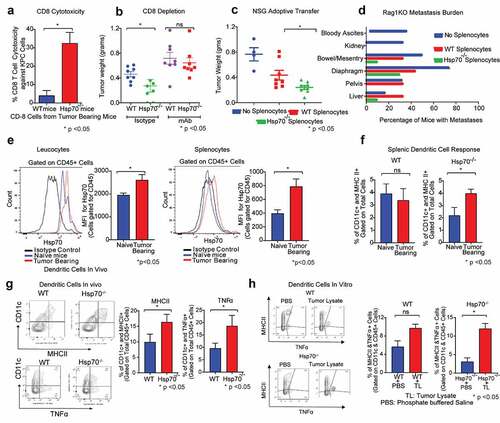
We further validated the role of Hsp70 in immune cells in shaping anticancer immune response using two separate immunodeficient models. Briefly, the effect of adoptive transfer of WT or Hsp70−/- splenocytes (Supplementary Figure 2D) on the tumor growth and metastases was evaluated () in immunodeficient NSG (NOD-scid IL2Rgammanull) mice, which lack both innate and adaptive immunity. As expected, NSG mice receiving WT splenocytes had smaller tumors compared to the mice that did not receive any splenocytes. Intriguingly, mice receiving splenocytes from Hsp70−/- donors had significantly smaller tumors compared to mice that received WT splenocytes. These findings were confirmed using another immunocompromised mouse model, the Rag1 KO (Rag1tmCitation1Mom) mice in an experiment using a similar experimental protocol (Supplementary Figure 2E). Furthermore, Rag1 KO mice with orthotopic KPC tumors demonstrate considerable intra-abdominal metastases and adoptive transfer of WT splenocytes in this model markedly decreased the intra-abdominal metastases. Indeed, the reduction of intra-abdominal metastases was much more marked with adoptive transfer of Hsp70−/- splenocytes, when compared to WT splenocytes (). These together suggest that lack of Hsp70 in immune cells energizes the anticancer immune response.
Given that our data suggest that the decreased tumor growth observed when Hsp70 is lacking in TME is dependent on immune cells, we evaluated the effect of cancer challenge on Hsp70 levels in immune cells. KPC orthotopic tumors were implanted in WT mice and after two weeks, splenocytes and leukocytes were harvested and Hsp70 expression was evaluated by flow cytometry. We observed that cancer challenge led to an increase in the expression of Hsp70 in both leukocytes and splenocytes, when compared to tumor-naïve mice (). Next, to further characterize the role of this increased Hsp70 expression in the resultant immune response, we evaluated the expression of MHCII in DC of tumor-bearing mice. As seen in (flow cytometric curve is shown in Supplementary figure s2F), orthotopic tumor challenge in WT mice did not significantly increase the expression of MHC-II, a marker of antigen presentation capacity, in the splenic dendritic cells. However, in Hsp70−/- mice, splenic dendritic cells had a significantly increased MHC-II response compared to tumor-naïve Hsp70−/- mice. This finding was further corroborated when we looked at the tumors themselves (). Dendritic cells from orthotopic KPC tumors growing in Hsp70−/- mice demonstrated significantly greater expression of antitumor molecules, MHC-II and TNF-α, suggesting that these cells initiated a more robust immune response. We found this to be true in vitro as well. WT and Hsp70−/- DCs were isolated from respective donor mice. They were then incubated with KPC cancer cells. WT dendritic cells did not significantly increase their expression of antitumor cytokines like TNF-α, while, in the absence of Hsp70, dendritic cells increased their expression of TNF-α and MHC-II almost five-fold (). We next investigated whether dendritic cells from Hsp70−/- mice can stimulate antitumor immunity in-vivo. Dendritic cells, isolated from tumor-bearing WT and Hsp70−/- mice, were adoptively transferred into tumor-bearing WT mice. While WT DCs failed to exert any antitumor effect upon adoptive transfer, Hsp70−/- DCs were able to show a strong trend toward decreased tumor burden, although it failed to reach statistical significance (p = .1). Thus, it appears that depletion of Hsp70 in dendritic cells leads to their increased activation and improved antitumor immune response. While the mechanism by which Hsp70 in DC affects their antigen presentation machinery is not known, it is possible that this may be mediated by induction of ER stress, as suggested by recent work by Cubillos-Ruiz and colleagues.Citation17
Another finding of note was that tumor-infiltrating macrophages (F4/80+ cells) too had greater expression of MHC-II in Hsp70−/- tumors (Supplementary figure s2G). However, when F4/80+ cells were selectively depleted in both WT and Hsp70−/- mice (Supplementary figure 2H), the difference in these tumors persisted, suggesting that macrophages were not the primary cells driving this phenomenon. These findings differ from the ones by Gabai and colleagues who show that Hsp70−/- macrophages exert an antitumorigenic effect in cancers.Citation18 This discrepancy may be a result of differences in cancers studied, models used and local factors such as differences in microbiome. Curiously, although the tumors from Hsp70−/- mice showed a disposition toward greater expression of TNFα in both CD4+ and CD8+ T cells, they did not reach statistical significance (Supplementary figure 2I) excluding T cells as primary mediators as well.
There are certain limitations to our work. Although our work highlights the broad role of antigen presenting cells in modulating the dysfunctional immune system in cancers, we have not looked at the exact molecular pathways that bring about the dysregulation of antitumor response in Hsp70+/+ dendritic cells. The next phase of our investigation will focus on studying this in greater detail.
In summary, our study suggests that Hsp70 in immune cells attenuates anticancer immune response and depletion of Hsp70 in TME leads to enhancement of anticancer immune response and decreased tumor growth. In our experiments, dendritic cells were shown to be significantly responsive to the absence of Hsp70 and reacted by exerting a robust antitumor response. We show that tumor mediates an enhanced heat shock response in immune cells, which, in turn, leads to blunted antitumor immune response. Fundamentally, like the role of immune checkpoint pathways,Citation19 our findings elucidate another mechanism by which tumors escape host immune response. Immunotherapy in pancreatic cancer suffers from poor response in patients.Citation1 Dendritic cell vaccinations against pancreatic cancer have shown some promise but lack efficacy.Citation20 Engineering Hsp70-deficient dendritic cells as tumor vaccination strategies either in combination with immunotherapy or as perioperative strategies could prove to be effective adjuncts to the treatment of pancreatic cancer and merit consideration.
Disclosure of Potential Conflicts of Interest
Dr. Ashok Saluja is one of the inventors of Minnelide (patented by the University of Minnesota), which has been licensed to Minneamrita Therapeutics. He has ownership interest in this company and is its Chief Scientific Officer and Cofounder. Minneamrita has contracted our group at the University of Miami to carry out translational studies of Minnelide, which are undertaken by our group. This relationship is managed by the Office of the Vice Provost for Research at the University of Miami. Rest of the authors have no conflict of interest to declare.
Author contributions
Conception: BGi, SR, AS, VD,
Experiments: BG, PSh, UV, TJ, SM, AF, BGa, SI, ZM, RS, HKCJ, JG, VS, PSa, EBa
Review of literature: BG, PSh, UV, TJ, SR, AS, VD
Acquisition and Validation: BGi, PSh, TJ, SM, UV, HKC, SM, RS, ZM, SI, BGa, VS, PSa, SR, AS, VD
Writing and editing: BG, PSh, TJ, UV, SR, AS, VD
Agreement and Accountability for integrity of work: All authors
Supplemental Material
Download ()Acknowledgments
The authors would like to thank the Dr. Eli Gilboa for MC-38 cell and the Department of Veterinary Resources for help with animal experiments at the University of Miami.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Torphy RJ, Zhu Y, Schulick RD. Immunotherapy for pancreatic cancer: barriers and breakthroughs. Annal Gastroenterol Surg. 2018;2:274–8. doi:10.1002/ags3.12176.
- Dougan SK. The pancreatic cancer microenvironment. Cancer J. 2017;23:321–325. doi:10.1097/PPO.0000000000000288.
- Bachem MG, Schünemann M, Ramadani M, Siech M, Beger H, Buck A, Zhou S, Schmid-Kotsas A, Adler G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–921. doi:10.1053/j.gastro.2004.12.036.
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi:10.1016/j.ccr.2012.01.007.
- Takahashi K, Ehata S, Koinuma D, Morishita Y, Soda M, Mano H, Miyazono K. Pancreatic tumor microenvironment confers highly malignant properties on pancreatic cancer cells. Oncogene. 2018;37:2757–2772. doi:10.1038/s41388-018-0144-0.
- Banerjee S, Modi S, McGinn O, Zhao X, Dudeja V, Ramakrishnan S, Saluja AK. Impaired synthesis of stromal components in response to minnelide improves vascular function, drug delivery, and survival in pancreatic cancer. Clin Cancer Res. 2016;22:415–425.
- Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P, Taddei A, Bencini L, Farsi M, Cappello P, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother. 2013;62:1249–1260. doi:10.1007/s00262-013-1429-3.
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences. 2013 Dec 10;110(50):20212-7.
- Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, Ban Y, Lavania S, Dawra R, Banerjee S, et al. NFκB in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via up-regulation of CXCL12. Gastroenterology. 2018;155:880–891.e888. doi:10.1053/j.gastro.2018.05.051.
- Nummer D, Suri-Payer E, Schmitz-Winnenthal H, Bonertz A, Galindo L, Antolovich D, Koch M, Büchler M, Weitz J, Schirrmacher V, et al. Role of tumor endothelium in CD4+CD25+ regulatory T cell infiltration of human pancreatic carcinoma. JNCI. 2007;99:1188–1199. doi:10.1093/jnci/djm064.
- Landry J, Bernier D, Chrétien P, Nicole LM, Tanguay RM, Marceau N. Synthesis and degradation of heat shock proteins during development and decay of thermotolerance. Cancer Res. 1982;42:2457–2461.
- Radons J. The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones. 2016;21:379–404.
- Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D, Sharif R, Dawra R, Lerch MM, Saluja A. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–625. doi:10.1158/0008-5472.CAN-06-1567.
- Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi:10.1136/gut.43.1.128.
- Rooijen NV, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi:10.1016/0022-1759(94)90012-4.
- Majumder K, Arora N, Modi S, Chugh R, Nomura A, Giri B, Dawra R, Ramakrishnan S, Banerjee S, Saluja A. A novel immunocompetent mouse model of pancreatic cancer with robust stroma: a valuable tool for preclinical evaluation of new therapies. J Gastrointestinal Surg. 2016;20:53–65.
- Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161:1527–1538. doi:10.1016/j.cell.2015.05.025.
- Gabai VL, Yaglom JA, Wang Y, Meng L, Shao H, Kim G, Colvin T, Gestwicki J, Sherman MY. Anticancer effects of targeting Hsp70 in tumor stromal cells. Cancer Res. 2016;76:5926–5932. doi:10.1158/0008-5472.CAN-16-0800.
- Escors D, Gato-Cañas M, Zuazo M, Arasanz H, García-Granda MJ, Vera R, Kochan G. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduction Targeted Ther. 2018;3:26. doi:10.1038/s41392-018-0022-9.
- Jiang N, Qiao G, Wang X, Morse MA, Gwin WR, Zhou L, Song Y, Zhao Y, Chen F, Zhou X, et al. Dendritic cell/cytokine-induced killer cell immunotherapy combined with S-1 in patients with advanced pancreatic cancer: a prospective study. Clin Cancer Res. 2017;23:5066–5073. doi:10.1158/1078-0432.CCR-17-0492.