
ABSTRACT
Combining natural killer (NK) cell adoptive transfer with tumor-sensitizing chemotherapy is an attractive approach against recurrent ovarian cancer (OC), as OC is sensitive to NK cell-mediated immunity. Previously, we showed that CD34+ hematopoietic progenitor cell (HPC)-derived NK cells can kill OC cells in vitro and inhibit OC tumor growth in mice. Here, we investigated the potential of HPC-NK cell therapy combined with chemotherapeutic gemcitabine (used in recurrent OC patients) against OC. We examined the phenotypical, functional, and cytotoxic effects of gemcitabine on HPC-NK cells and/or OC cells in vitro and in OC-bearing mice. To this end, we treated OC cells and/or HPC-NK cells with or without gemcitabine and analyzed the phenotype, cytokine production, and anti-tumor reactivity. We found that gemcitabine did not affect the phenotype and functionality of HPC-NK cells, while on OC cells expression of NK cell activating ligands and death receptors was upregulated. Although gemcitabine pre-treatment of OC cells did not improve the functionality of HPC-NK cells, importantly, HPC-NK cells and gemcitabine additively killed OC cells in vitro. Similarly, combined HPC-NK cell and gemcitabine treatment additively decreased tumor growth in OC-bearing mice. Collectively, our results indicate that combination therapy of HPC-NK cells and gemcitabine results in augmented OC killing in vitro and in vivo. This provides a rationale for exploring this therapeutic strategy in patients with recurrent OC.
INTRODUCTION
Ovarian cancer (OC) is estimated to have been diagnosed in 21,410 women in the United States in 2021.Citation1 OC treatment encompasses cytoreductive surgery and platinum/taxane chemotherapy. Nevertheless, the five-year survival is only 49% for all stages and 30% for advanced stage OC.Citation1 This is partly due to tumor recurrence after a median of 1.5–2 years.Citation2 Therefore, novel therapeutic strategies are urgently needed to improve patient outcome.
As ovarian tumors often downregulate MHC class I and express natural killer (NK) cell activating ligands, they are prone to NK cell-mediated immunity.Citation3,Citation4 However, autologous NK cells are often dysfunctional,Citation5 due to tumor-induced immunosuppression and remaining self MHC class I expression on tumor cells. Previously, we demonstrated that umbilical cord blood (UCB)-derived CD34+ hematopoietic progenitor cell (HPC)-NK cells are capable of killing OC monolayers and spheroids.Citation6 Furthermore, we showed that intraperitoneal infusion of HPC-NK cells in human OC-bearing mice significantly limits tumor progression and prolongs survival.Citation6 Currently, we are investigating the feasibility, safety, and toxicity of an intraperitoneal HPC-NK cell infusion in recurrent ovarian cancer patients (NCT:03539406). However, improvement of HPC-NK cell therapy is warranted to maximize the killing of OC cells.
Combining HPC-NK cells with gemcitabine could be a novel option for OC patients. Gemcitabine is a chemotherapy used in recurrent OC patients,Citation7 which inhibits DNA synthesis and thereby induces apoptosis of proliferating cells.Citation8,Citation9 Notably, gemcitabine also increases expression of natural killer group 2, member D (NKG2D) ligands in various cancer types, thereby providing activating signals to NK cells.Citation8,Citation10–14 Moreover, gemcitabine upregulates expression of death receptors in multiple cancers for which NK cells express the corresponding ligands, thereby sensitizing tumor cells to NK cell-mediated apoptosis.Citation10 In mice, gemcitabine was shown to promote NK cell-mediated immune responses after pancreatic tumor resection and to inhibit local recurrence of tumors.Citation15 These results suggest that NK cell therapy in combination with gemcitabine could be a promising treatment approach for recurrent OC patients.
The goal of this study was to investigate the phenotypical, functional, and cytotoxic effects of gemcitabine treatment on HPC-NK cells and/or OC cells in vitro and in OC-bearing mice. Our study showed that gemcitabine upregulates NK cell activating ligands and death receptors on OC cells. Moreover, HPC-NK cells and gemcitabine additively killed OC cells in vitro and in vivo. These data provide a rationale to explore combination therapy of HPC-NK cells and gemcitabine in patients with recurrent OC.
MATERIALS AND METHODS
HPC-NK cell culture
Umbilical cord blood was collected in cesarean sections after informed consent (approved by the Radboud University Medical Center Committee for Medical Research Ethics CMO 2014/226), in accordance with institutional guidelines and regulations, and the Declaration of Helsinki. CD34+ HPCs were isolated, cryopreserved and thawed for HPC-NK cell generation. CD34+ HPCs were expanded and differentiated into HPC-NK cells within 5–6 weeks as described previously.Citation16 HPC-NK cells (>70% CD56+) were used directly after culture or after cryopreservation and 5–8-day re-culture. HPC-NK cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM, Gibco, 21980–032) supplemented with 10% fetal calf serum (FCS, Corning, 35–079-CV or Integro, 5–45900) (IMDM10; HPC-NK cell functional assay) or Roswell Park Memorial Institute 1640 medium (RPMI, Gibco, 21875–034) with 10% FCS (RPMI10; phenotyping and killing assay). For experiments >24 h, medium was supplemented with 5 ng/ml recombinant human interleukin-15 (rhIL-15; Immunotools, 11340155) to support NK cell survival.
Tumor cell culture
Ovarian cancer (OC) cell lines IGROV-1, SKOV-3, and OVCAR-3 (RRID:CVCL_1304, RRID:CVCL_0532, and RRID:CVCL_0465, respectively) were cultured in RPMI10 (IGROV-1 and SKOV-3) or RPMI supplemented with 20% FCS and 1 µg/ml insulin (Merck, i0516) (OVCAR-3), using trypsin (Life Technologies, 25300054) for passaging. SKOV-3 was transduced with luciferase and green fluorescent protein (SKOV-3-luc-GFP) as described previously,Citation6 and used for mouse experiments. Leukemia cell-line K562 (RRID:CVCL_0004) was cultured in IMDM10. All cell lines were cultured for a maximum of 3 months and were mycoplasma free. SKOV-3 and K562 were purchased from the ATCC, and IGROV-1 and OVCAR-3 were kindly provided by Prof. Dr. OC Boerman, Department of Nuclear Medicine, Radboud University Medical Center, Nijmegen, the Netherlands. Primary patient-derived OC cell line ASC 009 was generated from ascites material obtained from the Gynecologic Oncology Biobank of the Radboud University Medical Center by NTRC in Oss, the Netherlands as described,Citation17 and kindly provided by Guido Zaman. This study was approved by the Radboudumc Committee for Medical Research Ethics (CMO 2018/4845), and written informed consent was obtained. ASC 009 was cultured in advanced RPMI 1640 (Life Technologies, 12633–020) supplemented with 10% FCS, 1% pen/strep (Life Technologies, 15140130) and 1% glutamax (Life Technologies, 35050–038).
Tumor nodule dissociation
SKOV-3-luc-GFP tumor nodules from mice were dissociated using the human tumor dissociation kit (Miltenyi Biotec, 130–095-929) and a gentleMACS Dissociator. A single-cell suspension was obtained using a 70 µm filter (Fisher Scientific, 10788201) before staining.
Drug treatment
OC cells were plated in 6-well (Corning, 3506), 24-well (Greiner CELLSTAR, 662160) cell culture plates, or T75 cell culture flasks (Greiner Bio-One, 658175). HPC-NK cells were plated in 24-well plates. Gemcitabine hydrochloride (Actavis/Aurobindo, 104283/104285, 40 mg/ml = 133 mM) was diluted in culture medium and added (immediately for HPC-NK cells and after 4 h for OC cells to first allow cell adherence) at concentrations ranging from 0 to 87.5 nM. When HPC-NK cells were co-cultured with OC cells, HPC-NK cells and gemcitabine were added 4 h after plating OC cells in 24-well plates. After 48 −72 h, supernatant was stored at −20°C for enzyme-linked immunosorbent assay (ELISA). HPC-NK cells were harvested by resuspending the remaining medium, OC cells were trypsinized (trypsin: Life Technologies, 25300054; or trypLE: Gibco, 12605028) and collected for further analyses.
Flow cytometry (FCM)-based assays
FCM samples were measured on the FC500, Gallios, or CytoFLEX (LX) (all Beckman Coulter). To determine percentage positivity, gates were placed based on a maximum of 5% background staining in a corresponding unstained or backbone sample.
HPC-NK cell functional assay
HPC-NK cells were stimulated for 4 h with K562 or SKOV-3 cells that were 48 h pre-treated with/without 8.8 nM gemcitabine at an effector-to-target (E:T) ratio of 1.5:1 in the presence of CD107a-PE-Cy7 (Biolegend, 328618), and brefeldin A (added after 1 h, BD Biosciences, 555029). Optionally, anti-DNAX Accessory Molecule-1 (DNAM-1) (mouse IgG1, Biolegend, 338302), and/or anti-NKG2D (mouse IgG1, Biolegend, 320813) blocking antibodies or isotype control (mouse IgG1, BioXcell, BE0083) were added at 10 μg/ml. After 4 h, surface staining was performed using CD56-BV510 (Biolegend, 318340), and intracellular staining using anti-interferon (IFN)γ-FITC (BD Biosciences, 554700) and anti-perforin-PE (Biolegend, 308106) using a fixation/permeabilization kit (eBioscience, 00–5521-00) and permeabilization buffer (eBioscience, 00–8333) following manufacturer’s instructions. Dead cells were excluded using fixable viability dye eFluor780 (eBioscience, 65–0865-14) staining. Unstimulated cells were used for gating on CD107a and IFNγ positive cells.
Phenotyping
OC cells and HPC-NK cells were immediately stained after drug treatment, or when staining HPC-NK cells for death-inducing ligands, after stimulation with K562 for 4 h. Staining was performed after 5–10 min incubation with nanogam (total human immunoglobulins; Sanquin, 8717185830262).
Parental OC cell lines, ASC 009, and dissociated SKOV-3-luc-GFP tumor nodules were stained using viability dye ViaKrome808 (Beckman Coulter, C36628) and the following antibodies: anti-ULBP2/5/6-PE (R&D Systems, FAB1298P), CD261(TRAIl-R1)-PE-Atto594 (Novus Biologicals, NBP2-31343PEATT594), CD112(Nectin-2)-PE-Cy7 (Biolegend, 337414), CD262(TRAIL-R2)-APC (Biolegend, 307407), CD155(PVR)-BV421 (Biolegend, 337631), and CD95(Fas)-BV785 (Biolegend, 305645); or anti-MICA/B-PE (Biolegend, 320906), anti-ULBP1-PE-Atto594 (Novus Biologicals, NBP2-11986PEATT594), anti-ULBP3-APC (R&D Systems, FAB1517A), anti-HLA-ABC-APC-Fire750 (Biolegend, 311443), and anti-HLA-E-BV421 (Biolegend, 342611).
HPC-NK cells were stained with CD56-BV510 (Biolegend, 318340), CD253(TRAIL)-APC (Biolegend, 308210), CD178(FasL)-PE (Biolegend, 306407), and fixable viability dye eFluor780; or CD45-BUV395 (BD Biosciences, 563792), CD56-BV421 (Biolegend, 318328), CD3-BV605 (Biolegend, 300460), CD159a(NKG2A)-APC (Beckman Coulter, A60797), CD158b-BV510 (BD Biosciences, 743452), CD158e1-AF700 (Biolegend, 312712), CD69-BUV737 (BD Biosciences, 612818), CD337(NKp30)-PE-Cy5 (Beckman Coulter, A66904), CD336(NKp44)-PE-Cy7 (Biolegend, 325116), CD335(NKp46)-PE-Dazzle594 (Biolegend, 331930), CD226(DNAM-1)-PE (Biolegend, 337106), CD314(NKG2D)-BV785 (Biolegend, 320830), and viability dye ViaKrome808 (Beckman Coulter, C36628).
Killing assay
After collecting HPC-NK cells and/or OC cells, viability dye 7-Aminoactinomycin D (7-AAD, Sigma, A9400) and, when measured on the FC500, 10 µL Flow-Count Fluorosphere Beads (Beckman Coulter, 7547053) were added, and the number of viable cells was analyzed. Percentage of gemcitabine-mediated death was calculated as follows: (1 – (number of viable cells treated with gemcitabine)/(number of viable cells treated without gemcitabine)) × 100%. Combined OC killing (by gemcitabine plus HPC-NK cells) compared to no gemcitabine was calculated as follows: 1 − (number of viable OC cells of an experimental condition)/(number of viable OC cells without gemcitabine and HPC-NK cells) × 100%.
ELISA
Supernatants were evaluated for IFNγ and granzyme B secretion following manufacturer’s instructions (IFNγ: Endogen, M700A; granzyme B: Mabtech, 3485–1 H-20). Absorbance at 450 nm was measured using the FLUOstar Omega Microplate Reader (BMG Labtech).
In vivo experiments
Animal experiments were approved by the Dutch central committee of animal experiments (CCD, 2015–0123) and supervised by the animal welfare committee of the Radboud University Medical Center, performed in accordance with institutional, national, and international guidelines, and comply with relevant legislation under university permit number 10300. Mice were inspected daily for general behavior, and weight was monitored at least twice/week. General humane endpoints were applied, whereupon mice were sacrificed using cervical dislocation or CO2 inhalation. In all the mouse experiments, 6–20 weeks old female NOD/SCID/IL2Rγnull (NSG) mice (Jackson Laboratories, in-house breeding) were injected intraperitoneally (i.p.) with 0.2 million SKOV-3-luc-GFP cells and divided into treatment groups based on block randomization after baseline bioluminescence imaging (BLI). BLI was performed following i.p. injection of 150 mg/kg D-luciferin (PerkinElmer, 122799), and isoflurane anesthesia. Images were taken using an In Vivo Imaging System and Living Image software (PerkinElmer). A region of interest was drawn around the torsos of the mice, and the integrated flux of photons (photons/second/cm2/steradian) was analyzed. Researchers involved in the experiments were blinded to treatment after baseline BLI.
For the gemcitabine titration, 20 mice (n = 5 per group) were injected i.p. with 0, 10, 20, or 40 mg/kg gemcitabine hydrochloride (diluted in PBS) starting 4 days after tumor injection, twice weekly for 2 weeks followed by 1-week rest (i.e. one cycle) for two cycles. For the first combination experiment, 44 mice (n = 11 per group) were treated with 40 mg/kg gemcitabine hydrochloride (diluted in PBS) or PBS, starting 4 days after tumor injection (day 0) for three cycles. Furthermore, i.p. HPC-NK cells (12 million/mouse/infusion) or PBS were infused on day 14, 35, 56, in combination with i.p. rhIL-15 or PBS injections between day 14–20, 35–41, 56–70 every other day. Mice were followed for survival. The second combination experiment had a similar setup with these adaptations: HPC-NK cells were infused on day 0, 21, 42 (17, 12, or 7 million/mouse, respectively); rhIL-15 between day 0–6, 21–27, 42–56; and gemcitabine from day 7 for two cycles. The gemcitabine phenotype experiment had a different design: 14 mice (n = 7 per group) were injected i.p. with gemcitabine hydrochloride or PBS starting 39 days after tumor injection for 2 weeks, whereupon the mice were sacrificed for tumor nodule assessment.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software version 5.03 or SPSS. The used statistical tests are indicated in the figure legends. Significance was defined as p < .05 (*), p < .01 (**) and p < .001 (***).
RESULTS
Gemcitabine increases expression of NK cell activating ligands and death receptors on OC cells
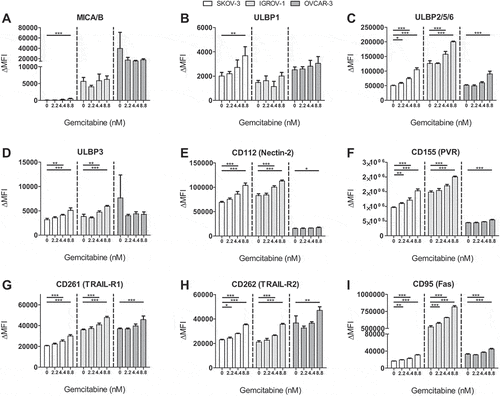
To investigate the phenotypical effects of gemcitabine treatment on OC cells and HPC-NK cells, we exposed these cells to varying gemcitabine concentrations for 48 h. Using similar concentrations as previous studies,Citation8,Citation12 gemcitabine (8.8 nM) significantly upregulated expression of activating NKG2D ligand ULBP2/5/6, activating DNAM-1 ligands CD112 (Nectin-2) and CD155 (PVR), and death receptors CD261 (TRAIL-R1), CD262 (TRAIL-R2) and CD95 (Fas) on OC cells (, Supplementary Figure S1). Similar effects were seen for a lower concentration of gemcitabine (4.4 nM). Inhibitory HLA-ABC expression was upregulated upon gemcitabine treatment on all OC cell lines, while HLA-E expression was enhanced on SKOV-3 only (Supplementary Figure S2). These results demonstrate that gemcitabine enhances both the percentage of OC cells that express NK cell activating ligands and death receptors, as well as the expression levels of these molecules.
Figure 1. Gemcitabine increases NK cell activating ligand and death receptor expression on OC cells. Phenotype of OC cell lines SKOV-3 (white), IGROV-1 (light gray) and OVCAR-3 (gray) treated with gemcitabine for 48 h (all n = 4). Delta Geometric Mean Fluorescence Intensity (ΔMFI) was determined based on the same sample stained with a live/dead dye only. Protein expression (mean + SD) of NKG2D ligands (a) MICA/B, (b) ULBP1, (c) ULBP2/5/6, (d) ULBP3, DNAM-1 ligands (e) CD112 (Nectin-2), (f) CD155 (PVR), and death receptors (g) CD261 (TRAIL-R1), (h) CD262 (TRAIL-R2), and (i) CD95 (Fas) is shown. Statistical significances were evaluated per cell line by One-way ANOVA with Bonferroni correction
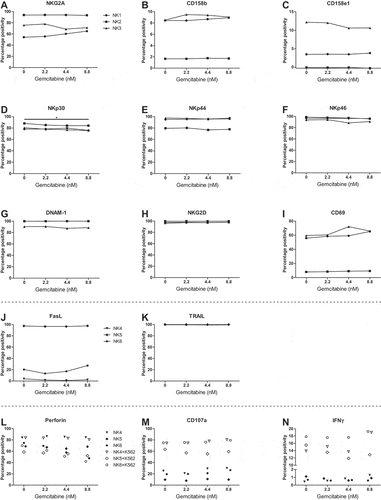
In contrast, even the highest concentration of gemcitabine (8.8 nM) did not alter the expression of activating and inhibitory receptors/ligands on HPC-NK cells (, Supplementary Figure S3). Moreover, gemcitabine did not affect perforin expression, degranulation (CD107a expression), and IFNγ production by HPC-NK cells upon 4 h stimulation with or without K562 (). Together, these data show that gemcitabine enhances expression of NK cell activating ligands and death receptors on multiple OC cell lines, while gemcitabine does not negatively impact HPC-NK cell phenotype nor inhibit functionality.
Figure 2. Gemcitabine does not negatively impact HPC-NK cell phenotype and functionality. A-K. Phenotype of HPC-NK cells treated with gemcitabine for 48 h. Percentage positivity corrected for background staining in the corresponding control sample is shown. Control samples did not contain the antibodies of interest. Protein expression of (a) NKG2A on viable CD56+CD3− cells, or (b) CD158b, (c) CD158e1, (d) NKp30, (e) NKp44, (f) NKp46, (g) DNAM-1, (h) NKG2D, (i) CD69 (all n = 3) on viable CD56+CD3−NKG2A+ cells, or (j) FasL, (k) TRAIL (both n = 3) on living CD56+ cells is shown. L-N. Functionality of HPC-NK cells treated with gemcitabine for 48 h, after 4 h stimulation with (white) or without (black) K562 (all n = 3). Percentage positivity as protein expression of (l) perforin on viable CD56+ cells, (m) CD107a (degranulation) and (n) IFNγ on viable CD56+ perforin+ HPC-NK cells is shown. Graphs show individual donors (NK1-6). Statistical significance was tested by repeated-measures One-way ANOVA for A-K and repeated-measures Two-way ANOVA for L-N, all ANOVAs with Bonferroni correction
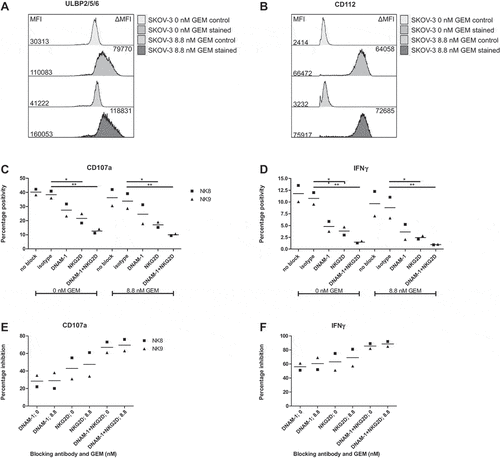
Pre-treatment of SKOV-3 cells with gemcitabine does not improve HPC-NK cell functionality
Based on the slight increases we found in the expression of NK cell activating ligands, we next investigated whether gemcitabine pre-treatment of SKOV-3 cells enhances SKOV-3 sensitivity to HPC-NK cell-mediated killing. First, we pre-treated SKOV-3 with 0 or 8.8 nM gemcitabine for 48 h. Again, a trend in upregulation of ULBP2/5/6 and CD112 expression, ligands of NKG2D and DNAM-1 on NK cells, respectively, was found on these SKOV-3 cells (). Next, we co-cultured HPC-NK cells with these pre-treated SKOV-3 cells for 4 h. There was no significant difference between HPC-NK cell degranulation () and IFNγ production () after co-culture with untreated vs pre-treated SKOV-3 cells (mean 40% vs 36%, and mean 12% vs 10%, respectively) (Supplementary Figure S4). To investigate whether gemcitabine sensitizes OC cells to DNAM-1 and/or NKG2D-mediated activation of HPC-NK cells, we used DNAM-1 and NKG2D blocking antibodies. First, we confirmed that DNAM-1 and NKG2D were expressed on HPC-NK cells (>85%, data not shown). Notably, DNAM-1 blockade as well as NKG2D blockade decreased HPC-NK cell degranulation and IFNγ production to a similar extent in response to untreated or pre-treated SKOV-3 cells. DNAM-1 and NKG2D co-blockade further diminished HPC-NK cell degranulation and IFNγ production to approximately 10–15% and 1%, respectively, both in untreated and pre-treated SKOV-3 cells. This indicates that NKG2D and DNAM-1 signaling is involved in SKOV-3-mediated HPC-NK cell activation. Compared to co-culture with untreated SKOV-3 cells, gemcitabine pre-treatment did not increase the percentage inhibition of HPC-NK cell degranulation and IFNγ production by the respective blocking antibodies relative to isotype control treatment (). These results indicate that OC cells can activate HPC-NK cells via expression of DNAM-1 and NKG2D ligands, yet upregulation of these ligands by gemcitabine does not enhance OC-reactivity of HPC-NK cells.
Figure 3. Pre-treatment of SKOV-3 cells with gemcitabine does not improve HPC-NK cell functionality. A-B. Phenotype of SKOV-3 pre-treated with or without 8.8 nM gemcitabine for 48 h in a representative experiment. Overlay of (a) ULBP2/5/6 and (b) CD112 (nectin-2) expression is shown. The control sample contained a live/dead dye only. The numbers on the left show the median fluorescence intensity (MFI), the numbers on the right the delta MFI (ΔMFI), which was determined based on the control sample. C-F. Functionality of HPC-NK cells after 4 h stimulation with SKOV-3 cells pre-treated 48 h with or without 8.8 nM gemcitabine (GEM, n = 2). C-D. Percentage positivity of (c) CD107a (degranulation) and (d) IFNγ expression on living CD56+ perforin+ HPC-NK cells is shown in the absence or presence of an isotype control antibody, DNAM-1 and/or NKG2D blocking antibody. E-F. Percentage inhibition of (e) CD107a (degranulation) and (f) IFNγ positivity by blocking antibodies relative to isotype after 4 h stimulation with pre-treated (8.8 nM GEM) or untreated (0 nM GEM) SKOV-3 cells. Graphs show NK cell donors (NK8-9) with mean in panel C-F. Statistical significance was tested for C-F by repeated-measures Two-way ANOVA with Bonferroni correction
HPC-NK cells and gemcitabine additively kill OC cells in vitro
Next, we determined the cytotoxic effects of gemcitabine on OC cell lines as well as HPC-NK cells after 48 h incubation. Gemcitabine dose-dependently killed OC cell lines and HPC-NK cells. A concentration of 2.2 nM gemcitabine induced 58% (SKOV-3), 43% (IGROV-1), or 20% (OVCAR-3) of OC cell death, and 24% HPC-NK cell death after 48 h (). We continued further experiments with SKOV-3 and IGROV-1, and used 2.2 nM gemcitabine as the highest concentration in next experiments to have a sufficient window of opportunity to study the anti-tumor effect of HPC-NK cell addition.
Figure 4. HPC-NK cells and gemcitabine additively kill OC cells in vitro. (a) Cell killing of OC cell lines (SKOV-3, white; IGROV-1, light gray; OVCAR-3, gray) or HPC-NK cells (dark gray) 48 h after gemcitabine incubation (n = 3). B-C. (b) SKOV-3 or (c) IGROV-1 cell killing 48 h after treatment with gemcitabine and/or HPC-NK cells (no HPC-NK cells, white; effector-to-target (E:T) ratio 0.3:1, light gray; 0.6:1/0.4:1, respectively, gray) (n = 3, n = 3–5, respectively). The stars above lines refer to significant differences as compared to no NK cells at the same gemcitabine concentration, the stars above bars reflect significant differences as compared to no gemcitabine at the same E:T ratio (ref). D-G. Fold change in HPC-NK cell (d-e) IFNγ (both n = 3), and (f-g) granzyme B (n = 2–3 and n = 3, respectively) secretion 48 h after co-culture with gemcitabine and (d, f) SKOV-3 or (e, g) IGROV-1 at an E:T ratio of 0.3:1 (light gray), or 0.6:1/0.4:1, respectively (gray) compared to 0 nM gemcitabine for each E:T ratio. (h) Cell killing of primary patient-derived OC cells ASC 009 72 h after gemcitabine incubation (one experiment, triplicates). (i) ASC 009 cell killing 72 h after treatment with gemcitabine and/or HPC-NK cells (no HPC-NK cells, white; E:T ratio 0.15:1, light gray; 0.3:1, gray; 0.6:1, dark gray) (n = 3 NK cell donors). The stars above lines refer to significant differences as compared to no NK cells at the same gemcitabine concentration, the stars above bars reflect significant differences as compared to no gemcitabine at the same E:T ratio (ref). Graphs show mean + SEM for A-G, I and mean + SD for H. Statistical significance was determined by (a) repeated-measures One-way ANOVA per cell type, (b and i) repeated-measures Two-way ANOVAs, (c) Two-way ANOVAs, (h) One-way ANOVA (all ANOVAs with Bonferroni correction), (f) a Kruskal–Wallis test with Dunn’s correction per E:T ratio, and (d-e and g) a Friedman test with Dunn’s correction per E:T ratio
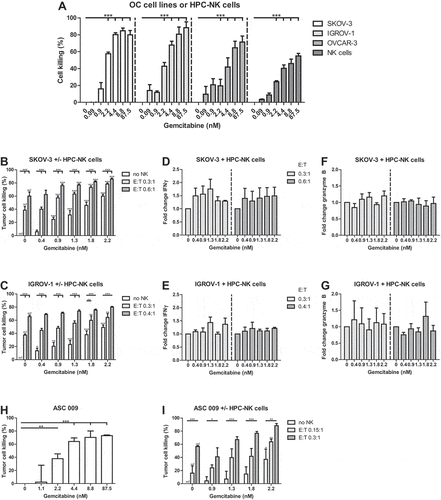
Importantly, HPC-NK cells (E:T ratio 0.3:1) combined with 2.2 nM gemcitabine induced significantly higher killing of SKOV-3 cells (78%) than monotreatment with 2.2 nM gemcitabine (60%) or HPC-NK cells (0.3:1, 39%) (). Furthermore, combination of HPC-NK cells (E:T 0.3:1) and 2.2 nM gemcitabine killed more IGROV-1 cells (64%), compared to monotherapy with 2.2 nM gemcitabine (49%) or HPC-NK cells (37%) (). The cell culture supernatant of HPC-NK and OC cells showed no difference in IFNγ and granzyme B secretion in the presence of gemcitabine (, Supplementary Figure S5).
Next, to confirm these results in primary cells, we performed similar experiments using primary patient-derived OC cells ASC 009. ASC 009 showed comparable phenotypical and cytotoxic effects upon 48–72 h exposure to gemcitabine as 48 h treatment of SKOV-3 and IGROV-1. ASC 009 C was incubated for 72 h in the killing assay because of a longer doubling time than 48 h (70 h). Gemcitabine (8.8 nM) slightly increased NK cell activating ligand and death receptor protein expression after 48 h (Supplementary Figure S6) and 2.2 nM gemcitabine killed 38% of OC cells after 72 h (). Combination therapy of HPC-NK cells (E:T 0.15:1) and 2.2 nM gemcitabine killed more OC cells (64%) than monotreatment with 2.2 nM gemcitabine (37%) and HPC-NK cells (0.15:1, 16%) (). These data demonstrate that gemcitabine and HPC-NK cells additively kill OC cells in vitro.
HPC-NK cells and gemcitabine additively decrease OC growth in SKOV-3 bearing NSG mice
To confirm the additive anti-OC effect of gemcitabine and HPC-NK cells seen in vitro, we next studied the effects of gemcitabine in NSG mice engrafted with i.p. SKOV-3-luc-GFP tumors. First, we examined the anti-tumor effect (using BLI) and toxicity of i.p. gemcitabine treatment. Mice were treated with gemcitabine twice weekly for 2 weeks followed by 1-week rest, for two cycles, which is similar to treatment of ovarian cancer patients.Citation18 Tumor growth dose-dependently decreased in mice receiving gemcitabine (). Since no dosage-related toxicity was observed, we continued with the highest dosage.
Figure 5. HPC-NK cells and gemcitabine additively decrease OC growth in SKOV-3 bearing NSG mice. (a) Radiance (photons/second/cm2/steradian) of tumors on day 34 after starting treatment (4–5 mice per group, excluding mice that did not show tumor growth over time) in SKOV-3 bearing NSG mice (0.2x106 tumor cells injected i.p. at day −4) treated i.p. with 0 (PBS), 10, 20 or 40 mg/kg gemcitabine (GEM) twice weekly for two weeks followed by one week rest (one cycle) for two cycles. (b) BLI images from the experiment depicted in panel A on day −1 (1 day before treatment) and day 34 after starting treatment. (c) Radiance (photons/second/cm2/steradian) of tumors over time (7–11 mice per group, excluding mis-injections) in SKOV-3 bearing NSG mice (0.2 million tumor cells injected i.p. 4 days before treatment) treated i.p. with PBS (control, gray) or 40 mg/kg gemcitabine (blue) for three cycles, HPC-NK cells together with rhIL-15 cytokine support (black) or a combination of gemcitabine, HPC-NK cells and rhIL-15 (red). HPC-NK cells and rhIL-15 were administered once or every other day, respectively during the weeks the mice were off gemcitabine. After the last HPC-NK cell injection, rhIL-15 was given for two weeks. All mice could be followed for tumor growth until day 55 due to saturation. (d) Survival of mice from the experiment depicted in panel C. Mice that were sacrificed solely due to a subcutaneous tumor >1000mm3 (n = 6/44) were censored in the survival analysis. E-F. Radiance (photons/second/cm2/steradian) of tumors over time (11 mice per group) in SKOV-3 bearing NSG mice (0.2 million tumor cells injected i.p. 4 days before treatment) treated i.p. with PBS (control, gray), 40 mg/kg gemcitabine for two cycles (blue), HPC-NK cells together with rhIL-15 cytokine support (black), or a combination of HPC-NK cells, rhIL-15 and gemcitabine (red). HPC-NK cells and rhIL-15 were administered (starting) one week before the first gemcitabine cycle and during the weeks the mice were off gemcitabine, once or every other day per week, respectively. After the last HPC-NK cell injection, rhIL-15 was given for two weeks. All mice could be followed for tumor growth until day 63 due to saturation. Statistics was performed on data from panel E and significant differences are shown in panel F. (g) BLI pictures from the experiment depicted in panels E-F on day 63 after starting treatment. (h) Survival of mice from the experiment depicted in panels E-G. Mice that were sacrificed solely due to a subcutaneous tumor >1000mm3 (n = 2/44) were censored in the survival analysis. Graphs show mean + SD for C and E. Statistical significance was tested for A by one-way ANOVA, for C by two-way ANOVA, for E (shown in F) by repeated-measures two-way ANOVA (all ANOVAs with Bonferroni correction), and for D and H by Cox regression analysis
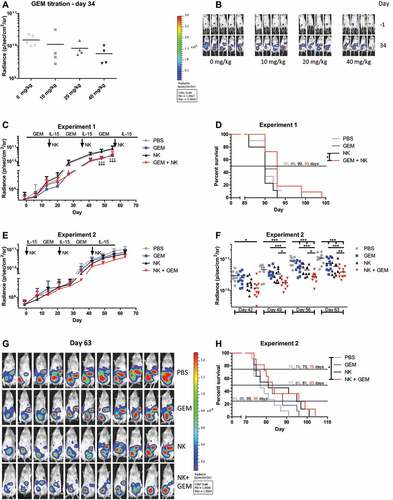
To analyze the phenotype of SKOV-3-luc-GFP tumor nodules, gemcitabine or PBS treatment was performed in a high tumor load model. In this model, one cycle of gemcitabine slightly enhanced the expression of NK cell activating ligands and death receptors on SKOV-3-luc-GFP tumor nodules, which is in agreement with our in vitro data (Supplementary Figure S7).
Subsequently, we investigated the combined anti-tumor effect of i.p. gemcitabine and i.p. HPC-NK cell co-treatment in NSG mice engrafted with i.p. SKOV-3-luc-GFP tumors. Since 40 mg/kg gemcitabine may be substantially higher than the concentrations used in vitro (initially 6 mM based on a mouse weight of 22.5 g and an injection volume of 0.5 ml, vs maximum 8.8 nM in vitro), HPC-NK cells might be severely harmed by gemcitabine. Hence, we separated HPC-NK cell and rhIL-15 injections from gemcitabine treatment, starting with the latter. Gemcitabine treatment significantly decreased tumor growth and increased survival compared to control mice (). Surprisingly, no (additional) anti-tumor effect was seen upon HPC-NK cell treatment, neither in HPC-NK only nor in gemcitabine plus HPC-NK cell treated animals. Possibly, the high tumor load at the time of HPC-NK cell therapy was not effective in killing OC cells. To further optimize our model, we switched the order of HPC-NK cell and gemcitabine administration. Here, tumor growth was decreased in gemcitabine-treated, HPC-NK-treated, and HPC-NK plus gemcitabine-treated mice compared to control mice (). Moreover, tumor growth was significantly lower in mice treated with the combination therapy of HPC-NK cells and gemcitabine compared to mice treated with HPC-NK cells or gemcitabine monotherapy, or PBS-treated mice. Survival analysis showed that the combination therapy slightly prolonged the survival of mice (median survival 83 days) compared to single treatment with gemcitabine or NK cells (median survival both 81 days), and that survival was significantly extended compared to PBS treated mice (median survival 77 days) (). Furthermore, 25% survival was reached at 96 days for combination therapy, 95 days for NK cell monotherapy, 88 days for gemcitabine monotherapy, and 84 days for PBS control therapy. Together, these experiments demonstrate that HPC-NK cells and gemcitabine additively decrease OC growth in vivo dependent on the timing of HPC-NK cell administration.
DISCUSSION
Since many OC patients experience recurrence after standard therapy, novel therapeutic approaches are urgently needed to combat and control relapse and improve survival for these patients. Because OC is prone to NK cell-mediated immunity, NK cell adoptive transfer could be an interesting therapeutic option. We previously showed that HPC-NK cells can kill human OC cells in vitro and in vivo.Citation6 However, improvement of HPC-NK cell therapy is warranted to further enhance anti-OC reactivity. This might be achieved by combining HPC-NK cells with gemcitabine, a drug already used in recurrent OC patients. Gemcitabine has been shown to increase expression of NKG2D ligands and death receptors in various cancers,Citation8,Citation10–14 thereby sensitizing tumor cells to NK cell-mediated killing. Therefore, we investigated the phenotypical, functional, and cytotoxic effects of gemcitabine treatment on HPC-NK cells and/or OC cells in vitro and in vivo.
First, we confirmed that gemcitabine promotes expression of NK cell activating ligands and death receptors on OC cells. This is in line with previous studies using various cancer types, where upregulation of NKG2D ligands (ULBP2/5/6 and MICA/B) was reported.Citation8,Citation10–14 Moreover, one of these studies reported a gemcitabine-mediated increase in CD95 and TRAIL-R2 expression.Citation10 Another study documented enhanced inhibitory HLA-ABC expression on lung cancer cells due to gemcitabine.Citation8 We show that gemcitabine increases expression of NKG2D ligand ULBP2/5/6, DNAM-1 ligands (CD112 (Nectin-2) and CD155 (PVR)), death receptors (CD261 (TRAIL-R1), CD262 (TRAIL-R2) and CD95 (Fas)), and inhibitory HLA-ABC molecules on OC cell lines and primary patient-derived OC cells. In contrast to other studies, we did not observe modulation of MICA/B in OC cells. This difference could be related to used cancer cell type.
Next, we found that gemcitabine did not negatively affect HPC-NK cell phenotype nor functionality at nanomolar concentrations. The literature on the impact of gemcitabine treatment on NK cells is very limited. Markasz et al. reported that gemcitabine pre-treatment (20 h, 2–250 µM) impaired NK-reactivity toward K562 cells, using NK cell lines.Citation19 However, this concentration is much higher than what we used for HPC-NK cell incubation (max 8.8 nM). For clinical translation, it is relevant to investigate the impact of higher gemcitabine concentrations on HPC-NK cell phenotype and functionality, since the mean plasma concentration of gemcitabine ranges between 30 and 85 µM directly after i.v. administration of 1000 mg/m2.Citation9,Citation20,Citation21 However, plasma clearance of gemcitabine is rapid with a half-life of 5–20 minCitation22,Citation23 and the gemcitabine concentration in the peritoneal cavity, the location where we would like to inject HPC-NK cells, will probably be lower. Therefore, the concentrations that we investigated are biologically relevant, especially if NK cells and gemcitabine are not administered at the same time.
Despite gemcitabine-mediated upregulation of NK cell activating ligands on OC cells, we did not observe that pre-treatment of OC cells with gemcitabine improved the functionality of HPC-NK cells on single cell level. Apparently, the increase in ULBP2/5/6 and CD112 expression on SKOV-3 cannot (further) enhance HPC-NK cell activation, which could be related to upregulated expression of inhibitory HLA class I (HLA-ABC and HLA-E) molecules on SKOV-3 by gemcitabine treatment, which may have balanced the activating and inhibitory effects. Altogether, this combined upregulation of both activating and inhibitory receptors may underly our observation that pre-treatment of SKOV-3 cells with gemcitabine did not enhance HPC-NK cell tumor reactivity. Another explanation might be that our HPC-NK cells are already highly activated in the presence of OC cells without gemcitabine treatment ().Citation6,Citation16,Citation24–28 In previous publications, we have shown that we can already approach nearly 100% HPC-NK cell-mediated OC cell death in the absence of gemcitabine using an E:T ratio of 10:1, which is lower compared to the E:T ratio that is needed for peripheral blood (PB)-NK cells to kill 100% of OC cells.Citation6,Citation16 However, to study possible additive effects of HPC-NK cells and gemcitabine in a more physiological setting, all combination experiments were performed at low E:T ratios. Furthermore, we previously demonstrated that our HPC-NK cells produce higher levels of granzyme B compared to PB-NK cells, and that our HPC-NK cell population contains serial killers, which kill two or more OC targets.Citation16 However, we cannot exclude HPC-NK cell exhaustion and/or OC immune escape because we did not perform long-term OC outgrowth experiments. Nevertheless, our results support that our HPC-NK cells are highly activated, and may therefore reach their maximum capacity already in the absence of gemcitabine. For gemcitabine monotherapy escape is a realistic scenario because even with our highest dose of 87.5 nM we did not reach 100% killing in OC cell lines. Instead, plateaus of maximum killing were observed. In contrast to HPC-NK cells, improvement of peripheral blood (PB)-NK cell functionality has been reported upon increased MICA/B or ULBP2/5/6 expression on tumor cells mediated by gemcitabine.Citation8,Citation11 Using NKG2D and DNAM-1 blocking antibodies, we show that NKG2D and DNAM-1 are important receptors for SKOV-3-mediated HPC-NK cell activation. Moreover, we demonstrate that upregulation of NKG2D and DNAM-1 ligands on OC cells by gemcitabine does not lead to increased HPC-NK cell activation.
Notably, in our study, we showed that all our OC cell lines were susceptible to gemcitabine, with the highest sensitivity for SKOV-3, followed by IGROV-1 and OVCAR-3. This is in contrast to another study showing that OVCAR-3 was most sensitive to gemcitabine, followed by IGROV-1 and SKOV-3.Citation29 This difference might be attributed to different incubation times (48 h in our study vs 5 days in the other study) or other differences in experimental conditions. Despite direct effects on OC survival, gemcitabine also negatively affects HPC-NK cell numbers. However, this did not lead to decreased IFNγ or granzyme B secretion in combination killing assays. A possible explanation for this observation might be that remaining HPC-NK cells treated with gemcitabine still produce substantial IFNg and granzyme B upon OC cell stimulation. Most importantly, we found that HPC-NK cells and gemcitabine additively killed OC cells, both well-established OC cell lines and primary patient-derived OC cells. Notably, in our view, the additive effect is not related to the upregulation of NKG2D and DNAM-1 ligands on ovarian cancer cells. Upregulation of NKG2D and DNAM-1 ligands is mostly found at a concentration higher than 2.2 nM, while the additive effects of gemcitabine and HPC-NK cells occur at concentrations of 2.2 nM or lower. Higher concentrations of gemcitabine were not investigated for additive effects because 4.4 nM gemcitabine already killed 80% of SKOV-3 and 68% of IGROV-1 cells, limiting the window of opportunity for NK cells to kill these cells in vitro.
Finally, we demonstrated that HPC-NK cells and gemcitabine additively decrease OC growth in vivo. We show an anti-tumor effect of i.p. gemcitabine administration in NSG mice bearing i.p. human OC. Our gemcitabine dosage (40 mg/kg twice weekly) and administration schedule is comparable to other studies in miceCitation13,Citation30–38 and lower than in patients (human equivalent dose in mg/kg = animal dose in mg/kg/(Km human/Km mouse) = 40/(37/3) = 3.24 mg/kg,Citation39 corresponding to 120 mg/m2 twice weekly (dose in mg/m2 = dose in mg * Km = 3.24 * 37),Citation39 which is ~4-fold lower than the recommended dosage of 1000 mg/m2 once weekly in patients).Citation18 However, since gemcitabine is administered i.v. in patients, the concentration in the peritoneal cavity of patients will likely be lower, decreasing the difference between the mouse and human dosage. We administered gemcitabine i.p. because the peritoneal cavity is the region of interest in our OC model. The concentration we injected was approximately 6 mM, which is considerably higher than our in vitro nanomolar concentrations, and the micromolar concentrations that can be reached in patients. Notably, gemcitabine injected i.p. rapidly disseminates to the blood, leading to a decreased i.p. concentration.Citation40 Nevertheless, we decided to separate gemcitabine injections from HPC-NK cell infusions in time, to avoid potential harmful effects of high gemcitabine concentrations on HPC-NK cell numbers, phenotype, and functionality. Since gemcitabine can enhance expression of NK cell activating ligands and death receptors, we started with gemcitabine treatment followed by HPC-NK cell treatment in our first combination experiment. Although we found that gemcitabine decreased tumor growth, HPC-NK cells did not unfortunately. This observation is in contrast to our previously published studies,Citation6,Citation16 where HPC-NK cells established anti-OC effects in vivo. An important difference between this study and our previous studies is the timing of HPC-NK cell treatment upon tumor administration, namely 4 days in previous studies, compared to 18 days in this study. Hoogstad-van Evert et al. showed that a higher tumor load decreases the antitumor efficacy of HPC-NK cells.Citation6 Probably, in these 18 days, the tumor mass has grown too large for HPC-NK cells to effectively kill the tumor cells. Likewise, combination treatment did not lead to an extra antitumor effect compared to gemcitabine monotherapy, indicating that even in gemcitabine-treated animals the tumor load was too high for effective HPC-NK cell tumor reactivity. Therefore, in the next experiment, we started with HPC-NK cell treatment 4 days after tumor injection followed by gemcitabine treatment. Here, we observed effective anti-tumor responses in mice treated with either gemcitabine or HPC-NK cell monotherapy, and mice treated with both gemcitabine and HPC-NK cells demonstrated additive killing of OC cells in vivo. The second experiment shows that a combination of gemcitabine and HPC-NK cells has potential, especially when the tumor load is low, which resembles the situation in OC patients early during recurrence when bulk disease is not yet present. This suggests that HPC-NK cells clear the tumor cells, while or before bulky tumor is established. In follow-up studies, we will further evaluate the anti-tumor effect of a single HPC-NK cell injection 4 days after tumor injection vs. multiple HPC-NK cell injections in the presence or absence of subsequent gemcitabine cycles for possible clinical translation to patients. Moreover, additional gemcitabine and HPC-NK cell cycles or optimizing the treatment schedule might further improve the anti-OC effect.
Collectively, our data indicate that combination therapy of HPC-NK cells and gemcitabine improves ovarian cancer killing in vitro and in vivo. We show that gemcitabine promotes the expression of NK cell activating ligands and death receptors on OC cells. Furthermore, we demonstrate that gemcitabine and HPC-NK cells additively kill OC cells in vitro and in vivo. Based on our encouraging preclinical data, we provide a rationale for clinical translation to combine HPC-NK cell adoptive transfer with gemcitabine in OC patients early during recurrence. In conclusion, HPC-NK cells and gemcitabine are promising candidates for combination therapy in recurrent OC patients.
Author contributions
JvdM designed and performed experiments, analyzed data and wrote the manuscript; PdJ performed experiments, analyzed data and provided advice; AG and JM designed and performed experiments and analyzed data; JdK and MV performed experiments; AvdW, JB, RM, NS, JHvE, PO and JJ provided advice; WH and HD designed and supervised the research; and all authors read and approved the manuscript.
ORCIDs
Jolien M.R. Van der Meer 0000-0001-9288-1500, Paul K.J.D. de Jonge 0000-0002-1228-5002, Anniek B. van der Waart 0000-0001-5986-3060, Alexander C. Geerlings 0000-0002-9146-8164, Jurgen P. Moonen not available, Jolanda Brummelman 0000-0003-3581-4816, Janne de Klein not available, Malou C. Vermeulen not available, Ralph J.A. Maas 0000-0002-3185-5226, Nicolaas P.M. Schaap 0000-0001-7696-4752, Janneke S. Hoogstad-van Evert 0000-0001-9838-0977, Petronella B. Ottevanger 0000-0001-8209-487X, Joop H. Jansen 0000-0001-9459-568X, Willemijn Hobo 0000-0002-8206-8185, Harry Dolstra 0000-0002-3998-687X
Supplemental Material
Download ()Acknowledgments
The authors thank the Radboud University Medical Center Central Animal Laboratory for assistance. We also thank Dr. Guido Zaman (Netherlands Translational Research Center B.V.) for providing primary patient-derived ovarian cancer cells ASC 009.
Disclosure statement
None of the authors have any competing interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–13. doi:10.3322/caac.21654.
- Gadducci A, Cosio S, Zola P, Landoni F, Maggino T, Sartori E. Surveillance procedures for patients treated for epithelial ovarian cancer: a review of the literature. International Journal of Gynecological Cancer: Official Journal of the International Gynecological Cancer Society. 2007;17(1):21–31. doi:10.1111/j.1525-1438.2007.00826.x.
- Norell H, Carlsten M, Ohlum T, Malmberg KJ, Masucci G, Schedvins K, Altermann W, Handke D, Atkins D, Seliger B, et al. Frequent loss of HLA-A2 expression in metastasizing ovarian carcinomas associated with genomic haplotype loss and HLA-A2-Restricted HER-2/ neu-specific immunity. Cancer Res. 2006;66(12):6387–6394. doi:10.1158/0008-5472.CAN-06-0029.
- Li K, Mandai M, Hamanishi J, Matsumura N, Suzuki A, Yagi H, Yamaguchi K, Baba T, Fujii S, Konishi I, et al. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: high expression of ULBP2 is an indicator of poor prognosis. Cancer Immunology, Immunotherapy: CII. 2009;58(5):641–652. doi:10.1007/s00262-008-0585-3.
- Felices M, Chu S, Kodal B, Bendzick L, Ryan C, Lenvik AJ, Boylan KLM, Wong HC, Skubitz APN, Miller JS, et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol Oncol. 2017;145(3):453–461. doi:10.1016/j.ygyno.2017.02.028.
- Hoogstad-van Evert JS, Cany J, van den Brand D, Oudenampsen M, Brock R, Torensma R, Bekkers RL, Jansen JH, Massuger LF, Dolstra H. Umbilical cord blood CD34(+) progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rg(null) mice. Oncoimmunology. 2017;6(8):e1320630. doi:10.1080/2162402X.2017.1320630
- Fruscella E, Gallo D, Ferrandina G, D’Agostino G, Scambia G. Gemcitabine: current role and future options in the treatment of ovarian cancer. Crit Rev Oncol Hematol. 2003;48(1):360–371. doi:10.1016/S1040-8428(03)00119-7.
- Okita R, Wolf D, Yasuda K, Maeda A, Yukawa T, Saisho S, Shimizu K, Yamaguchi Y, Oka M, Nakayama E, et al. Contrasting effects of the cytotoxic anticancer drug gemcitabine and the EGFR tyrosine kinase inhibitor gefitinib on NK cell-mediated cytotoxicity via regulation of NKG2D ligand in non-small-cell lung cancer cells. PloS One. 2015;10(10):e0139809. doi:10.1371/journal.pone.0139809.
- Kroep JR, Giaccone G, Voorn DA, Smit EF, Beijnen JH, Rosing H, van Moorsel CJ, van Groeningen CJ, Postmus PE, Pinedo HM et al. Gemcitabine and Paclitaxel: pharmacokinetic and Pharmacodynamic Interactions in Patients With Non–Small-Cell Lung Cancer . Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 1999;17(7):2190–2197. doi:10.1200/JCO.1999.17.7.2190.
- Gravett AM, Dalgleish AG, Copier J. In vitro culture with gemcitabine augments death receptor and NKG2D ligand expression on tumour cells. Sci Rep. 2019;9(1):1544. doi:10.1038/s41598-018-38190-2.
- Morisaki T, Onishi H, Koya N, Kiyota A, Tanaka H, Umebayashi M, Ogino T, Nagamatsu I, Katano M. Combinatorial cytotoxicity of gemcitabine and cytokine-activated killer cells in hepatocellular carcinoma via the NKG2D-MICA/B system. Anticancer Res. 2011;31(7):2505–2510
- Miyashita T, Miki K, Kamigaki T, Makino I, Nakagawara H, Tajima H, Takamura H, Kitagawa H, Fushida S, Ahmed AK, et al. Low-dose gemcitabine induces major histocompatibility complex class I-related chain A/B expression and enhances an antitumor innate immune response in pancreatic cancer. Clin Exp Med. 2017;17(1):19–31. doi:10.1007/s10238-015-0394-x.
- Xie X, Zhou Y, Wang X, Guo J, Li J, Fan H, Dou J, Shen B, Zhou C. Enhanced antitumor activity of gemcitabine by polysaccharide-induced NK cell activation and immune cytotoxicity reduction in vitro/vivo. Carbohydr Polym. 2017;173:360–371. doi:10.1016/j.carbpol.2017.06.024.
- Shimizu T, Tomogane M, Miyashita M, Ukimura O, Ashihara E. Low dose gemcitabine increases the cytotoxicity of human Vγ9Vδ2 T cells in bladder cancer cells in vitro and in an orthotopic xenograft model. Oncoimmunology. 2018;7(5):e1424671. doi:10.1080/2162402X.2018.1424671.
- Gurlevik E, Fleischmann-Mundt B, Brooks J, Demir IE, Steiger K, Ribback S, Yevsa T, Woller N, Kloos A, Ostroumov D, et al. Administration of gemcitabine after pancreatic tumor resection in mice induces an antitumor immune response mediated by natural killer cells. Gastroenterology. 2016;151(2):338–50.e7. doi:10.1053/j.gastro.2016.05.004.
- Van der Meer JMR, Maas RJA, Guldevall K, Klarenaar K, de Jonge P, Evert JSH, van der Waart AB, Cany J, Safrit JT, Lee JH et al. IL-15 superagonist N-803 improves IFNγ production and killing of leukemia and ovarian cancer cells by CD34(+) progenitor-derived NK cells. Cancer Immunology, Immunotherapy: CII. 2021;70(5):1305–1321. doi:10.1007/s00262-020-02749-8
- Grobben Y, De Man J, Van Doornmalen AM, Muller M, Willemsen-Seegers N, Vu-Pham D, Mulder WR, Prinsen MBW, De Wit J, Sterrenburg JG, et al. Targeting Indoleamine 2,3-Dioxygenase in cancer models using the novel small molecule inhibitor NTRC 3883-0. Front Immunol. 2020;11:609490. doi:10.3389/fimmu.2020.609490.
- Abushahin F, Rose PG. Role of gemcitabine in the treatment of ovarian cancer. Women’s Health. 2007;3(3):279–290. doi:10.2217/17455057.3.3.279.
- Markasz L, Stuber G, Vanherberghen B, Flaberg E, Olah E, Carbone E, Eksborg S, Klein E, Skribek H, Szekely L, et al. Effect of frequently used chemotherapeutic drugs on the cytotoxic activity of human natural killer cells. Mol Cancer Ther. 2007;6(2):644–654. doi:10.1158/1535-7163.MCT-06-0358.
- Lanz C, Früh M, Thormann W, Cerny T, Lauterburg BH. Rapid determination of gemcitabine in plasma and serum using reversed-phase HPLC. J Sep Sci. 2007;30(12):1811–1820. doi:10.1002/jssc.200600534.
- Conte PF, Gennari A, Donati S, Salvadori B, Baldini E, Bengala C, Pazzagli I, Orlandini C, Danesi R, Fogli S, et al. Gemcitabine plus epirubicin plus taxol (GET) in advanced breast cancer: a phase II study. Breast Cancer Res Treat. 2001;68(2):171–179. doi:10.1023/A:1011945623464.
- Wong A, Soo RA, Yong WP, Innocenti F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab Rev. 2009;41(2):77–88. doi:10.1080/03602530902741828.
- Ciccolini J, Serdjebi C, Peters GJ, Giovannetti E. Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: an EORTC-PAMM perspective. Cancer Chemother Pharmacol. 2016;78(1):1–12. doi:10.1007/s00280-016-3003-0.
- Spanholtz J, Tordoir M, Eissens D, Preijers F, van der Meer A, Joosten I, Schaap N, de Witte TM, Dolstra H. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PloS One. 2010;5(2):e9221. doi:10.1371/journal.pone.0009221
- Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, Schaap N, Dolstra H. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PloS One. 2011;6(6):e20740. doi:10.1371/journal.pone.0020740.
- Cany J, van der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, De Vries J, Boerman O, Schaap N, van der Voort R, Spanholtz J, et al. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow-residing human leukemia cells in NOD/SCID/IL2Rgnull Mice. PloS One. 2013;8(6):e64384. doi:10.1371/journal.pone.0064384.
- Cany J, van der Waart AB, Spanholtz J, Tordoir M, Jansen JH, van der Voort R, Schaap NM, Dolstra H. Combined IL-15 and IL-12 drives the generation of CD34+-derived natural killer cells with superior maturation and alloreactivity potential following adoptive transfer. Oncoimmunology. 2015;4(7):e1017701. doi:10.1080/2162402X.2015.1017701.
- Roeven MW, Thordardottir S, Kohela A, Maas F, Preijers F, Jansen JH, Blijlevens NM, Cany J, Schaap N, Dolstra H, et al. The aryl hydrocarbon receptor antagonist stemRegenin1 improves in vitro generation of highly functional natural killer cells from CD34+hematopoietic stem and progenitor cells. Stem Cells Dev. 2015;24(24):2886–2898. doi:10.1089/scd.2014.0597.
- Beaufort CM, Helmijr JC, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, Murtaza M, Van Ijcken WFJ, Heine AAJ, Smid M, et al. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PloS One. 2014;9(9):e103988. doi:10.1371/journal.pone.0103988.
- Mondal G, Almawash S, Chaudhary AK, Mahato RI. EGFR-targeted cationic polymeric mixed micelles for codelivery of gemcitabine and miR-205 for treating advanced pancreatic cancer. Mol Pharm. 2017;14(9):3121–3133. doi:10.1021/acs.molpharmaceut.7b00355.
- Kattel K, Mondal G, Lin F, Kumar V, Mahato RI. Biodistribution of self-assembling polymer–gemcitabine conjugate after systemic administration into orthotopic pancreatic tumor bearing mice. Mol Pharm. 2017;14(5):1365–1372. doi:10.1021/acs.molpharmaceut.6b00929.
- Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Self-assembling, amphiphilic polymer–gemcitabine conjugate shows enhanced antitumor efficacy against human pancreatic adenocarcinoma. Bioconjug Chem. 2013;24(7):1161–1173. doi:10.1021/bc400032x.
- Raki M, Kanerva A, Ristimaki A, Desmond RA, Chen DT, Ranki T, Sarkioja M, Kangasniemi L, Hemminki A. Combination of gemcitabine and Ad5/3-Δ24, a tropism modified conditionally replicating adenovirus, for the treatment of ovarian cancer. Gene Ther. 2005;12(15):1198–1205. doi:10.1038/sj.gt.3302517.
- Raki M, Sarkioja M, Desmond RA, Chen DT, Butzow R, Hemminki A, Kanerva A. Oncolytic adenovirus Ad5/3-Δ24 and chemotherapy for treatment of orthotopic ovarian cancer. Gynecol Oncol. 2008;108(1):166–172. doi:10.1016/j.ygyno.2007.09.013.
- Garcia-Cremades M, Pitou C, Iversen PW, Troconiz IF. Characterizing gemcitabine effects administered as single agent or combined with carboplatin in mice pancreatic and ovarian cancer xenografts: a semimechanistic pharmacokinetic/pharmacodynamics tumor growth-response model. J Pharmacol Exp Ther. 2017;360(3):445–456. doi:10.1124/jpet.116.237610.
- Gallo D, Fruscella E, Ferlini C, Apollonio P, Mancuso S, Scambia G. Preclinical in vivo activity of a combination gemcitabine/liposomal doxorubicin against cisplatin-resistant human ovarian cancer (A2780/CDDP). International Journal of Gynecological Cancer: Official Journal of the International Gynecological Cancer Society. 2006;16(1):222–230. doi:10.1111/j.1525-1438.2006.00304.x.
- Zajchowski DA, Biroc SL, Liu HL, Chesney SK, Hoffmann J, Bauman J, Kirkland T, Subramanyam B, Shen J, Ho E, et al. Anti-tumor efficacy of the nucleoside analog 1-(2-deoxy-2-fluoro-4-thio-?-D-arabinofuranosyl) cytosine (4?-thio-FAC) in human pancreatic and ovarian tumor xenograft models. International Journal of Cancer. 2005;114(6):1002–1009. doi:10.1002/ijc.20835.
- Kawaguchi H, Terai Y, Tanabe A, Sasaki H, Takai M, Fujiwara S, Ashihara K, Tanaka Y, Tanaka T, Tsunetoh S, et al. Gemcitabine as a molecular targeting agent that blocks the Akt cascade in platinum-resistant ovarian cancer. J Ovarian Res. 2014;7(1):38. doi:10.1186/1757-2215-7-38.
- Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep. 1966;50:219–244.
- Veerman G, Ruiz van Haperen VW, Vermorken JB, Noordhuis P, Braakhuis BJM, Pinedo HM, Peters GJ. Antitumor activity of prolonged as compared with bolus administration of 2′,2′-difluorodeoxycytidine in vivo against murine colon tumors. Cancer Chemother Pharmacol. 1996;38(4):335–342. doi:10.1007/s002800050492.