
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Serum autoantibodies against tumor-associated antigen have important value in the early diagnosis of hepatocellular carcinoma (HCC), but the mechanism of autoantibody production is poorly understood. We previously showed that autoantibodies against the centromere protein F (CENPF) may be useful as an early diagnostic marker for HCC. Here we explored the mechanism of cell apoptosis-based CENPF autoantibody production and verified the correlation of CENPF autoantibody level with HCC development. We demonstrated that CENPF was overexpressed and aberrantly localized throughout the nuclei and cytoplasm in human HCC cells compared with hepatic cells. CENPF overexpression promoted the production of CENPF autoantibodies in a manner that correlated with tumor growth of mouse HCC model. During apoptosis of HCC cells, CENPF protein translocated to apoptotic vesicles and relocalized at the cell surface. Through isolating apoptotic components, we found apoptotic body and blebs with lower CD31 and CD47 expression more effectively induced DC phagocytosis and maturation compared with apoptotic intact cells in vitro, and this DC response was independent of CENPF expression. Moreover, injection of mice with apoptotic bodies and blebs effectively induced an immune response and the production of CENPF-specific antibodies. Our findings provide a first elucidation of mechanisms underlying the CENPF autoantibody production via cell apoptosis-induced CENPF translocation, and demonstrate a direct correlation between CENPF autoantibody levels and HCC progression, suggesting the potential of CENPF autoantibody as an HCC diagnostic marker.
Introduction
Primary liver cancer is one of the most common malignant tumors and has the most rapidly increasing incidence in both men and women worldwide.Citation1 The estimated 5-y relative survival rate for patients diagnosed between 2008 and 2014 is only 18%.Citation2 Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and accounts for 90% of cases.Citation2 The poor sensitivity, specificity, or compliance of current screening and diagnostic methods for HCC detection could lead to decreased chance of early diagnosis and thus poor clinical outcomes, highlighting the requirement for more effective approaches.Citation3,Citation4 Tumors can stimulate the production of autoantibodies against tumor-associated antigens (TAAs).Citation5 Compared with the antigens themselves, autoantibodies have good stability and signal amplification effect in serum, thus can be detected in peripheral blood before clinical symptoms appearance. These properties suggest that serum autoantibodies could be promising early diagnostic markers for HCC.Citation6 It has been reported that autoantibodies against HCC1, CDKN2A, p53, CIP2A, and survivin could indicate the presence of HCC prior to clinical diagnosis.Citation7 In a previous study, we performed a high-throughput screening of serum autoantibodies and a clinical correlation analysis for early diagnosis of HCC using a protein microarray, and found that the centromere protein F (CENPF) autoantibodies showed a great value of early diagnosis and screening of HCC.Citation8 It had an area under the curve (AUC) in receiver operating characteristic (ROC) curve analysis of 0.826, higher than that of AFP (0.749), for detection of HCC. In addition, the sensitivity of anti-CENPF autoantibody for detecting HCC was 76.2%, which was also far superior to that of AFP (49.5%). We also found that 73.6% of patient with early AFP− HCC were positive for serum CENPF autoantibodies.Citation8
CENPF is a member of the centromere protein family, which is involved in formation of the nuclear matrix and regulates chromosome segregation during mitosis.Citation9 It is expressed in a cell cycle-dependent manner and is mainly localized to mitotic structures. CENPF gradually accumulates during the cell cycle until it reaches peak levels in the G2/M phase and is rapidly degraded upon completion of mitosis.Citation10–12 It has been reported that CENPF was highly expressed in many malignant tumors, such as HCC,Citation13,Citation14 breast cancer,Citation15 and colorectal cancer,Citation16 and was an independent prognostic factor for HCCCitation13 and breast cancer.Citation17 In our previous study, we found that CENPF is distributed throughout the nucleus and cytoplasm of HCC cells. Moreover, we detected a positive association between tumor expression levels of CENPF and serum CENPF autoantibody levels in HCC patients.Citation8 Hence we hypothesize that overexpression and abnormal localization of CENPF are important factors in promoting autoantibody production in HCC.
Apoptotic cells have been proposed as a possible source of autoantigens.Citation18 During apoptosis, the cell membrane forms cytoplasmic vesicles, some of which are shed as apoptotic bodies or blebs. Casciola-Rosen et al. have shown that UV-induced apoptosis of keratinocytes leads to redistribution of several autoantigens to apoptotic blebs.Citation19 In certain autoimmune diseases, such as systemic lupus erythematosus (SLE), the nuclear membrane, chromatin fragments, and other nuclear antigens are translocated, concentrated, and exposed on the cell surface membrane of apoptotic bodies or blebs.Citation20 Such translocation and exposure promote the phagocytosis of apoptotic bodies and blebs, which in turn increase autoantigen uptake and processing by antigen-presenting cells (APCs) such as dendritic cells (DCs), resulting in autoantibody production.Citation21–23 Autoantigen translocation and autoantibody production in cancer sera may also be linked to the process of apoptosis.Citation5,Citation24 Data obtained by screening cDNA expression libraries with autoantibodies in cancer sera suggested that apoptosis may also be involved in the autoantibody response in breast cancer.Citation25
In the present study, we sought to investigate the mechanism by which CENPF autoantibodies are produced in HCC and to determine their reliability as a potential HCC diagnostic marker. Using HCC cell lines in vitro and a mouse model of HCC, we investigated the correlation between abnormal CENPF expression and localization in HCC cells and the level of CENPF autoantibody production. We also examined the capacity of different components of apoptotic HCC cells with or without CENPF knockdown to induce CENPF exposure and DC maturation in vitro, and autoantibody production in vivo. Our results suggest that HCC development is associated with elevated serum levels of CENPF autoantibodies that may be induced by HCC cell apoptosis, followed by translocation and exposure of highly expressed CENPF in apoptotic body and blebs, phagocytosis by DCs, and stimulation of CENPF-specific immune response. These data advance our understanding of the pathways by which CENPF is delivered to the immune system and promotes autoantibody production in HCC.
Materials and methods
Cell culture and patient samples
Human HCC cell line Hep 3b and mouse HCC cell line H22 were purchased from the Cell Resource Center of Chinese Academy of Medical Sciences (Beijing, China). The human hepatic cell line QSG-7701 and HCC cell lines BEL-7402, HuH-7, Li-7, PLC/PRF/5, SK-Hep-1 were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in 5% CO2 at 37°C in RPMI 1640 (Hyclone Thermo Scientific, Waltham, MA) supplemented with phenol red, 10% fetal bovine serum (FBS; Hyclone Thermo Scientific), 100 U/mL penicillin, and 100 U/mL streptomycin. Cells in the exponential growth phase were used in the experiments.
Bone marrow-derived DCs were obtained by culturing bone marrow from BALB/c mice. Briefly, bone marrow was flushed from femur and tibia of mice, and erythrocytes were excluded using the blood cell lysis buffer (Beckman Coulter, Krefeld, Germany). Bone marrow precursors were seeded in 6-well plates at a concentration of 1 × 106 cells/well in complete RPMI 1640 medium containing 10% FBS, 3 ng/ml GM-CSF, and 2.5 ng/ml IL-4 (PeproTech, London, UK) and used at days 6–9 of culture.
HCC specimens and adjacent tissues were acquired from the Liver Research Center of Beijing Friendship Hospital, Capital Medical University. All of the patients provided written informed consent, and this study was approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University (Number 2015-P2-019-01).
Lentiviral vector construction and transfection
Lentivirus vector for mouse CENPF small hairpin RNA (shRNA) encoding a green fluorescent protein (GFP) sequence was constructed by GENECHEM (Shanghai, China). The target shRNA sequence is 5ʹ-ACTGACCAAACTTGTGATAAA-3ʹ (mouse CENPF gene GenBank accession no. XM_006497119.4). The lentivirus vector was confirmed by sequencing. Negative control shRNA was provided by GENECHEM. Lentivirus-encoded shRNA against CENPF and control were prepared and titered to 2 × 108 (TU/mL) as the manufacturer’s protocol. H22 Cells (1 × 104 cells/well) were seeded in six-well plates overnight before transfection. The virus (0.1 mL) was mixed with 0.1 mL complete medium containing polybrene (8 mg/mL) and added to cells and incubated for 1 h at 37°C. Then the cells were incubated in fresh complete medium containing polybrene for 24 h, followed by incubation for 48 h in complete RPMI-1640 medium. The cells were then harvested and expanded for subsequent studies.
RNA extraction and quantitative real-time PCR
Total RNA was isolated using Trizol reagent (Promega) according to the manufacturer’s instructions, and 2 μg of total RNA was reverse transcribed into cDNA with Molony murine leukemia virus reverse transcriptase (Promega, Madison, WI). GAPDH was used as an internal control. Primers for CENPF and GAPDH were used as follows: human CENPF: forward: 5ʹ-TACTGAGTTTGAGCCAGAGGGACT-3ʹ, reverse: 5ʹ-CATGGTTGTTCTTCGCAGGATAT-3ʹ; human GAPDH: forward: 5ʹ-CCATCACCATCTTCCAGG-3ʹ, reverse: 5ʹ-ATGAGTCCTTCCACGATAC-3ʹ; mouse CENPF: forward:5ʹ-AAGCAGAAGGTTGAAGACGGA-3ʹ, reverse: 5ʹ- TTTGCATGAGCTCAGTTGGC-3ʹ; mouse GAPDH: forward: 5ʹ- TGGCCTTCCGTGTTCCTAC-3ʹ, reverse: 5ʹ-GAGTTGCTGTTGAAGTCGCA-3ʹ. Gene expression levels were evaluated by real-time quantitative PCR kinetics with SYBR Master Mix (Takara, Japan). Real-time PCR was performed on the ABI 7500 Real-Time PCR System (Applied Biosystems) with 1.0 μL of appropriate diluted cDNA, 0.5 μL (5μM) of forward and reverse primers specific for CENPF and GAPDH, 10 μL of SYBR PremixEx taq, and 8.0 μL of water. The cycling conditions were as follows: 95°C for 10 min; 40 cycles of 95°C for 30 sec, and 60°C for 1 min. A uniform amplification of the products was rechecked by analyzing the melting curves of the amplified products. All reactions were carried out in triplicate to access the reproducibility. The relative quantitation of mRNA expression was calculated with the comparative Ct (the threshold cycle) method using the following formula:
Western blot analysis
Cells or tumors were lysed in ice-cold lysis buffer containing 100 mM Tris-HCl (pH 6.8), 4% sodium dodecyl sulfate (SDS), 20% glycerol, 2% hydroxyethanal, and clarified by centrifugation at 15000 rpm for 15 min. Equal amounts of cell lysate protein (20 μg of protein per lane) were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinyl difluoride (PVDF) membranes (0.45 μm; Millipore, Bedford, MA). For immunoblot analysis, the membranes were probed with specific primary antibodies, anti-CENPF (Abcam, Cambridge, MA), anti-cyclinD1 (Abcam, Cambridge, MA), anti-c-Myc (Abcam, Cambridge, MA), anti-CDK2 (Cell Signaling Technology, Beverly, MA), anti-CDK4 (Cell Signaling Technology, Beverly, MA), anti-CDK6 (Cell Signaling Technology, Beverly, MA), and anti-β-actin antibody (Cell Signaling Technology, Beverly, MA), and secondary antibodies conjugated to HRP (ZhongShan JinQiao, Beijing, China). Immunocomplexes on the membrane were visualized using Image Quant LAS 4000 (GE Healthcare, Tokyo, Japan) with Immobilon® Western HRP Substrate luminol reagent and peroxide solution (Millipore).
Confocal microscopy
CENPF localization and translocation
The QSG-7701, HuH-7, Li-7, Hep 3b cells grown on coverslips were washed 3 times in PBS, fixed in 4% paraformaldehyde/PBS (PFA/PBS) for 30 min, and permeabilized in 0.5% Triton X-100/PBS for 5 min. Nonspecific binding sites were blocked by 1 h incubation in 5% BSA/PBS at room temperature. Then cells were incubated with the primary antibody against CENPF (Abcam, Cambridge, MA) at 4°C overnight. After three washes with PBS, cells were stained with anti-rabbit Alexa Fluor 647 conjugated secondary antibody (Abcam, Cambridge, MA) and mounted with an aqueous mounting medium containing DAPI (Molecular Probe, USA). Cells were examined and photographed with a laser confocal microscope (FV 300, Olympus, Tokyo, Japan).
Apoptotic cells (HuH-7 and Hep 3b) on coverslips were generated by serum-starved culture for 48 h and labeled with antibody against CENPF as described above. The CENPF translocation in apoptotic cells was observed with the confocal microscope.
DCs distribution in HCC tumor tissues
Frozen sections of tumor and paratumor tissues from HCC patients or mouse model were prepared, fixed with 4% PFA/PBS for 10 minutes at room temperature and blocked with 5% FBS/PBS for 1 hour at room temperature. Then the sections were incubated with primary antibody against CD11c (Abcam, Cambridge, MA) at 4°C overnight. After 3 washes with PBS, the sections were stained with goat anti-mouse Alexa Fluor 488 conjugated secondary antibody (Abcam, Cambridge, MA) and mounted with a mounting medium containing DAPI. The images were captured with the confocal microscope.
Cell proliferation, apoptosis, and necrosis assay
The Cell Counting Kit-8 (CCK-8) (Yeasen Biotechnology, Shanghai, China) was used to determine the proliferation rate of HCC cells with or without CENPF knockdown according to the manufacturer’s protocol. Cells were seeded in 96-well plates, and at certain time incubated with 20 μL of CCK-8 at 37°C for 1 h in a humidified, 5% CO2 atmosphere. The supernatant culture medium was collected, and the absorbance at 450 nm was recorded using a 96-well plate reader.
HCC WT and KD cells were resuspended in the binding buffer (10 mM Hepes/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). FITC-Annexin V and propidium iodide (PI) (eBioscience, Vienna, Austria) were added followed by 10 min incubation at room temperature, and then cells (5 × 105) were subjected to flow cytometry (BD Bioscience, San Jose, CA). Acquired data were analyzed by FlowJo software (Treestar, Inc.). Apoptotic cells were FITC positive (Q2 and Q3 gate). Necrotic cells were PI positive and FITC negative (Q1 gate). Healthy cells were double negative (Q4 gate).
Induction and measurement of apoptosis and isolation of different apoptotic cell components
HCC cells of HuH-7 and Hep 3b were cultivated in RPMI 1640 medium containing 8%, 6%, 4%, 0% FBS, respectively, for 72 h, and collected with trypsin without EDTA. Cells were labeled with CENPF antibody and APC conjugated secondary antibody (Abcam, Cambridge, MA). After washed with PBS, cells were incubated with FITC-Annexin V and PI, and then subjected to flow cytometry. Acquired data were analyzed by FlowJo software.
H22 cells were cultivated in RPMI 1640 medium without FBS for 72 h and collected. Intact cells were isolated from apoptotic cell culture by centrifugation for 5 min at 100 g at room temperature. Subsequently, apoptotic body and blebs were isolated from the resulting supernatant by centrifugation for 15 minutes at 1550 g at room temperature. The concentrations of intact cells and apoptotic body and blebs were determined with the bicinchoninic acid protein determination assay (Sigma-Aldrich, USA) and expressed as protein equivalents.
Phagocytosis and maturation assay of DCs
H22 cells were labeled with 10 μM DiD (Sigma-Aldrich, USA) after the induction of apoptosis and serial centrifugation. DiD-labeled intact cells or apoptotic body and blebs (50 μg/mL) were added to 1 × 106/mL DCs with a final volume of 500 μL and subsequently incubated at 4°C or 37°C for 2 hours. Next, DCs were collected and labeled with FITC conjugated anti-CD11c antibody (BioLegend, San Diego, CA). Internalization of intact cells or apoptotic body and blebs by DCs was visualized with confocal microscope. In addition, the percentage of FITC/DiD double-positive cells was analyzed by flow cytometry.
For the analysis of DC maturation, 5 × 105 immature DCs were incubated in medium alone or medium supplemented with intact cells or apoptotic body and blebs. Addition of 1 μg/mL lipopolysaccharide (LPS) (Sigma-Aldrich, USA) was used as a positive control for the maturation of DCs. After 16 hours of incubation, cells were harvested and stained with FITC conjugated anti-CD11c antibody and PE conjugated anti-CD86 antibody (BioLegend, San Diego, CA). Cells were subjected to flow cytometry and acquired data were analyzed by FlowJo software.
Mouse HCC model with H22 cell inoculation
Five-week-old female BALB/c mice were maintained in the Experimental Animal Center at the Beijing Friendship Hospital, Capital Medical University (Beijing, China) under specific pathogen-free conditions. All mice received sterilized food and water. All the animal experiments in this study were carried out in accordance with the approved guidelines and approved by the committee on Animal Care and Use of Beijing Friendship Hospital, Capital Medical University. Mice were acclimatized to laboratory conditions for 1 week prior to experiments. H22 cells (1 × 106) stably transfected with lentivirus-encoded shRNA against CENPF (KD) or control (WT) and suspended in 200 μL matrigel/PBS were injected subcutaneously into the mice. Tumor volume and body weight were monitored every week. The serum was collected weekly and analyzed for CENPF autoantibody level with the CENPF antibody Elisa kit (Shanghai Mlbio Biotechnology Co, Shanghai, China) according to the manufacturer’s protocol. Tumor volumes were estimated by measuring the largest diameter (a) and its perpendicular (b) according to the formula (a× b2)/2. The curve of tumor growth was drawn according to tumor volume and time of cell implantation. The mice were sacrificed on day 21 after cell inoculation. The tumor and spleen tissues were excised from the mice and weighed.
Immunohistochemistry
The tumor tissues were fixed with 4% paraformaldehyde (PFA), dehydrated and embedded in paraffin. Heat-induced antigen retrieval in citrate buffer (pH 6.0) was undertaken before quenching of endogenous peroxidase using H2O2 and blocking with 2.5% horse serum. The tumor sections were then incubated with Ki-67 primary antibody (Abcam, Cambridge, MA) and goat anti-rabbit IgG (HRP) antibody (ZhongShan JinQiao, Beijing, China). DAB was used as the chromogen. The sections were counterstained with hematoxylin. For TUNEL staining, the tumor sections were stained with Colorimetric TUNEL Apoptosis Assay Kit (Beyotime Biotechnology, Shanghai, China) according to manufacturer’s instructions. The images were taken using an Olympus BX53F microscope equipped with a digital camera (Olympus, Tokyo, Japan), and three representative slides for each group were reviewed. The frequency of Ki-67 or TUNEL positive cells was measured by counting the total number of cells and the number of positively stained cells.
BALB/c mice immunizations
Five-week-old female BALB/c mice were maintained as described previously. Intact cells and apoptotic body and blebs were generated by serum-starved culture of H22 cells and serial centrifugation. The cell lysate was generated by three freeze–thaw cycles. Starting at week 1, 10 μg protein equivalents of intact cells, apoptotic body and blebs, cell lysate or saline were injected intravenously into mice, respectively, every week. The serum was obtained weekly and analyzed for CENPF autoantibody level with the CENPF antibody Elisa kit (Shanghai Mlbio Biotechnology Co, Shanghai, China) according to the manufacturer’s protocol. The mice were sacrificed at week 7. The spleen tissues were excised from mice and weighed.
Frozen sections were prepared with spleen tissues stored in liquid nitrogen, fixed with 4% paraformaldehyde (PFA) for 10 minutes at room temperature and blocked with 5% FBS for 1 hour at room temperature. Then the sections were stained with 5 µg/ml of B220-Alexa Fluor 488 overnight at 4°C and mounted. The images were captured with a confocal microscope (FV 300, Olympus, Tokyo, Japan). Paraffin sections were prepared with spleens tissues fixed in 4% PFA. These spleen tissues were dehydrated and embedded in paraffin. Paraffin sections were prepared and stained with hematoxylin and eosin (H&E). The images were taken using an Olympus BX53F microscope. Three representative slides for each group of immunized mice were reviewed.
Statistical analysis
All experiments were carried out at least three times. Data are represented as mean ± standard error. Differences between groups were calculated by Student’s t-test. Statistical significance of the results was defined as a P-value (*P < .05 and **P < .01) for a two-tailed test.
Results
CENPF is overexpressed and diffusely distributed in whole HCC cells compared with non-neoplastic liver cells
In a previous study, we performed an immunohistochemical staining analysis on the pathological sections of HCC patients with different CENPF autoantibody titers in the serum, and found that CENPF was diffusely distributed in nucleus and cytoplasm of tumor cells, and high expression of CENPF in tumor cells was observed in HCC patients with high serum autoantibody level.Citation8 To confirm and extend the results of our previous study, we first analyzed CENPF expression levels in representative samples of HCC tumor and adjacent non-tumor liver tissues and in a panel of human HCC cell lines and normal hepatic cells. qRT-PCR analysis revealed significantly higher CENPF mRNA levels in HCC tumor tissues compared with adjacent non-tumor tissues (). Similarly, qRT-PCR and western blot analysis of the normal human hepatic cell line QSG-7701 and the human HCC cell lines Hep 3b, HuH-7, BEL-7402, Li-7, PLC/PRF/5, and SK-Hep-1 demonstrated significantly higher CENPF expression in the tumor cell lines at both the mRNA and protein levels (). To examine the localization of CENPF in the cell lines, we performed immunofluorescence staining followed by laser confocal microscopy. CENPF was highly expressed only in the mitotic nucleus of QSG-7701 cells but was abundant in both the cytoplasm and nucleus of the HCC cell lines HuH-7, Li-7, and Hep 3b (). Thus, CENPF is overexpressed and exhibits aberrant subcellular localization in human HCC cell lines.
Figure 1. Expression and localization of CENPF in HCC cells and tissues. (a) qRT-PCR analysis of CENPF mRNA levels in five representative tumor and adjacent non-tumor tissue samples from HCC patients. Levels were normalized to GAPDH mRNA. (b) qRT-PCR (upper) and Western blot (lower) analysis of CENPF mRNA and protein expression, respectively, in the normal hepatic cell line QSG-7701 and six HCC cell lines. β-Actin was probed as a loading control. (c) Confocal fluorescence micrographs of CENPF localization (white arrows) in QSG-7701 and HCC cell lines. Red represents CENPF and blue represents nucleus revealed with DAPI (blue). **, p < .01
CENPF autoantibody level is positively correlated with CENPF expression and tumor progression in a mouse HCC model
Next, we investigated the ability of tumor-derived CENPF to induce autoantibody production and the correlation between CENPF autoantibody production and tumor growth in vivo. For this, we infected H22, a mouse HCC cell line, with lentiviruses encoding CENPF-specific shRNA or control scrambled shRNA and selected stable transfectants (H22KD and H22WT, respectively). As shown in , levels of CENPF mRNA and protein were significantly reduced in H22KD compared with H22WT cells (p < .01). The cell lines were subcutaneously injected into BALB/C mice, and tumor growth and serum CENPF autoantibody levels were monitored. The subcutaneous tumors were excised at 3 weeks. We found that tumors derived from H22KD cells grew much more slowly and were much smaller than tumors derived from H22WT cells (). Moreover, mice with H22KD tumors had significantly lower serum CENPF autoantibody levels than the control mice, and a positive trend was observed between tumor size and serum CENPF autoantibody level, especially at 2 weeks after tumor cell injection (). Interestingly, mice with H22KD tumors also had spleens that weighed significantly less than those from mice with H22WT tumors, revealing a lower immune response (). These results suggest that elevated CENPF expression in HCC cells may be an important factor in tumor growth and CENPF autoantibody production, and that serum CENPF autoantibody levels may correlate with tumor progression and have potential predictive value.
Figure 2. Tumor growth and CENPF autoantibody production in a mouse HCC model. (a) qRT-PCR (left) and western blot (right) analysis of CENPF mRNA and protein expression, respectively, in H22 cells stably expressing a control shRNA (WT) or CENPF-specific shRNA (KD). (b) Tumor volume in mice injected subcutaneously with H22 WT and KD cell lines. (c) Images of subcutaneous tumors excised on day 21 after H22 cell injection. (d) Weight (left) and appearance (right) of spleens from mice on day 21 after H22 cell injection. (e) Serum CENPF AAb level in mice on the indicated days after H22 cell injection. **, p < .01 vs. WT group. WT, wild type. KD, knockdown
To explore the effect of CENPF expression on HCC cell and tumor growth and its mechanism, we analyzed the proliferation curve of H22WT and H22KD cells during 4 d. We found H22KD cells grew more slowly than H22WT cells especially at day 4 (). We also detected the apoptotic (FITC+) and necrotic (FITC−, PI+) rate of H22WT and H22KD cells by flow cytometry. The results suggested that the apoptotic or necrosis rate of H22WT and H22KD cells had no significant difference (). We further performed immunohistochemical staining of proliferation marker Ki-67, TUNEL staining and H&E staining on tumor sections with or without CENPF knockdown (), and calculated the positive rates of Ki-67 and TUNEL (). The Ki-67 positive rate of H22WT tumor was dramatically higher than that of H22KD tumor (almost three times), while there was no significant difference in TUNEL positive rate or necrosis level between these two groups (). These results indicated that CENPF knockdown slowed proliferation of H22 cells and tumors but did not affect the apoptosis and necrosis. We further detected the expression of proliferation and cell cycle-related proteins in H22WT and H22KD cells and tumors. Western blot results showed that CENPF knockdown suppressed the expression of cyclinD1, c-Myc, CDK2, CDK4 significantly, while CDK6 expression was not affected ().
Figure 3. The effect of CENPF expression on HCC cell proliferation, apoptosis and necrosis. (a) Relative proliferation rate of H22 WT and KD cells during 4 d was measured by CCK-8. The apoptotic rate (b) or necrosis rate (c) of H22 WT and KD cells on day 2 or 4 were detected by flow cytometry. (d) Immunohistochemical staining of Ki-67, TUNEL staining and H&E staining of tumor biopsies obtained from mice injected subcutaneously with H22 WT or KD cells. Scale bar 50 μm. The rate of Ki-67 (e) or TUNEL (f) positive cells was calculated. The expression of proliferation related proteins in H22 cells (g) or tumors (h) with or without CENPF knockdown was analyzed by western blot. *, p < .05. **, p < .01. WT, wild type. KD, knockdown
HCC cell apoptosis induces CENPF protein translocation and autoantigen exposure
The question that how highly expressed intracellular protein CENPF exposed to the immune system and induced autoantibody production was unclear. We detected the CENPF protein content in the intercellular fluid of the tumor tissues and adjacent normal tissues of HCC patients using CENPF protein ELISA quantitative assay and found that the CENPF content was very low (Figure S1). There was no difference in the free CENPF content between tumor and non-tumor intercellular fluid. The results suggested that CENPF was not secreted into the extracellular matrix of the HCC tumor environment. It has been reported that apoptotic cells could be a possible source of autoantigens in autoimmune response.Citation18 Therefore, we determined whether CENPF protein may be relocalized to the cell surface during apoptosis of HCC cells and be exposed to the plasma membrane of apoptotic cells.
HuH-7 and Hep 3b cell lines were serum-starved in medium containing 8%, 6%, 4%, 0% FBS, respectively, to induce apoptosis, and then incubated with annexin V-FITC and propidium iodide (PI) to detect cells at various stages of apoptosis. The cells were also stained with anti-CENPF antibody. Flow cytometric analysis of the cells showed that very few healthy (FITC−, PI−; Q4 gate) and necrotic (FITC−, PI+; Q1 gate) cells were also CENPF+, but the proportion of CENPF+ cells was significantly higher among cells in early apoptosis (FITC+, PI−; Q3 gate) and late apoptosis (FITC+, PI+; Q2 gate) in both HuH-7 () and Hep 3b () cultures. Notably, the proportion of CENPF+ and apoptotic HCC cells correlated positively for both cell lines (R2 > 0.9) (). We also examined the distribution of CENPF protein in apoptotic HuH-7 and Hep 3b cells by laser confocal microscope. This analysis showed that CENPF protein was dispersed in the cytoplasm and nuclei of healthy HCC cells. However, apoptotic HCC cells displayed a large number of vesicles (apoptotic body and blebs) on the plasma membrane, and CENPF staining was concentrated in these areas (). Collectively, these results suggest that apoptosis of HCC cells is associated with translocation and exposure of CENPF at the plasma membrane. This interpretation also suggests a mechanism by which an anti-CENPF immune response and autoantibody production may be stimulated during HCC.
Figure 4. Apoptosis induced CENPF translocation and exposure in HCC cells. HuH-7 and Hep 3b cells were cultured under different serum starvation conditions (8%, 6%, 4%, 0% FBS respectively for 72 h), to induce apoptosis and then stained with Annexin V-FITC, PI, and CENPF antibody. (a, b) The CENPF positive rate of apoptotic cells, necrotic cells and normal cells was detected by flow cytometry (left), and the correlation of CENPF positive rate with apoptosis rate was analyzed (right). (c) Confocal images of apoptotic HCC cells stained with anti-CENPF antibody (red) showing CENPF redistribution into apoptotic vesicles (white arrows). Nuclei were stained with DAPI (blue)
HCC cell apoptotic body and blebs are taken up by and promote the maturation of DCs in vitro
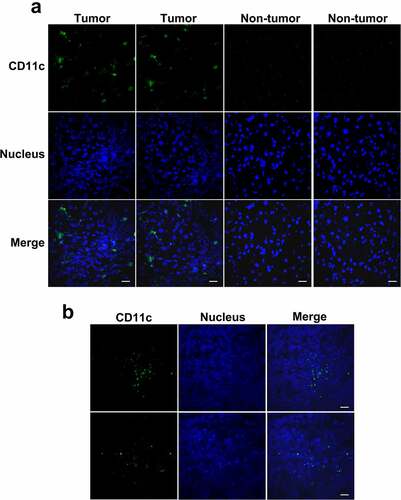
Through examine the immunofluorescence staining of human and mouse HCC tumor frozen sections with anti-CD11c antibody, we found that DCs were distributed throughout the tumor tissue but not the normal liver tissue (). DCs may play a role in antigen recognition and presentation during CENPF autoantibody production in HCC.
Figure 5. Distribution of DCs in HCC tumor tissues from patients or mice. Frozen sections of tumor and paratumor tissues from HCC patients (a) or mice injected with the murine H22 HCC cell line (b) were stained with anti-CD11c antibody and Alexa Fluor 488 conjugated secondary antibody (green) to detect DCs, counterstained with DAPI (blue) to reveal nucleus, and observed by confocal microscope. Scale bar 20 μm
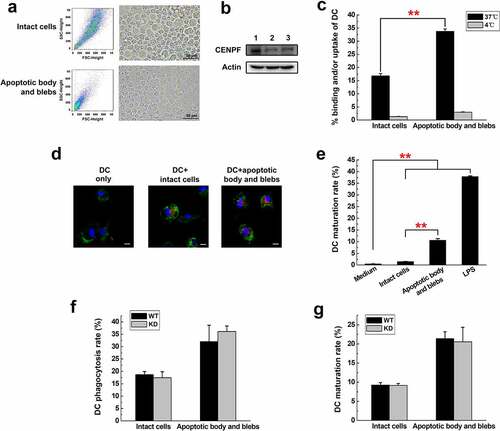
Next, we investigated the possibility that apoptotic bodies or blebs with highly expressed CENPF may be immunogenic. To prepare apoptotic cell components, H22 cells were serum-starved to induce apoptosis and then subjected to serial centrifugation to separate intact cells from apoptotic body and blebs. shows by forward/side scatter and light microscopy that our separation yielded a preparation of enriched intact cells and apoptotic body/blebs. Forward- and side-scatter plots showed that apoptotic body and blebs had a much lower scatter than intact cells. Phase contrast microscope showed their smaller size (). Western blot analysis demonstrated that the CENPF expression of apoptotic body and blebs was a little bit higher than that of intact cells (). The fractions were then incubated with the lipophilic fluorescent dye DiD to label the apoptotic components. We prepared DCs by culturing bone marrow cells isolated from mouse femurs and tibias with granulocyte-macrophage colony stimulating factor (GM-CSF) and interleukin-4 (IL-4), and the DCs were then incubated with DiD-labeled intact cells or apoptotic body/blebs to determine whether they stimulated DC phagocytosis and maturation. After incubation, DCs were detected by staining with anti-CD11c antibody. Flow cytometry and fluorescence microscopy showed that DCs more readily phagocytosed small-sized apoptotic body and blebs compared with the larger intact cells (). Apoptotic body and blebs were phagocytosed almost twice as much as intact cells (p < .01). Co-incubation of DCs and apoptotic components at 4°C resulted in little to no uptake by DCs, excluding the possibility that the results reflected nonspecific binding of apoptotic components to the DCs. Apoptotic body and blebs were preferred targets of DC phagocytosis. Moreover, apoptotic body and blebs promoted DC maturation much more effectively than did intact H22 cells, as reflected by the proportion of DCs expressing the costimulatory protein CD86 (). The DC maturation rate of apoptotic body and blebs group was almost seven times higher than that of intact cells. To explore if CENPF expression of H22 cells had any effect on DC phagocytosis and maturation, we incubated DCs with intact cells and apoptotic body and blebs from H22WT or H22KD cells, respectively, and analyzed the phagocytosis rate and maturation rate of DCs. Flow cytometry results showed that apoptotic cell components from CENPF KD cells induce DC phagocytosis and maturation to a level similar to that of apoptotic components from H22 WT cells. There was no significant difference in the rate of DC phagocytosis and maturation between the WT and KD groups of apoptotic cell components ().
Figure 6. Phagocytosis and maturation of DCs induced by apoptotic cell components. (a) Flow cytometry (forward and side scatter) plots (left) and phase contrast micrographs (right) of H22 cells after serum starvation, showing intact cells and apoptotic body and blebs. The fractions were separated and labeled with DiD before incubation with DCs. (b) Western blot analysis of CENPF expression in healthy H22 cells (lane 1), intact cells (lane 2), apoptotic body and blebs (lane 3). (c) Flow cytometric analysis of the proportion of CD11c+ DCs that had ingested/bound DiD-labeled apoptotic cell components. Binding (4°C) and uptake/binding (37°C) were quantified as the percentage of FITC/DiD double-positive cells. (d) Fluorescence microscopy of CD11c+ DCs (green) incubated alone or with DiD-labeled intact cells or apoptotic body and blebs (red). The nucleus was revealed with DAPI (blue). Scale bar 10 μm. (e) Proportion of mature DCs (CD86+) after incubation with intact cells, apoptotic body and blebs, or LPS (positive control). Proportion of phagocytotic (f) or mature (g) DCs after incubation with intact cells or apoptotic body and blebs from H22 WT or KD cells respectively. **, p < .01
CENPF autoantibody production in vivo is preferentially induced by HCC cell apoptotic body and blebs
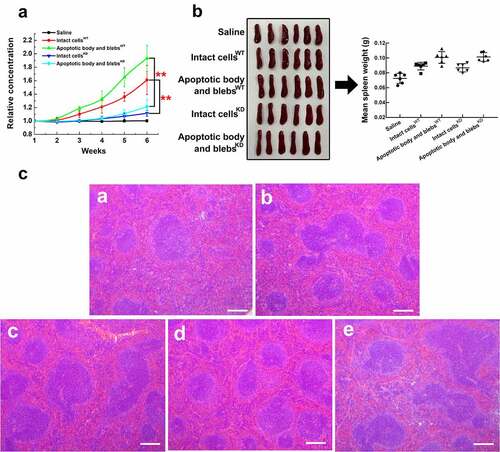
Finally, we compared the ability of H22 apoptotic cell components to induce CENPF antibody production in mice. Saline (control), intact apoptotic H22 cells, apoptotic body and blebs, or cell lysates were injected intravenously into groups of BALB/c mice once a week for 7 weeks. Blood samples were collected weekly, and serum CENPF antibody levels were measured by ELISA kit. At week 7, the spleens were excised, and sections were prepared for analysis. The results showed that serum CENPF antibody levels progressively increased between week 3 and 7 in mice injected with intact cells and, to a significantly greater extent, in mice injected with apoptotic body and blebs, whereas antibody levels were virtually undetectable in mice injected with saline or cell lysate (). Consistent with these findings, apoptotic body and blebs induced a greater immune response than did intact cells, as reflected by increased spleen weights (), white pulp, and lymphoid follicular area in spleen sections (). To directly explore the effect of CENPF expression in apoptotic cell components on serum CENPF antibody production in vivo and exclude the crossover of antigens caused by similar sequence, we injected BALB/c mice intravenously with saline, intact cellsWT, apoptotic body and blebsWT, intact cellsKD, or apoptotic body and blebsKD respectively once a week and measured serum CENPF antibody levels weekly. The results showed that serum CENPF antibody levels increased more slowly after CENPF expression was knocked down, and CENPF antibody levels of KD mice groups were significantly lower than that of WT groups between week 3 and 6 (). However, there were no significant differences in spleen weight or white pulp and lymphoid follicular area between WT and KD groups injected with the same sort of apoptotic cell components (). These results suggest that CENPF knockdown in apoptotic cell components suppressed the serum CENPF antibody production but did not affect the immune response in vivo.
Figure 7. HCC apoptotic cell components induce anti-CENPF antibody production in BALB/c mice. Groups of mice were injected with saline, H22 apoptotic intact cells, H22 apoptotic body and blebs, or H22 cell lysate once a week for 7 weeks. (a) Serum CENPF antibody levels measured by Elisa every week. (b) Images and weights of mouse spleens at week 7. (c) Immunofluorescence staining of the B cell marker B220-Alexa Fluor 488 (left) and H&E staining (right) of sections of spleens obtained from mice injected with saline (a, b), cell lysate (c, d), intact cells (e, f), and apoptotic body and blebs (g, h). Scale bar 150 μm. *, p < .05. **, p < .01
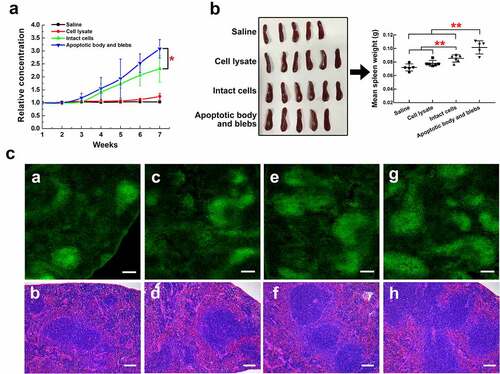
Figure 8. Anti-CENPF antibody production induced by HCC apoptotic cell components with or without CENPF knockdown in BALB/c mice. Groups of mice were injected with saline, apoptotic intact cells or apoptotic body and blebs from H22 WT or KD cells once a week for 6 weeks. (a) Serum CENPF antibody levels measured by Elisa every week. (b) Images and weights of mouse spleens at week 6. (c) H&E staining of sections of spleens obtained from mice injected with saline (a), intact cellsWT (b), apoptotic body and blebsWT (c), intact cellsKD (d), and apoptotic body and blebsKD (e). Scale bar 150 μm. **, p < .01
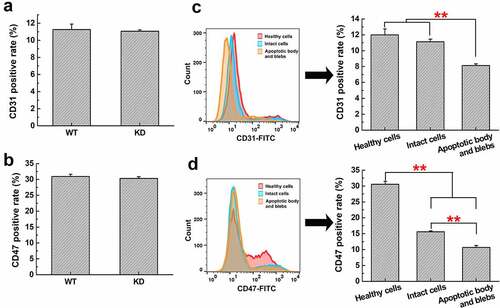
CD31 (platelet endothelial cell adhesion molecule-1, PECAM-1) and CD47 (integrin-associated protein, IAP) are well-known ‘don’t eat me’ markers on healthy cell membrane which inhibit phagocytosis by sending negative engulfment signals to DCs. Thus, we detected the CD31 and CD47 expression on H22 cells with or without CENPF knockdown by flow cytometry and found that there was no significant difference in the expression of CD31 and CD47 between WT and KD groups (). We further isolated the apoptotic cell components and detected CD31 and CD47 expression among different apoptotic components. As shown in , CD31 level of apoptotic body and blebs was lower than that of healthy cells and apoptotic intact cells, but there was little difference between the healthy cells and intact cells. A more significant difference was found in the CD47 expression of these three groups. Apoptosis dramatically decreased CD47 expression on cell surface compared with healthy cells, and CD47 level of apoptotic body and blebs was also much lower than that of intact cells ().
Figure 9. Expression of CD31 and CD47 in H22 WT and KD cells, or different apoptotic cell components. The CD31 (a) or CD47 (b) positive rate in H22 WT and KD cells was analyzed by flow cytometry. The CD31 (c) or CD47 (d) positive rate in healthy H22 cells, intact cells, apoptotic body and blebs was analyzed. **, p < .01
Discussion
Patients with advanced HCC typically only survive for 3–6 months, which can mainly be attributed to late diagnosis and the lack of effective nonsurgical treatments.Citation3,Citation4 At the early stage of tumor, low level of TAAs cannot be detected using conventional protein detection method. However, the immune system can detect the presence of these specific proteins, trigger the immune reaction and produce antibody with a signal amplification effect, thus distinguishing between normal human and patients. Compared with the antigen detection method, the autoantibody detection method has a higher sensitivity, and serum autoantibodies may become a very promising marker for the early diagnosis of HCC.Citation5,Citation26 In recent years, researchers have used phage display technology, protein microarray, protein serological analysis, and other methods to find some HCC-related TAA autoantibodies, such as those antibodies against p53, HSP60, HCC1, survivin, respectively.Citation6,Citation7 These TAA autoantibodies from different sources have the potential to be used as detection indicators for screening and diagnosis of early HCC.
In the previous study, we performed a high-throughput screening of serum autoantibodies and a clinical correlation analysis for diagnosis of HCC from other liver diseases using serum immune proteomics and protein microarrays, and found that CENPF autoantibodies showed a great value of early diagnosis and screening of HCC.Citation8 Homology analysis was performed on CENPF dominant antigen epitopes in NCBI protein database, and no homology sequences were found with other proteins, which could exclude the crossover of antigens caused by the same sequence.Citation8 In addition, Wu Mengchao at Shanghai Oriental Hepatobiliary Hospital has also verified the presence of specific CENPF autoantibodies in HCC using T7 phage display technology.Citation27 Here using HCC tumor transplanted mice models, we confirmed that CENPF autoantibody was a good predictor of HCC tumor progression and could be detected at the early stage of disease.
CENPF is a large (approximately 350 kDa) coiled-coil protein whose transient expression and subcellular localization patterns resemble that of a ‘chromosome passenger protein’.Citation9,Citation12 However, it has recently been reported that CENPF was highly expressed in several human malignancies, such as HCC and breast cancer, and was identified as a marker of cell proliferation and prognosis.Citation13,Citation17 CENPF silencing with small interfering RNA can impair the proliferation and clonal formation of HCC cells in vitro and inhibit the growth of transplanted tumors in nude mice.Citation13 Our study indicated that in the hepatic cell QSG-7701 CENPF was highly expressed only in nucleus of dividing cells, while in HCC cell lines it was overexpressed in both the cytoplasm and nucleus of all cells. In vivo study showed its overexpression was closely related to tumor growth and CENPF autoantibody production in mouse HCC model. Our mechanism studies showed that CENPF knockdown inhibited the HCC cell proliferation through suppressing expression of cyclinD1, c-Myc, CDK2, CDK4, and had no effect on cell apoptosis and necrosis. Other studies also reported that TAAs, such as HSP60,Citation28 p53,Citation29 cyclin B1,Citation30 showed abnormal expression, accumulation, and distribution in specific tumor cells, which could lead to the production of autoantibodies. HSP60 is widely distributed in the cytoplasm and membrane of tumor cells, while in normal cells it is located in mitochondria.Citation28 Cyclin B1 is located in cytoplasm of tumor cells, while in normal cells it is mainly distributed in the nucleus.Citation30 These results suggest that the overexpression and abnormal localization of CENPF autoantigens were important contributing factors in the production of CENPF autoantibodies in HCC.
In some liver cancer, colorectal cancer, breast cancer, lung cancer cell line and tumor tissues, centromere protein A (CENPA) presents abnormal high expression level, but only in part of the tumor patients, serum CENPA autoantibodies could be detected.Citation31–33 The researchers speculated that overexpression of CENPA was not enough to induce autoantibody producing, and there may be other activation mechanism of the immune system.Citation33 They speculated that autoantibodies may be produced by a mechanism similar to anti-DNA antibodies, in which the immune system activates antibodies in recognition of antigens exposed to apoptotic vesicles.Citation34 Intracellular antigenic proteins trigger the immune system to produce autoantibodies, which must first be directed to the cell surface or released into extracellular matrix. In the present study, we detected the CENPF protein content in the intercellular fluid of the tumor tissues and adjacent normal tissues of HCC patients. The results suggested that CENPF exposure was not through the secretion into extracellular matrix.
An important difference between apoptosis (programmed cell death) versus necrotic death (accidental/toxic death) is that apoptosis results in the ordered fragmentation of the cell leading to rapid phagocytosis by neighboring cells and/or professional phagocytes.Citation35 Apoptotic cells have been proposed as a possible source of autoantigens. It has been reported that in some autoimmune diseases, such as SLE and Sjogren’s syndrome (SS), autoantigen in nucleus or cytoplasm could cluster and translocate into apoptotic blebs and bodies.Citation20,Citation36 Autoantigen translocation may have the effect of turning dying cells ‘inside out’. By exposing autoantigen on surface, apoptotic cells, especially their fragments as apoptotic bodies or blebs, could consequently promote phagocytosis and antigen presentation of APCs, and autoantibody production.Citation23,Citation37 The translocation and exposure of autoantigen in breast cancer were also reported to be linked to the process of apoptosis.Citation24,Citation25 Here we found that the CENPF antigen exposure was in positive correlation with cell apoptosis rate. During HCC cell apoptosis, CENPF protein was translocated, condensed into the apoptotic vesicles and exposed to the outer surface of plasma membrane. Compared with cell lysates, apoptotic cell components including intact cells, apoptotic body and blebs, were immunogenic and could induce antibody production. The DC phagocytosis rate of apoptotic body and blebs was twice as much as that of intact cells, while the DC maturation rate was almost seven times higher than the intact cell group. We hypothesize that apoptotic blebs and bodies are the right size for DC uptake and antigen presentation. The uptake of a much larger-sized dying cell would be more difficult. CENPF expression level of apoptotic cell components had no significant effect on phagocytosis and maturation of DCs in vitro and the immune response in vivo but was positively correlated with serum CENPF antibody level of mice. Therefore, CENPF expression was not the key influencing factor of DC-mediated immune response. Engulfment of apoptotic cells by DCs is regulated by ‘eat me’ and ‘don’t eat me’ signals on the cell surface.Citation38 CENPF did not provide an ‘eat me’ signal to DCs. CD31 and CD47 are well-known ‘don’t eat me’ markers on healthy cell membrane which discourage activated DCs from attempting to uptake those cells.Citation38,Citation39 Our study showed that there was no significant change in CD31 and CD47 expression with or without CENPF knockdown in healthy H22 cells, while apoptosis decreased the expression of these two markers. The CD31 and CD47 expression level of apoptotic body and blebs were dramatically lower than that of healthy cells and intact cells, and this promoted the DC uptake of apoptotic body and blebs. Most APCs present autoantigens from apoptotic components in either a tolerogenic or immunogenic way, depending on the activation state of APCs and the surrounding inflammatory environment.Citation40,Citation41 Maybe more costimulatory signals are expressed on the surface of apoptotic body and blebs than intact cells, which contributing to much more DC maturation and antibody production. Autoantigen-rich apoptotic body and blebs that are not efficiently cleared might have two important properties that determined their subsequent immune fate: 1) high concentration of autoantigens and 2) adjuvant effect on DCs, and that needs to be verified by further studies.
Conclusions
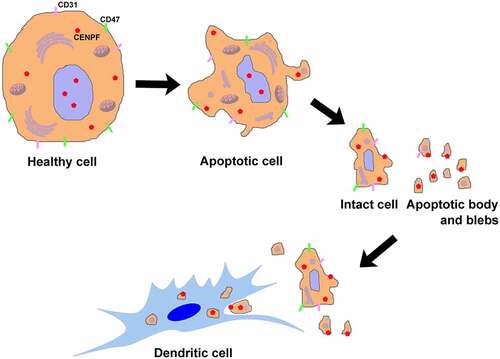
Taken together, our findings reveal the mechanism of CENPF exposure and autoantibody production for the first time from the perspective of cell apoptosis induced autoantigen translocation in HCC (), and verify the reliability of CENPF autoantibody as a promising marker that predicting the HCC tumor progression.
Figure 10. Apoptosis plays a role in CENPF autoantigen translocation, exposure, and antigen-presentation of DCs mainly through apoptotic body and blebs
Abbreviations
| AFP | = | Alpha-fetoprotein |
| APC | = | Antigen-presenting cell |
| AUC | = | Area under curve |
| CDKN2A | = | Cyclin dependent kinase inhibitor 2A |
| CENPA | = | Centromere protein A |
| CENPF | = | Centromere protein F |
| CIP2A | = | Cancerous inhibitor of protein phosphatase 2A |
| DC | = | Dendritic cell |
| ELISA | = | Enzyme linked immunosorbent assay |
| GFP | = | Green fluorescent protein |
| GM-CSF | = | Granulocyte-macrophage colony stimulating factor |
| HCC | = | Hepatocellular carcinoma |
| H&E | = | Hematoxylin and eosin |
| IL-4 | = | Interleukin-4 |
| KD | = | Knockdown |
| PI | = | Propidium iodide |
| qRT-PCR | = | Quantitative real-time PCR |
| ROC | = | Receiver operating characteristic curve |
| SDS | = | Sodium dodecyl sulfate |
| shRNA | = | Small hairpin RNA |
| SLE | = | Systemic lupus erythematosus |
| SS | = | Sjogren’s syndrome |
| TAA | = | Tumor-associated antigen |
| WT | = | Wild type |
Consent for publication
Consent to publish has been obtained from the participants.
Ethics approval and consent to participate
All the animal experiments in this study were carried out in accordance with the approved guidelines and approved by the committee on Animal Care and Use of Beijing Friendship Hospital, Capital Medical University. HCC specimens and adjacent tissues were acquired from the Liver Research Center of Beijing Friendship Hospital, Capital Medical University. All of the patients provided written informed consent, and this study was approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University (Number 2015-P2-019-01).
Availability of data and material
The datasets supporting the conclusions of this article are included within the article and additional files.
Authors’ Contributions
Jian Huang conceptualized and supervised the study. Xiaojin Li designed the research study, analyzed the data, and wrote the manuscript. Yanmeng Li performed the research and prepared the figures. Anjian Xu, Donghu Zhou, Bei Zhang, Saiping Qi, Zhibin Chen, and Xiaoming Wang collected data and performed the experiments. Xiaojuan Ou, Bangwei Cao, and Chunfeng Qu designed and analyzed the experiments. All authors revised and approved the manuscript final version.
Supplemental Material
Download ()Acknowledgments
We thank the Liver Research Center of Beijing Friendship Hospital, Capital Medical University for providing HCC specimens and adjacent tissues.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Henley SJ, Ward EM, Scott S, Ma J, Anderson RN, Firth AU, Thomas CC, Islami F, Weir HK, Lewis DR, et al. Annual report to the nation on the status of cancer, part I: national cancer statistics. Cancer. 2020;126:2225–15. doi:10.1002/cncr.32802.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi:10.3322/caac.21551.
- Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastro Hepat. 2019;16:589–604. doi:10.1038/s41575-019-0186-y.
- Honsová E. Histopathological differential diagnosis of primary liver tumors. Rozhl Chir. 2014;93:170–175.
- Zaenker P, Gray ES, Ziman MR. Autoantibody production in cancer–the humoral immune response toward autologous antigens in cancer patients. Autoimmun Rev. 2016;15:477–483. doi:10.1016/j.autrev.2016.01.017.
- Hong Y, Huang J. Autoantibodies against tumor-associated antigens for detection of hepatocellular carcinoma. World J Hepatol. 2015;7:1581–1585. doi:10.4254/wjh.v7.i11.1581.
- Koziol JA, Imai H, Dai L, Zhang J, Tan EM. Early detection of hepatocellular carcinoma using autoantibody profiles from a panel of tumor-associated antigens. Cancer Immunol Immunother. 2018;67:835–841. doi:10.1007/s00262-018-2135-y.
- Hong Y, Long J, Li H, Chen S, Liu Q, Zhang B, He X, Wang Y, Li H, Li Y, et al. An analysis of immunoreactive signatures in early stage hepatocellular carcinoma. EBioMedicine. 2015;2:438–446. doi:10.1016/j.ebiom.2015.03.010.
- Varis A, Salmela AL, Kallio MJ. Cenp-F (mitosin) is more than a mitotic marker. Chromosoma. 2006;115:288–295. doi:10.1007/s00412-005-0046-0.
- Hussein D, Taylor SS. Farnesylation of Cenp-F is required for G2/M progression and degradation after mitosis. J Cell Sci. 2002;115:3403–3414. doi:10.1242/jcs.115.17.3403.
- Rattner JB, Rao A, Fritzler MJ, Valencia DW, Yen TJ. CENP-F is a .ca 400 kDa kinetochore protein that exhibits a cell-cycle dependent localization. Cell Motility Cytoskeleton. 1993;26:214–226. doi:10.1002/cm.970260305.
- Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol. 1995;130:507–518. doi:10.1083/jcb.130.3.507.
- Dai Y, Liu L, Zeng T, Zhu YH, Li J, Chen L, Li Y, Yuan Y-F, Ma S, Guan X-Y, et al. Characterization of the oncogenic function of centromere protein F in hepatocellular carcinoma. Biochem Biophys Res Commun. 2013;436:711–718. doi:10.1016/j.bbrc.2013.06.021.
- Kim HE, Kim DG, Lee KJ, Son JG, Song MY, Park YM, Kim -J-J, Cho S-W, Chi S-G, Cheong HS, et al. Frequent amplification of CENPF, GMNN and CDK13 genes in hepatocellular carcinomas. PLoS One. 2012;7:e43223. doi:10.1371/journal.pone.0043223.
- Brendle A, Brandt A, Johansson R, Enquist K, Hallmans G, Hemminki K, Lenner P, Försti A. Single nucleotide polymorphisms in chromosomal instability genes and risk and clinical outcome of breast cancer: a Swedish prospective case-control study. Eur J Cancer. 2009;45:435–442. doi:10.1016/j.ejca.2008.10.001.
- Chen WB, Cheng XB, Ding W, Wang YJ, Chen D, Wang JH, Fei R-S. Centromere protein F and survivin are associated with high risk and a poor prognosis in colorectal gastrointestinal stromal tumours. J Clin Pathol. 2011;64:751–755. doi:10.1136/jcp.2011.089631.
- Sun JB, Huang JZ, Lan J, Zhou K, Gao Y, Song ZG, Deng Y, Liu L, Dong Y, Liu X, et al. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int. 2019;19:264. doi:10.1186/s12935-019-0986-8.
- Herrera-Esparza R, Herrera-van-Oostdam D, López-Robles E, Avalos-Díaz E. The role of apoptosis in autoantibody production. Reumatismo. 2007;59:87–99.
- Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi:10.1084/jem.179.4.1317.
- Frisoni L, McPhie L, Colonna L, Sriram U, Monestier M, Gallucci S, Caricchio R. Nuclear autoantigen translocation and autoantibody opsonization lead to increased dendritic cell phagocytosis and presentation of nuclear antigens: a novel pathogenic pathway for autoimmunity? J Immunol. 2005;175:2692–2701. doi:10.4049/jimmunol.175.4.2692.
- Fransen JH, Hilbrands LB, Ruben J, Stoffels M, Adema GJ, van der Vlag J, Berden JH. Mouse dendritic cells matured by ingestion of apoptotic blebs induce T cells to produce interleukin-17. Arthritis Rheum. 2009;60:2304–2313. doi:10.1002/art.24719.
- Racanelli V, Prete M, Musaraj G, Dammacco F, Perosa F. Autoantibodies to intracellular antigens: generation and pathogenetic role. Autoimmun Rev. 2011;10:503–508. doi:10.1016/j.autrev.2011.03.001.
- Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–392. doi:10.1084/jem.188.2.387.
- Hansen MH, Nielsen HV, Ditzel HJ. Translocation of an intracellular antigen to the surface of medullary breast cancer cells early in apoptosis allows for an antigen-driven antibody response elicited by tumor-infiltrating B cells. J Immunol. 2002;169:2701–2711. doi:10.4049/jimmunol.169.5.2701.
- Fernández Madrid F. Autoantibodies in breast cancer sera: candidate biomarkers and reporters of tumorigenesis. Cancer Lett. 2005;230:187–198. doi:10.1016/j.canlet.2004.12.017.
- Macdonald IK, Parsy-Kowalska CB, Chapman CJ. Autoantibodies: opportunities for early cancer detection. Trends Cancer. 2017;3:198–213. doi:10.1016/j.trecan.2017.02.003.
- Liu H, Zhang J, Wang S, Pang Z, Wang Z, Zhou W, Wu M. Screening of autoantibodies as potential biomarkers for hepatocellular carcinoma by using T7 phase display system. Cancer Epidemiol. 2012;36:82–88. doi:10.1016/j.canep.2011.04.001.
- Cappello F, Angileri F, de Macario EC, Macario AJ. Chaperonopathies and chaperonotherapy. Hsp60 as therapeutic target in cancer: potential benefits and risks. Curr Pharm Des. 2013;19:452–457. doi:10.2174/138161213804143653.
- Anderson KS, LaBaer J. The sentinel within: exploiting the immune system for cancer biomarkers. J Proteome Res. 2005;4:1123–1133. doi:10.1021/pr0500814.
- Ersvaer E, Zhang JY, McCormack E, Olsnes A, Anensen N, Tan EM, Gjertsen BT, Bruserud Ø. Cyclin B1 is commonly expressed in the cytoplasm of primary human acute myelogenous leukemia cells and serves as a leukemia-associated antigen associated with autoantibody response in a subset of patients. Eur J Haematol. 2007;79:210–225. doi:10.1111/j.1600-0609.2007.00899.x.
- Li YM, Zhu Z, Zhang SH, Yu DH, Yu HY, Liu LN, Cao X, Wang L, Gao H, Zhu M, et al. ShRNA-targeted centromere protein A inhibits hepatocellular carcinoma growth. PLoS One. 2011;6:e17794. doi:10.1371/journal.pone.0017794.
- Tomonaga T, Matsushita K, Yamaguchi S, Oohashi T, Shimada H, Ochiai T, Yoda K, Nomura F. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res. 2003;63:3511–3516.
- Perosa F, Prete M, Di Lernia G, Ostuni C, Favoino E, Valentini G. Anti-centromere protein A antibodies in systemic sclerosis: significance and origin. Autoimmun Rev. 2016;15:102–109. doi:10.1016/j.autrev.2015.10.001.
- Suber T, Rosen A. Apoptotic cell blebs: repositories of autoantigens and contributors to immune context. Arthritis Rheum. 2009;60:2216–2219. doi:10.1002/art.24715.
- Savill J, Fadok V, Henson P, Haslett C. Phagocyte recognition of cells undergoing apoptosis. Immunol Today. 1993;14:131–136. doi:10.1016/0167-5699(93)90215-7.
- Ohlsson M, Jonsson R, Brokstad KA. Subcellular redistribution and surface exposure of the Ro52, Ro60 and La48 autoantigens during apoptosis in human ductal epithelial cells: a possible mechanism in the pathogenesis of Sjögren’s syndrome. Scand J Immunol. 2002;56:456–469. doi:10.1046/j.1365-3083.2002.01072_79.x.
- Nakamura H, Kawakami A, Eguchi K. Mechanisms of autoantibody production and the relationship between autoantibodies and the clinical manifestations in Sjogren’s syndrome. Transl Res. 2006;148:281–288. doi:10.1016/j.trsl.2006.07.003.
- Grimsley C, Ravichandran KS. Cues for apoptotic cell engulfment: eat-me, don’t eat-me and come-get-me signals. Trends Cell Biol. 2003;13:648–656. doi:10.1016/j.tcb.2003.10.004.
- Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol. 2006;79:896–903. doi:10.1189/jlb.1005550.
- Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi:10.1038/32183.
- Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi:10.1038/15200.