
ABSTRACT
Co-stimulatory 4–1BB receptors on tumor-infiltrating T cells are a compelling target for overcoming resistance to immune checkpoint inhibitors, but initial clinical studies of 4–1BB agonist mAbs were accompanied by liver toxicity. We sought to engineer a tri-specific antibody-based molecule that stimulates intratumoral 4–1BB and blocks PD-L1/PD-1 signaling without systemic toxicity and with clinically favorable pharmacokinetics. Recombinant fusion proteins were constructed using scMATCH3 technology and humanized antibody single-chain variable fragments against PD-L1, 4–1BB, and human serum albumin. Paratope affinities were optimized using single amino acid substitutions, leading to design of the drug candidate NM21-1480. Multiple in vitro experiments evaluated pharmacodynamic properties of NM21-1480, and syngeneic mouse tumor models assessed antitumor efficacy and safety of murine analogues. A GLP multiple-dose toxicology study evaluated its safety in non-human primates. NM21-1480 inhibited PD-L1/PD-1 signaling with a potency similar to avelumab, and it potently stimulated 4–1BB signaling only in the presence of PD-L1, while exhibiting an EC50 that was largely independent of PD-L1 density. NM21-1480 exhibited high efficacy for co-activation of pre-stimulated T cells and dendritic cells. In xenograft models in syngeneic mice, NM21-1480 induced tumor regression and tumor infiltration of T cells without causing systemic T-cell activation. A GLP toxicology study revealed no evidence of liver toxicity at doses up to 140 mg/kg, and pharmacokinetic studies in non-human primates suggested a plasma half-life in humans of up to 2 weeks. NM21-1480 has the potential to overcome checkpoint resistance by co-activating tumor-infiltrating lymphocytes without liver toxicity.
Introduction
Immune checkpoint inhibitors (ICIs) have revolutionized cancer therapy, inducing durable responses in cancer patients diagnosed with various tumor types.Citation1 The most widely established ICIs inhibit the interaction of PD-1 (programmed cell death-1) with its ligand PD-L1; inhibitors of PD-L1/PD-1 are currently in use for ~20 types of solid and hematologic malignancies, and they have indications for tumors with high microsatellite instability (MSI-H) or high tumor mutational burden (TMB-H) independent of the tumor’s anatomic origin.Citation2 ICIs targeting other immune checkpoint pathways, especially CTLA-4 (cytotoxic T lymphocyte-associated protein 4), are also in use, but less commonly and often in combination with PD-L1/PD-1 inhibitors. Despite the clinical success of PD-L1/PD-1 inhibitors, the majority of malignancies―even those expressing high levels of PD-L1―exhibit inadequate responses to these agents or other ICIs. The mechanisms underlying the limited efficacy of ICIs are not well understood, but downregulation of effector function of tumor-infiltrating lymphocytes (TILs) is a likely cause, at least in some ICI-nonresponsive tumor phenotypes.Citation3,Citation4 Heterogeneous tumor expression of PD-L1, or lack of PD-L1 expression, is likely another contributing factor.Citation2
Activation of co-stimulatory receptors on T cells has emerged as a promising strategy for stimulating T-cell effector function and improving rates of tumor response to ICIs. A compelling co-stimulatory receptor is 4–1BB (CD137), a member of the tumor necrosis factor receptor superfamily (TNFRSF) expressed on antigen-experienced T cells but not on resting T cells.Citation5–7 This expression profile may help to restrict the activating signal to tumor-reactive T cells.Citation5,Citation8,Citation9 In preclinical models, monoclonal antibodies (mAbs) that act as agonists of 4–1BB can restore proliferation and cytokine secretion of CD8+ T cells in the tumor microenvironment, and produce anti-tumor responses.Citation10–12 Furthermore, synergistic anti-tumor effects of agonist α4–1BB mAbs and αPD-1 mAbs have been reported in tumor models.Citation13–15 Despite the promise of agonist α4–1BB mAbs in mechanistic and preclinical studies, clinical studies of α4-1BB mAbs have exposed significant challenges for their use in cancer immunotherapy. The fully human agonist α4-1BB mAb urelumab showed encouraging evidence of efficacy in early-stage clinical trials, but also dose-dependent liver toxicity.Citation16 Doses sufficiently low to avert liver toxicity had disappointing efficacy.Citation5 The humanized agonist α4-1BB mAb utomilumab exhibited less liver toxicity than urelumab, but it also exhibited less activity than urelumab in preclinical and clinical studies.Citation17,Citation18
Recent advances in our understanding of the mechanics of 4–1BB signaling, as well as the mechanisms of urelumab-induced liver toxicity, provide opportunities to improve on the targeting and avert the problems of early 4–1BB agonists. First, the epitope on 4–1BB to which an agonist binds affects the strength of 4–1BB stimulation. For example, urelumab and utomilumab have similar affinities for 4–1BB but they bind to different epitopes, and that difference accounts for at least part of the difference in their agonist activity.Citation19 Second, stimulation of 4–1BB signaling requires membrane crosslinking by its agonist.Citation19–21 For example, its endogenous agonist, 4–1BBL, exists in both a soluble and membrane-anchored forms; soluble 4–1BBL has little agonist activity, whereas membrane-anchored (crosslinked) 4–1BBL is a much more effective agonist.Citation22 Importantly, a variety of moieties can replace the membrane anchor of endogenous 4–1BBL―moieties such as Fcγ or antibody scFv fragments against membrane antigens.Citation23,Citation24 Indeed, urelumab-induced liver toxicity is at least partly explained by cross-linking of its Fc domain to FcγR expressed by resident liver myeloid and sinusoidal endothelial cells, thereby allowing 4–1BB stimulation of immune cells in the liver.Citation19,Citation25–27 However, the ability to use different crosslinking moieties opens the possibility of constructing a 4–1BB agonist employing a crosslinking moiety that averts liver toxicity and improves its tumor selectivity. That strategy has been used to construct bi-specific antibodies that stimulate T-cell 4–1BB in specific tumor types such as HER2-expressing breast cancers or EGFR-expressing tumors, or when the tumor stroma expresses fibroblast activation protein (FAP).Citation27–31 We have employed this strategy by constructing and characterizing a trispecific molecule combining a 4–1BB agonist domain with a PD-L1-binding domain, whereby PD-L1 co-ligation in the tumor microenvironment facilitates 4–1BB stimulation on tumor-infiltrating lymphocytes. The third binding domain interacts with serum albumin and enables an extended serum half-life.
Stimulation of 4–1BB signaling also requires clustering of at least three 4–1BB molecules in the cell membrane.Citation19,Citation20 Indeed, 4–1BBL binds as a homotrimer to 4–1BB and induces the formation of a three-receptor complex, which represents the basic unit of signaling.Citation20 This requirement has been interpreted by some to imply that an ideal 4–1BB agonist should be bi- or tri-valent to promote receptor clustering and to increase avidity (and hence selectivity). However, avidity-based binding (as opposed to affinity-based binding) is dependent on target density;Citation32 thus, the potency (and hence clinical dosing) of a bi- or tri-valent tumor-targeting drug varies according to the target density in the tumor. For example, a bivalent αHER2 T cell-engaging bispecific antibody had an anti-tumor potency that was highly dependent on tumor HER2 expression level, and it had a very narrow therapeutic window.Citation32 In contrast, bi-specific antibodies monovalent for 4–1BB have the potential for better on-target 4–1BB specificity and have a potency that is not dependent on tumor target density.Citation33–35 According to this principle, an αPD-L1/α4-1BB bispecific antibody that exhibits ultra-high affinity monovalent binding for PD-L1 should stimulate 4–1BB with a potency that is dependent on PD-L1 expression per se, but is independent of the density of PD-L1 expression. Furthermore, the affinity-based binding of monovalent antibodies allows systematic and precise calibration of binding affinities, in contrast to the avidity-based binding of multivalent antibodies in which binding properties depend on extrinsic factors (e.g., target density). As a consequence, the individual paratopes of a multispecific monovalent antibody can be engineered to achieve overlapping dose dependence and a broad window for clinical dosing, essential features of a successful multispecific drug.Citation32,Citation36
These advances in our understanding of valency, 4–1BB signaling, and off-tumor toxicity led us to use a rational design strategy to engineer a novel drug that combines monovalent 4–1BB agonism with monovalent, ultra-high affinity PD-L1 blockade. At the outset, we identified several essential properties that the final drug candidate should exhibit. First, the drug should potently block the PD-L1/PD-1 pathway. Second, it should concomitantly and potently stimulate the 4–1BB pathway in a manner that a) requires PD-L1 expression in the tumor microenvironment but b) has consistent and predictable dosing that is independent of (non-zero) tumor PD-L1 expression level and c) maximally stimulates 4–1BB in the same dose range as maximal PD-L1 antagonism. Third, stimulation of the 4–1BB pathway should be restricted to the tumor microenvironment, with minimal or no systemic activation of T cells or effects on non-tumor tissues. Furthermore, the drug should have an advantageous plasma half-life, allowing infrequent administration in clinical settings. This article describes the molecular engineering and proof-of-concept studies leading to the development of a tri-specific drug candidate satisfying these requirements, and with potential applicability across numerous tumor types.
Materials and methods
Ethics
All mouse experiments and protocols were approved by the animal welfare body at CR Discovery Research Services Germany and North Carolina and the associated local authorities and were conducted according to all applicable international, national and local laws and guidelines. All experiments using cynomolgus macaques were in compliance with applicable animal welfare acts and were approved by the local Institutional Animal Care and Use Committee (IACUC). For studies using human PBMCs, donors provided written informed consent and blood samples were collected, anonymized, and used according to ethical approval from York Research Ethics Committee (Harrogate, UK; Approval numbers: 05/Q1107/86; 05/Q1107/87; 05/Q1107/90; 05/Q1107/91; 06/Q1107/6; 06/Q1107/7) and West Midlands – Black Country Research Ethics Committee (West Midlands, UK; Approval number 19/WM/0260), both affiliated with the U.K. National Health Service.
Molecule design and production
CDRs from rabbit antibodies targeting human PD-L1, serum albumin, and 4–1BB ECD were grafted onto a stability promoting human variable domain acceptor scaffold, in which framework region 4 (FR4) of the kappa-type light chain was substituted by a lambda-type FR4 (see Egan et al.)Citation37 resulting in λcapped™ antibody Fvs, single-chain Fvs (scFvs). The three target-specific variable domains were assembled into a trispecific single-chain diabody fused to a single-chain Fv (scDb-scFv; herein termed scMATCH3™).
Expression of scFv and scMATCH3 molecules was performed using CHO cells and were purified from the clarified harvest by standard capture, polishing platform processes. All produced molecules had a purity > 95%.
The control molecules were either ordered at evitria AG (avelumab, urelumab, utomilumab) or purchased via a pharmacy (nivolumab).
4-1BB variants were transiently expressed as Fc-fusion proteins. For complex formation with 38–27-A11, residues 24–160 of 4–1BB (Uniprot: Q07011) were expressed. For complex formation with 38–02-A04, residues 119–160 were expressed. Both variants were mutated to remove a free cysteine as well as a glycosylation motif (C121S and N149Q). A N-terminal secretion sequence and a C-terminal hingeless Fc-tag preceded by an IdeS cleavage siteCitation38 were added. The fusion protein was transiently expressed in CHO cells and captured from the cell supernatant using protein A resin (Cytiva). The Fc-tag was cleaved by IdeS at 37°C and removed using protein A beads (Sino Biological).
Co-crystallization
For complex crystallization of both proteins, scFv and 4-BB ECD variants were incubated at an equimolar ratio followed by size exclusion chromatography polishing (S75 column by Cytiva) in 50 mM HEPES, 100 mM NaCl, pH 6.7. The complexes were concentrated to 10 mg/ml and 12 mg/ml for 38–27-A11 in complex with 4–1BB and 38–02-A04 in complex with 4–1BB ECD, respectively.
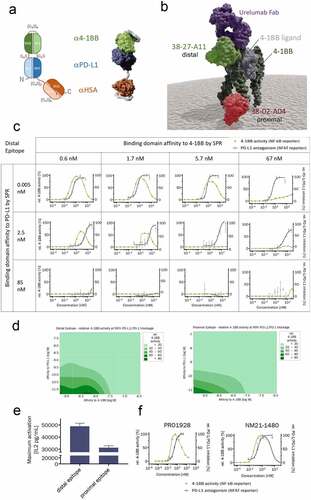
Figure 1. Engineering of the molecular scaffold and calibration of the 4–1BB and PD-L1 binding affinities. (a) Schematic representation (left) of the trispecific scMATCH3 molecular scaffold and a structural model (right) derived from X-ray crystallography structures. αPD-L1 (VL1/VH1) in blue, α4-1BB (VH2/VL2) in green, αHSA (VL3/VH3) in orange, and Gly4-Ser peptide linkers in red. (b) Surface representation of a structural model based on X-ray crystallography complex structures showing the trimer of 4–1BB extracellular domain (dark green), its natural trimeric ligand, 4–1BB-L (gray), and the bound 4–1BB-binding domains of urelumab as an Fab fragment (purple), as scFv targeting the proximal part of 4–1BB (38–02-A04 in red) and an scFv targeting the distal part of 4–1BB (38–27-A11 in light green). (c) Drug concentration–response curves for 12 scMATCH3 analogues with the indicated α4-1BB and αPD-L1 affinities. Responses were measured from Jurkat cells containing NFAT (PD-L1, blue) or NF-κB (4–1BB, green) reporters. Markers indicate mean (± SD) of two technical replicates of one experiment. (d) Contour plots showing the percentage activity of scMATCH3 constructs to stimulate 4–1BB activity measured by NF-ĸB (in shades of green) at 99% relative PD-L1 antagonistic activity. Data are shown for distal 4–1BB epitope-targeting scMATCH3 molecules (left) and proximal 4–1BB epitope-targeting molecules (right). The contour plot was generated using the distance method for interpolation in Minitab. (e) Maximum secretion of IL-2 from SEA pre-activated human PBMCs measured by ELISA after addition of trispecific scMATCH3 molecules binding to the membrane distal or membrane-proximal epitope of 4–1BB (mean ± SD of four independent experiments). (f) Concentration–response curves for stimulation of 4–1BB signaling in Jurkat NF- κB reporter cells (green) and PD-L1 antagonism in Jurkat NFAT reporter cells (blue) of a bivalent, bispecific mAb (PRO1928) employing the same α4-1BB and αPD-L1 paratopes as NM21-1480 (left graph) and NM21-1480 (right graph). Mean (± SD) of one representative experiment of two independent experiments
For 38–27-A11 in complex with 4–1BB ECD, initial crystals were grown by sitting drop vapor diffusion at 20°C in 0.1 M sodium acetate, pH 5.5, 22% PEG2000 MME, 0.17 M to 0.23 M calcium acetate and an equal volume of mother liquor and complex. Crystals were crushed using SeedBeads (Hampton Research) and used as crystal seeds in subsequent crystallization screens. Final crystals were grown at 20°C in 0.1 M Tris-Acetate pH 8.5, 1 M Na-Formate, 25% PEG2000 MME with a mother-liquor-to protein-to-seed ratio of 1:1:0.125. Crystals were cryoprotected in 80 mM tris-acetate pH 8.5, 0.8 M sodium formate, 22.4% PEG2000 MME, 20% ethylene glycol, and frozen in liquid nitrogen.
For 38–02-A04 in complex with 4–1BB ECD, crystals were grown at 20°C in 0.1 M tris-acetate pH 8.4 to pH 8.5, 0.95 M Li2SO4, 0.01 M NiCl2 with a mother-liquor-to-protein ratio of 1:1. Crystals were cryoprotected by adding 100% ethylene glycol to the crystallization drop (approx. 25% v/v). After a short incubation, the crystal was frozen in liquid nitrogen. A native dataset of the complexes with 4–1BB ECD was collected at the Swiss Light Source at the Paul-Scherrer Institute, Villigen, Switzerland.
The complex of 38–27-A11 with 4–1BB crystallized in space group I222 and was processed using XDS to a resolution of 2.2 Å. The complex of 38–02-A04 with 4–1BB crystallized in space group I422 and was processed using XDS to a resolution of 1.7 Å.Citation39 The structure was solved by molecular replacement with Phaser using a previously solved in-house scFv structure and refined using Refmac.Citation40,Citation41 Manual model building was executed in Coot.Citation42
Design of affinity variants
For the 4–1BB binding domains targeting the proximal and distal epitope, the affinity toward 4–1BB ECD was modified by mutating antigen interacting CDR residues based on the complex crystal structure. After purification, the affinity was determined by SPR. The affinity toward PD-L1 was modified by an alanine scan of CDR3 residues in the light and heavy chain of the scFv. Based on the scFv affinity data various scMATCH3 were designed to cover a broad range of affinities and affinity ratios for both PD-L1 and 4–1BB.
Potency to activate 4-1BB signaling in Jurkat reporter cell line
The potency of scMATCH3 molecules to activate 4–1BB signaling was assessed by using the GloResponse™ NF-ĸB-luc2/4.1BB Jurkat reporter gene assay cells by Promega (J2332) and the Bio-Glo™ Luciferase Assay System (Promega, G7940). In this Jurkat reporter cell line, clustering of 4–1BB leads to NF-κB activation, which in turn induces expression of the luciferase reporter gene. The cell lines tested were the cancer cell lines HCC827 (lung adenocarcinoma, CRL-2868™, ATCC), HCC1954 (ductal carcinoma, ATCC, CRL-2338™) expressing PD-L1 at high levels, MDA-MB-231 (breast adenocarcinoma, ATCC, HTB-26™) at medium levels and HT-29 (colorectal adenocarcinoma, DSMZ, ACC 299) at low levels and the PD-L1 negative CHO K1 (ATCC, CCL-61™) cells. Cancer cells stimulated for 24 h with 10 ng/ml IFNγ to boost PD-L1 expression were seeded at 25,000 cells per well in 96-well culture plates. Serial dilutions of the scMATCH3 were added. Subsequently, 40,000 4–1BB expressing Jurkat reporter gene cells per well diluted in assay buffer with human SA (final concentration of 25 mg/ml) were added and plates were incubated for 6 h and 24 h at 37°C and 5% CO2. Luciferase expression was read by a chemiluminescent reaction. IC50 and AUC values were calculated by fitting the concentration–response curves with a four parameter logistic regression model using Prism v.6.0 software (GraphPad Software).
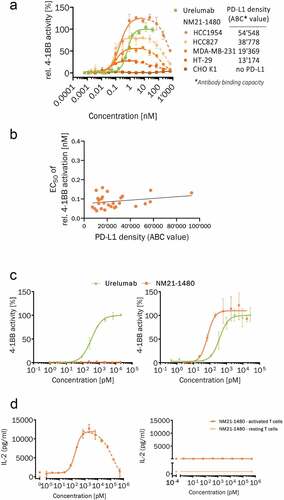
Figure 2. NM21-1480 stimulation of 4–1BB requires PD-L1, but its EC50 is largely independent of PD-L1 density. (a) Concentration–response curve for NM21-1480-induced stimulation of 4–1BB relative to maximal urelumab-induced activation in a NF-κB Jurkat reporter cell line co-incubated with IFN-γ pre-stimulated cancer cell-lines expressing different PD-L1 densities. PD-L1 density on the cancer cell lines is represented as the average αPD-L1 antibody-binding capacity quantified by flow cytometry. Markers represent mean (± SD) of one representative experiment of ≥ 4 independent experiments. (b) EC50 values for NM21-1480 stimulation of 4–1BB measured by NF-κB Jurkat reporter cells plotted against PD-L1 density of cell lines co-incubated within the assay. Every cell line was tested in ≥ 4 independent experiments and all obtained individual EC50 values were plotted against PD-L1 density. A linear regression model is shown as solid line to indicate constant EC50. (c) Normalized concentration–response curve for NM21-1480 or urelumab stimulation of NF-κB in Jurkat reporter cells co-incubated with PD-L1-negative CHO cells (left) or PD-L1-positive HCC827 cells (right). Markers represent mean (± SD) of one representative experiment of ≥ 4 independent experiments. (d) Human PBMCs were pre-activated with an αCD3 antibody then exposed to NM21-1480. Concentration–response curves for IL-2 secretion in response to NM21-1480 stimulation in the presence of PD-L1-positive HCC827 cells (left) or PD-L1-negative CHO cells (right). No measurable Il-2 secretion was observed in non-pre-activated PBMCs. Mean (± SD) of one representative experiment of 3 independent experiments
Determination of PD-L1 cell surface expression
Antibody binding capacity (ABC) as a measure to quantify the PD-L1 cell surface expression was assessed using Quantum Simply Cellular beads anti-human IgG (Bangs Laboratories, Inc., 816) and Alexa Fluor 488 labeled avelumab analog according to the manufacturer’s protocol. The ABC was derived from geometric means for the four Quantum Simply Cellular bead populations using the NovoExpress software (ACEA Biosciences) and the PD-L1 densities were calculated according to the manufacturer’s instructions.
Potency to induce IL-2 secretion in human PBMCs co-stimulated with anti-CD3 antibody
To assess the potency of NM21-1480 to induce IL-2 secretion in PBMCs, HCC827 (ATCC®, CRL-2868) cells expressing PD-L1 or PD-L1 negative CHO cells as control (CHO-K1, ATCC, CCL-61™) were seeded at a density of 10,000 cells per well on 96-well culture plates pre-coated with 2.0 µg/ml of an agonistic anti-human CD3 antibody (BD Pharmingen, 551916). The plates were incubated overnight at 37°C, 5% CO2. On the next day, peripheral blood mononuclear cells (PBMC) were isolated from fresh human blood by means of density gradient centrifugation. 100,000 PBMC per well were added to the plate, followed by the addition of three-fold serial dilutions of NM21-1480 in assay medium (RPMI-1640, 10%FCS) at concentrations ranging from 40,000 to 0.08 ng/ml or from 40,000 to 0.23 ng/ml, in duplicates or in triplicates. HSA was added to each well to a final concentration of 1 mg/ml. Additionally, NM21-1480 at 40,000 ng/ml or at 40,000 and 100 ng/ml was added to microplate wells that were not pre-coated with the anti-human CD3 antibody. After 76 hours of incubation, cell supernatants were harvested. Human interleukin-2 (IL-2) levels in the culture supernatants were quantified using the IL-2 human ELISA MAX assay from Biolegend (431801), according to kit instructions. IL-2 concentrations were plotted against NM21-1480 concentrations. The curves were fitted using a four parameter logistic regression model in GraphPad prism to calculate EC50 values and maximum levels of IL-2 secretion by activated T cells.
Neutralization of PD-L1/PD-1 and PD-L1/B7.1 interaction by competition ELISA
Potency to neutralize PD-L1 binding to PD-1 and B7.1 was assessed in competition ELISAs where 4 µg/ml PD-1-Fc chimera or B7.1-Fc chimera (R&D systems, 1086-PD or 140-B1) was coated. After simultaneous incubation with serial dilutions of NM21-1480 and biotinylated PD-L1 at a concentration of 1 ng/ml or 40 ng/ml, the plate was washed and bound biotinylated PD-L1 was detected by a streptavidin-polyHRP40 – TMB detection system. IC50 values were calculated by fitting the concentration–response curves with a four parameter logistic regression model using Prism 6.0 software (GraphPad Software).
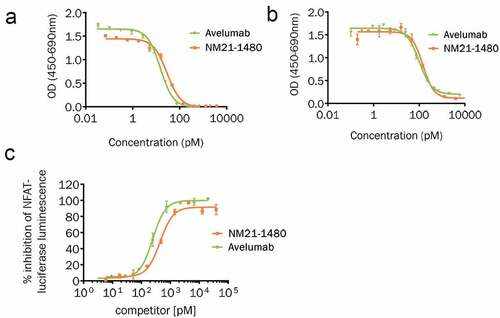
Figure 3. NM21-1480 potently inhibits PD-L1 interactions and signaling. (a) Competition ELISA quantifying PD-L1/PD-1 interaction in the presence of different concentrations of NM21-1480 or avelumab. Mean (± SD) of two technical replicates of one experiment. (b) Competition ELISA quantifying PD-L1/B7.1 interaction in the presence of different concentrations of NM21-1480 or avelumab. Markers represent mean (± SD) of two technical replicates of one experiment. (c) Jurkat NFAT reporter cells co-incubated with PD-L1-positive CHO cells and a T-cell receptor activator were exposed to different concentrations of NM21-1480 or avelumab. Both agents inhibited NFAT expression with similar EC50. Representative results from 9 independent experiments
Neutralization of PD-L1/PD-1 interaction by NFAT reporter gene assay
CHO cells stably expressing PD-L1 and a TCR activator (BPS Bioscience, cat. 60536) were incubated with Jurkat T cells stably expressing PD-1 and a reporter gene to monitor T cell activation (BPS Bioscience, 60535). T cell activation via TCR engagement is monitored by NFAT-induced expression of firefly luciferase. The interaction between PD-L1 and PD-1 negatively regulates TCR signaling and decreases firefly luciferase expression. 35,000 CHO/PD-L1/TCR activator cells were seeded in 96-well cell culture plates and incubated with threefold serial dilutions of scMATCH3 molecules. 20,000 effector Jurkat cells/well diluted in assay buffer containing 2 mg/ml human SA (1 mg/ml human SA final concentration) were added and the plates were incubated for 6 hours at 37°C and 5% CO2. Luciferase expression was read by a chemiluminescent reaction. IC50 values were calculated by fitting the concentration–response curves with a four parameter logistic regression model using Prism 6.0 software (GraphPad Software).
Potency to activate T-cells ex-vivo by SEA PBMC assay
The synergistic effect of concomitant inhibition of PD-L1 and 4–1BB co-stimulation was assessed using a SEA PBMC assay. The assay consisted of human PBMCs that were stimulated with superantigen SEA to induce expression of PD-L1 on APCs and T cells as well as 4–1BB on T cells. Human PBMC were isolated from human whole blood by density gradient centrifugation and depleted of natural killer cells (NK cells) using anti-CD56 antibody and MACS cell separation kit (Miltenyi Biotec). 200,000 human PBMC per well were seeded in a 96-well plate, followed by the addition of five-fold serial dilutions of NM21-1480 and combinations of clinical-stage anti-PD-1/PD-L1 (avelumab, produced from published sequence data (CAS number 1537032–82-8); pembrolizumab, Merck & Co., Inc.; nivolumab, Bristol‑Myers Squibb Company) and anti-4-1BB antibodies (urelumab, produced from published sequence data (CAS number 934823–49-1); utomilumab, produced from published sequence data (CAS number 1417318–27-4)) ranging from 40,000 to 0.1 ng/ml in assay buffer (RPMI-1640 with 10% FCS, 1 mg/ml HSA, 10 ng/ml SEA or without SEA). After 96 hours of incubation human IL-2 levels in the culture supernatants were quantified using the IL-2 human ELISA MAX kit (BioLegend, 431801).
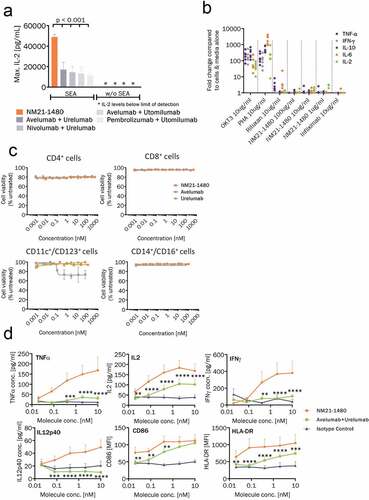
Figure 4. Potent stimulation of 4–1BB and immune cell co-stimulation by NM21-1480. (a) Human PBMCs were pre-stimulated with SEA to induce T- cell activation and expression of PD-L1 and 4-1BB expression. IL-2 secretion was then measured by ELISA in response to NM21-1480 or combinations of αPD-L1/PD-1 mAbs plus α4-1BB mAbs (p < .001 by one-way ANOVA and Tukey’s multiple comparisons test). In non-pre-stimulated PBMCs, IL-2 secretion was near or below the lower limit of detection. Bars indicate mean of maximum IL-2 levels ± SD from ≥3 independent experiments. (b) Cytokine release from quiescent human PBMCs in response to 3 concentrations of NM21-1480 and positive (OKT3, PHA, Rituxan) and negative (infliximab) controls. Colors indicate cytokine, and individual data points for each condition and cytokine represent each of 6 healthy donors. (c) Viability of 4 indicated classes of SEA pre-stimulated PBMCs after exposure to NM21-1480, avelumab, or urelumab. Viability was assessed by annexin V-APC staining. Mean (± SD) of three technical replicates of one experiment), representative of 2 experiments. (d) Markers of T-cell activation (TNFα, IL-2, IFNγ) or DC activation (IL12p40, CD86, HLA-DR) in response to stimulation in co-cultures of T cells and monocyte-derived dendritic cells (MoDCs). MoDCs were prepared from CD14+ cells cultured for 7 days. MoDCs were then cultured together with T cells from a separate donor for 5 days in the presence of NM21-1480, avelumab plus urelumab, or IgG1 plus IgG4. Supernatants and cells were collected at the end of culture (or 48 hours for IL2) and cytokine production was measured by ELISA or cell surface markers measured by flow cytometry. Data are presented as mean ± SEM (n = 6 biological replicates). **p˂0.01, ***p˂0.001, ****p˂0.0001 comparing to NM21-1480, as determined using a repeated measures two-way ANOVA with Dunnett’s multiple comparisons test
Analysis of viability of T cells, APCs and monocytes in SEA PBMC assay
In the same set of experiments using SEA stimulated PBMCs described above, the effects of NM21-1480 on viability of monocytes, APCs, CD4 + T cells and CD8 + T cells was investigated. Viability of CD4 + T cells, CD8 + T cells, monocytes, CD11c+/CD123+ APCs and CD11c+ /CD86+ APCs was assessed by flow cytometry using antibodies specific for each marker cited above, anti-CD8-FITC (BioLegend, 300906), anti-TCR ɑ/β-APC (BioLegend, 306718), anti-CD11c-PE/Cy7 (BioLegend, 344710), anti-CD123-PE (BioLegend, 306006), anti-CD14-FITC (BioLegend, 325604), anti-CD16-APC/Cy7 (BioLegend, 302018). Apoptotic cells were quantified using Annexin V-APC (BioLegend, 640941).
Assessment of potential in vitro cytokine release by NM21-1480 in human PBMCs
The potential in vitro cytokine release from PBMCs after 24-hour incubation with either NM21-1480 or controls was assessed in the liquid-phase setup using PBMCs isolated from six donors. The analysis of cytokine release (IFNγ, IL-2, IL6, IL-10, and TNFα) was performed on the Luminex Lx200 platform. The R&D HS cytokine kits (R&D systems, LHSCM000) were used along with the Luminex Performance Assay Control Set 889 (R&D systems, QC11). All samples were stimulated with the following test conditions; 10 µg/mL PHA (Sigma, L8902), 10 µg/mL anti-CD3 OKT3 (Biolegend, 317325), 10 µg/mL rituximab (Kays Medical, 40526423), 1, 10, and 100 µg/mL NM21-1480, media only and 10 µg/mL infliximab (Janssen, 1000004176912).
Investigation into the effect of NM21-1480 in an allogeneic mixed leukocyte reaction assay
Human PBMCs from six healthy donors (n = 6) were prepared from buffy coats by density gradient centrifugation over Lymphoprep™. Immature monocyte-derived dendritic cells (MoDCs) were prepared by isolating CD14+ cells using immunomagnetic separation (positive selection) (Stemcell™) and resuspended in MoDC differentiation media (Miltenyi Biotec) at a density of 1 × 106 cells/mL and cultured for 7 days. Responder CD3 + T cells were prepared from cryopreserved PBMCs on day 7, from a different donor to that used for the MoDCs, using an immuno-magnetic isolation kit (negative selection) (Stemcell™) and resuspended at 2×106/mL in culture medium. 1 × 104 iMoDCs and 1 × 105 T cells were added per well to a 96-well round-bottomed plate in a total volume of 100 µL. NM21-1480 and reference antibodies were added in 100 µL. Cells were cultured for five days. Supernatants were sampled at 48 hours for immediate assessment of IL-2 by ELISA (Invitrogen™, 88–7025-88). At the end of culture, cells were collected for flow cytometry assessment and supernatants were collected for subsequent assessment of IFN-γ by ELISA (Invitrogen™, 88–7316-77) and of IL-12p40, IL-6, TNF-α and IL-1β by multiplex (Invitrogen™ PPX-04-MXTZ9ZT). Cells were stained for viability, CD3 (BioLegend, 300420), CD11b (BioLegend, 301318), CD11c (Invitrogen™, 48–0116-42), CD40 (BioLegend, 334334), CD80 (Invitrogen™, 12–0809-42), CD86 (BioLegend, 305430), CD206 (BioLegend, 321110) and HLA-DR (Invitrogen™, 45–9956-42).
Anti-tumor activity in HSC engrafted humanized mouse tumor model
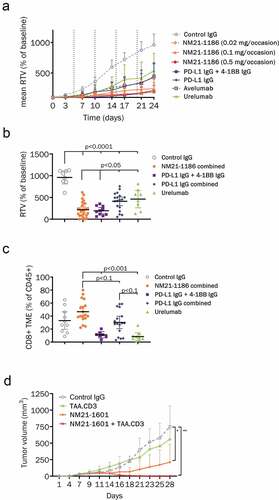
Female 5- to 7-week-old NOG mice (NOD.Cg-Prkdcscid Il2rgtm1Sug/ JicTac) engrafted with human umbilical cord blood (UCB)-derived hematopoietic stem cells (HSC) (huNOG) were purchased from Taconic Denmark and were injected subcutaneously (s.c) with 5 × 106 (total injection volume 100 μl, 50% cell suspension in PBS with 50% Matrigel®) HCC827 non-small cell lung cancer (NSCLC) cells. After tumor establishment to a volume of 80 to 100 mm3 mice were randomized into treatment groups (n = 10 each) on day 0 of the study. Randomization also aimed at a comparable distribution of the five individual HSC donors between the different treatment groups wherever possible. Mice were treated intraperitoneally (i.p.) with palivizumab (0.1 mg), proprietary anti-PD-L1 IgG (0.1 mg), avelumab analog (0.1 mg), urelumab analog (0.1 mg), NM21-1186 at 3 different dose levels (0.02 mg, 0.1 mg and 0.5 mg) or PD-L1 IgG + 4–1BB IgG combination (same binding domains as NM21-1186, 0.1 mg each) on day 0, 5, 10, 15 and 20. Tumor growth and body weight were recorded twice weekly. Tumors were harvested at the end of the study on day 29 and day 30, and processed for flow cytometry. Tumors were measured with a caliper in two dimensions. Tumor volume was calculated using the following formula V = (length x width2) x 0.5. The RTVs are shown only up to day 24, as the tumors started to regress spontaneously from day 24 on in the negative control group treated with palivizumab.
Figure 5. Activity of NM21-1480 in xenograft models. (a), (b) and (c), CD34+ stem cell-substituted NOG mice were engrafted with HCC-827 NSCLC cells. Mice were randomized at day 0 when tumor volumes reached between 80 and 100 mm3, and were treated on days 0, 5, 10, 15, and 20 as indicated by the dotted vertical lines. (a) Relative tumor volume at the indicated times after randomization. Each marker represents mean of 10 animals. (b) Relative tumor volume at the end of the study. Horizontal bars indicate mean (±95% CI). Statistical analysis was performed by one-way ANOVA with Bonferroni correction. The results are representative of two experiments. (c) At the end of the study, tumors were harvested and assessed for infiltration of human T cells by flow cytometry. Each colored marker represents the normalized CD8 + T-cell count in the tumor microenvironment of one mouse (n = 10). Horizontal bars indicate mean (±95% CI). Statistical analysis was performed by one-way ANOVA with Bonferroni correction. (d) NCG mice were injected subcutaneously (s.c.) with H292 cells and human PBMCs on day 0. Mice were treated on days 5, 10, 15, 20 and 25 with a control IgG, a CD3.TAA scMATCH3 molecule, NM21-1601 or the combination of CD3.TAA and NM21-1601. The graph displays tumor volume at the indicated times after randomization. Each point represents mean and standard deviation of 8 animals. Statistical analysis was performed using a two-way ANOVA with Tukey’s multiple comparison test, * p < .05; ** p < .005 compared to control IgG treatment group at day 28
Flow cytometry for assessment of tumor T cell infiltration
Tumor tissue processing into single cell suspension for flow cytometry analysis was performed according to the Miltenyi human tumor dissociation kit instructions. Single-cell suspension was treated with FcR-Block in a PBS-based buffer containing 2% of fetal bovine serum to avoid unspecific staining. Fluorochrome-conjugated mAbs to the following human antigens were used: CCR7/CD197 (353244), CD4 (300518), hCD223/LAG-3 (369314), hCD8 (301012) from BioLegend; hCD279/PD-1 (46–2799-42) and hFoxp3 (17–4776-42) from eBioscience; hCD45 (MHCD4530TR) from Thermo Fisher and hCD25 (555431), CD45RO (563722) hGrB (561142) from BD Biosciences. Attune NXT Acoustic Focusing Cytometer (Thermo Fisher Scientific) was used for cell acquisition and data analysis was carried out using the FlowJo software (TreeStar Inc.).
Anti-tumor activity of combination treatment with a T cell engager in a xenograft model of lung cancer
Nine-week-old NCG mice were injected subcutaneously (s.c.) in the right flank with a 0.2 mL cell suspension containing 1 × 107 H292 cells combined with 1 × 107 human PBMCs. This day was designated as Day 1 of the study and mice were sorted into nine groups of eight mice per group. Tumor growth was monitored three times a week for the duration of the study. Tumors were measured with a caliper in two dimensions. All therapeutic agents were given intravenously (i.v.) every five days (q5d) to the end of the study beginning on Day 5.
Tissue distribution of NM21-1601 in MDA-MB-231 bearing mice
200 μg of NM21-1601 was incubated with 2800 μCi (104 MBq) of 125I-SIB in a coupling reaction mixture for 30 minutes at room temperature. The radiolabeling yield was determined by instant thin-layer chromatography (ITLC) with 10% TCA as eluent and the distribution of radioactivity on the strips was analyzed using gamma counter (Wizard2 2470, Perkin Elmer). The resultant 125ISIB-NM21-1601, was diluted with unlabeled NM21-1601 to reach a specific activity at 1.5 mCi/mg and a concentration of 200 μg/mL.
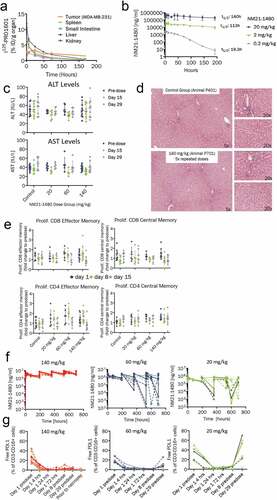
Figure 6. Preclinical pharmacology of NM21-1480 and GLP toxicology study in cynomolgus monkey. (a) Organ distribution of 125I-labeled NM21-1601 at the indicated times after administration to BalbC nude mice bearing a PD-L1-positive tumor (MDA-MB-231). Each marker represents the scintillation count (mean ± SD) per gram of tissue from the indicated organ of 3 animals. (b) Serum concentration (mean ± SD) of NM21-1480 from a single-dose pharmacokinetic study of 3 dose levels in 3 cynomolgus monkeys. C-G, A GLP toxicology study in cynomolgus monkeys was conducted with 5 doses of NM21-1480 administered weekly at doses levels of 0, 20, 60 and 140 mg/kg. (c) Serum transaminase (ALT and ALT) levels before first dose and at days 15 and 29. (d) Representative liver photomicrographs at the end of the study. (e) Memory cell populations from blood samples measured by flow cytometry on the indicated days. Each marker represents an individual animal. Horizontal bars indicate mean ± SD. (f) Plasma levels of NM21-1480 during repeated dosing. Each marker represents an individual animal. (g) Unoccupied PD-L1 binding sites on circulating NK and B cells before the first dose of NM21-1480 and at the indicated times during repeated dosing. Each marker represents an individual animal
Mice were injected subcutaneously with 6 × 106 MDA-MB-231 cells in 100 μL of sterile PBS. At the day of 125ISIB-NM21-1601 injection, the mean tumor volume was 266 ± 46 mm3; and corresponding to a growth period of 3–4 weeks after cell inoculation. Anesthetized mice were injected intravenously with drug at a dose level of 1 mg/kg.
At the given timepoints, blood samples were collected and the radioactivity in blood, and separately plasma, was measured in a Gamma counter. At terminal time points, the organs of interest were harvested, rinsed, and weighed. Tissue radioactivity was measured in an automatic gamma counter (Wallace Wizard 2470 – Perkin Elmer).
Cyno PK study
The PK study was performed with cynomolgus macaques of Asian (Cambodia) origin obtained through Covance Research Products, Inc. To determine the half-life of NM21-1480 in cynomolgus monkeys, blood was drawn at different timepoints from animals following a single intravenous (i.v.) injection of NM21-1480 at three concentrations (low dose = 0.2 mg/kg, mid-dose = 2 mg/kg, high dose = 20 mg/kg). Serum samples were analyzed by an in-house developed pharmacokinetic (PK)-ELISA. Noncompartmental analysis was applied to the individual serum NM21-1480 concentration data using Phoenix WinNonlin (Certara USA, Inc.) to estimate pharmacokinetic parameters.
GLP toxicology study in cynomolgus monkeys
A 4-week repeated-dose toxicology study with a 4-week recovery phase has been performed under GLP conditions in cynomolgus monkeys of African (Mauritius) origin obtained through Covance Research Products, Inc. The animals were dosed weekly for 5 doses by IV infusion at 0, 20, 60, and 140 mg/kg of NM21-1480 (3/sex/group and 5/sex/group for 0 and 140 mg/kg dose groups including recovery animals). For the evaluation of toxicity, standard toxicology parameters such as body weight, food consumption, clinical observations, hematology, and blood chemistry were evaluated over the duration of the study. Anatomical pathology and histopathology assessments were performed at the end of the study.
Immunophenotyping of peripheral blood was performed using a validated method to assess peripheral lymphocyte populations (T cells, B cells and natural killer (NK) cells) as well as the induction of proliferation of central memory (CM) and effector memory (EM) CD4+ and CD8 + T cell subpopulations. For this purpose, blood samples were stained on the indicated study days over the course of the study with antibodies against CD45 (BD Biosciences, 563530), CD3 (BD Biosciences, 557757), CD4 (BD Biosciences, 562842), CD8 (BD Biosciences, 560662), CD16 (BD Biosciences, 562874), CD20 (BD Biosciences, 556632), CD95 (BD Biosciences, 561633), CD28 (Biolegend, 302912) and Ki67 (Miltenyi Biotec, 130–120-417) and analyzed using a FACSCanto II flow cytometer (BD Biosciences, USA) and FACSDiva (BD Biosciences, USA) and GraphPad Prism software.
Serum levels of NM21-1480 were measured using a validated PK-ELISA developed in-house at indicated time points and PK parameters were calculated using noncompartmental analysis as described above for the PK study. PD-L1 receptor occupancy on CD3-CD16+ NK cells and CD3-CD20 + B cells was analyzed at indicated time points using a method developed and validated at Covance Laboratories. The method determined free PD-L1 ligand on the surface on NK cells and B cells in cynomolgus monkey whole blood in the presence of NM21-1480.
Results
An scMATCH™3 molecular scaffold allows engineering of trispecific drug candidates with monovalence for PD-L1, 4-1BB, and HSA
The basis for the molecule development described here is a recombinant fusion protein constructed using scMATCHTM3 technologyCitation37 and consisting of three stabilized and humanized antibody single-chain variable fragments (Fvs) directed against PD-L1, 4–1BB, and HSA. Initial sequences for each domain were derived from rabbit monoclonal antibodies, and complementarity determining regions (CDRs) were grafted onto human variable domain acceptor frameworks. The frameworks were engineered from human consensus and germline sequences for favorable stability and solubility using λ-capTM technology.Citation37 The three Fv domains were assembled into a single protein chain using (Gly4Ser) linkers of varying length as shown in ). Drug candidates employing this structure were able to simultaneously bind all three targets irrespective of the binding sequence (Supplementary Figure S1). We chose monovalent binding for each target to allow affinity-based engineering and calibration of each paratope, as described in the Introduction and in the following sections.
Monovalent binding to the membrane-distal epitope of 4-1BB in combination with ultra-high affinity for PD-L1 allows overlapping maximal efficacy for 4-1BB stimulation and PD-L1 inhibition
On 4–1BB, two agonistic-binding sites have been identified to date that are distinct from the 4–1BBL binding pocket.Citation19 Utomilumab and CTX-471 bind a membrane-proximal epitope corresponding to cysteine-rich domains 3–4 (CRD3-4) of 4–1BB;Citation19,Citation43 this interaction inhibits 4–1BBL binding despite lack of overlap with the 4–1BBL binding site. Urelumab binds a membrane-distal epitope on the N terminus of CRD1 and allows concurrent 4–1BBL binding.Citation19 ) shows a surface representation of 4–1BB binding with its ligand 4–1BBL. In conjunction with this are two engineered human scFv fragments binding to either a membrane-distal or membrane-proximal epitope on 4–1BB (scFv fragments 38–27-A11 and 38–02-A04, respectively). Note that, like urelumab, the membrane-distal scFv fragment does not block the 4–1BBL binding pocket.
As part of the molecular engineering process, we sought to optimize the affinities of PD-L1 and 4–1BB paratopes so the chosen drug candidate would exhibit maximal 4–1BB stimulation in a dose range that also enables maximal inhibition of PD-L1/PD-1 signaling. Using single amino acid substitutions in the 4–1BB and PD-L1 paratopes, we constructed an array of analogues with varying affinities for PD-L1 and 4–1BB for either the membrane-proximal or membrane-distal epitopes of 4–1BB (Supplementary Figure S2) (as verified by surface plasmon resonance; SPR).
Concentration–response curves for each analog were obtained using Jurkat cells containing NFAT or NF-ĸB reporters, for PD-L1 antagonism and 4–1BB stimulation, respectively. Results from 12 such analogues targeting the distal epitope are shown in ). Note that the 4–1BB stimulation curves are bell-shaped in most instances, with peak stimulation spanning 1–2 log units of drug concentration. We believe that 4–1BB stimulation declines at drug concentrations greater than those at which binding to both targets is saturated and therefore preventing bi-specific binding (see Discussion). The top-left graph of ) shows the results for an analog with 4–1BB binding affinity of 0.6 nM and PD-L1 binding affinity of 0.005 nM. For all PD-L1 affinities (rows), declining 4–1BB affinities (columns) produced a lower peak of 4–1BB stimulation. At any given 4–1BB affinity, declining PD-L1 affinity also produced a lower peak of 4–1BB stimulation and separation of the maxima for the two concentration–response curves. The magnitude of maximal PD-L1 antagonism was minimally affected by 4–1BB affinity, as expected. Similar results were observed with analogues targeting the membrane-proximal epitope (Supplementary Figure S3a).
Also evident in these plots is that ultra-high affinity PD-L1 binding is essential to achieve overlapping concentration–activity curves. The overlapping maximal activity of PD-L1 antagonism and 4–1BB agonism―evident in the first row of )―is lost when PD-L1 affinity is reduced to the single-digit nanomolar range, as in the second row.
To monitor the potential of drug analogues to concomitantly modulate both pathways (PD-L1 and 4–1BB), we used contour plots of the percentage of 4–1BB stimulation (NF-ĸB) at 99% of PD-L1 antagonism (NFAT) ()). We also generated contour plots of the intersection between the 4–1BB stimulation curve and the PD-L1 antagonism curve (Supplementary Figure S3b). These plots illustrate that analogues binding to the membrane-distal 4–1BB epitope had much greater flexibility with regard to 4–1BB affinity than analogues binding to the membrane-proximal epitope.
We then compared the best-performing distal or proximal 4–1BB epitope containing scMATCH3 molecules for their efficacy in inducing IL-2 secretion from human PBMCs pre-activated with the superantigen staphylococcal enterotoxin A (SEA), which induces PD-L1 expression on antigen-presenting cells (APCs) and T cells, and 4–1BB expression on T-cells. The membrane-distal analog stimulated IL-2 release significantly more than the membrane-proximal analog ()) despite their similar affinities. Based on these data, subsequent drug development utilized 4–1BB binding domains that bound to the membrane-distal epitope.
On the basis of these results, an optimal balance of binding affinities for the final drug candidate, which is designated NM21-1480, was implemented. NM21-1480 incorporates a monovalent ultra-high affinity PD-L1–binding domain and a monovalent domain that binds to the distal epitope of 4–1BB. Its binding affinity for PD-L1 is approximately 100-fold higher than its affinity for 4–1BB. This asymmetry in affinities is also associated with optimal broadening of the bell-shaped 4–1BB stimulation curve, which should facilitate dose-finding in clinical settings. Also note that affinity-optimized NM21-1480 has PD-L1 binding affinity 10–400-fold higher than other PD-L1-targeting antibodies in clinical use.Citation44 In our experiments, this ultra-high affinity for PD-L1 was essential to achieve both maximal 4–1BB stimulation and overlap of the dose–response curves for 4–1BB stimulation and PD-L1 inhibition. As shown below, our monovalent approach enabled a consistent potency of 4–1BB stimulation that was independent of (non-zero) PD-L1 target density.
Monovalent 4-1BB binding by NM21-1480 evokes 4-1BB stimulation as potently as a bivalent analogue
Bivalent agonist α4-1BB mAbs can efficiently trigger 4–1BB immobilization and clustering, which are requisite precursors for 4–1BB cell signaling.Citation19,Citation45 The 4–1BB clustering induced by urelumab has been attributed in part to its bivalence, as well as to its distal epitope binding and crosslinking with FcγR.Citation19 We quantified the stimulation of 4–1BB signaling by the monovalent NM21-1480 in relation to a bivalent, bispecific 4–1BB agonist mAb. The bivalent mAb (designated PRO1928) employed the same 4–1BB–binding Fvs used in NM21-1480 as well as PD-L1–binding domains, and was constructed using a Morrison-H format.Citation46 As shown in ), the concentration–response curve for NM21-1480 stimulation of 4–1BB signaling is similar to that of the bivalent bi-specific mAb. Thus, the monovalent (for 4–1BB) format of NM21-1480 stimulates 4–1BB signaling as effectively as a bivalent 4–1BB agonist. Similarly, NM21-1480 inhibits PD-L1 with the same potency as the bivalent bi-specific PD-L1 antagonist ()).
4-1BB stimulation by NM21-1480 requires PD-L1 expression but has a potency (EC50) unrelated to PD-L1 target density
NM21-1480 was engineered to be monovalent for PD-L1 binding, with the expectation that this affinity-based design would allow 4–1BB stimulation to have a potency that was relatively insensitive to (non-zero) PD-L1 expression levels (see Introduction). To test this design feature, we characterized NM21-1480-induced stimulation of NF-ĸB signaling and its dependence on PD-L1 density using Jurkat reporter cells co-cultured with various interferon-γ-stimulated cancer cell lines expressing PD-L1 at different densities ()). The maximal stimulation of NF-ĸB signaling, as well as the AUC of the concentration–response curve, were dependent on PD-L1 density in the co-cultured cells. Notably, however, the EC50 for NM21-1480 stimulation of NF-ĸB signaling showed little or no dependence on PD-L1 density ()).
Despite the desire for a consistent EC50 of 4–1BB stimulation regardless of PD-L1 target density, we sought to restrict 4–1BB stimulation to tissues expressing PD-L1, such as the tumor microenvironment. The anti-PD-L1 domain of NM21-1480 is one design feature intended to support that goal. Its effectiveness was tested in Jurkat cells employing an NF-ĸB reporter gene, which were incubated for 24 hours with either PD-L1-positive HCC827 cells or PD-L1-negative CHO-K1 cells in the presence of different concentrations of NM21-1480 or urelumab. As shown in ) (left), urelumab, but not NM21-1480 stimulated NF-ĸB signaling in the presence of PD-L1-negative cells. In contrast, both 4–1BB-stimulating agents (urelumab and NM21-1480) activated NF-ĸB signaling in a concentration-dependent manner in the presence of PD-L1-expressing cells (), right). Related experiments demonstrated that IL-2 secretion from pre-activated human PBMCs was stimulated by NM21-1480 in a dose-dependent manner when the PBMCs were co-incubated with PD-L1-expressing HCC827 cells (), left), but not when the PBMCs were co-incubated with PD-L1-negative CHO cells (), right). No IL-2 secretion was observed in the absence of pre-activation.
Together, these results support the concept that NM21-1480 is not expected to cause systemic activation of T cells, and that its 4–1BB agonist activity should be localized to PD-L1-expressing tissues, such as the tumor microenvironment. Furthermore, as long as PD-L1 is present in the target tissue, the EC50 for 4–1BB stimulation is largely independent of PD-L1 density. As a consequence, it is expected that clinical dosing of NM21-1480 will be relatively consistent and predictable across patients, independent of PD-L1 expression levels in the tumor.
The PD-L1 moiety of NM21-1480 potently inhibits checkpoint signaling
NM21-1480 blockade of PD-L1/PD-1 and PD-L1/B7.1 interactions was assessed by competition ELISA and compared with the bivalent αPD-L1 mAb avelumab. NM21-1480 inhibited PD-L1/PD-1 interactions with an EC50 of 29 pM, comparable to that of avelumab (15 pM; )). Because PD-L1 also interacts with B7.1 to inhibit T-cell proliferation,Citation47 we also studied NM21-1480 blockade of this interaction ()) and found similar EC50 values for NM21-1480 (143 pM) and avelumab (98 pM). The ability of NM21-1480 to inhibit PD-L1–induced PD-1 signaling was studied in cell-based assays using a transgenic NFAT-luciferase reporter Jurkat cell line expressing PD-1. PD-1 signaling was stimulated by co-cultivation of the reporter Jurkat cells with CHO cells expressing PD-L1 and an activator of the T-cell receptor (TCR). Addition of NM21-1480 inhibited NFAT-luciferase expression with an IC50 of 0.44 nM ()), comparable to that of avelumab (0.25 nM). These studies demonstrate the functional activity of NM21-1480 as a monovalent immune checkpoint inhibitor, with potency and efficacy comparable to the potency and efficacy of the bivalent αPD-L1 mAb avelumab.
NM21-1480 has high efficacy for co-activation of T cells and dendritic cells and for preserving immune cell viability
In human PBMCs pre-stimulated with SEA, incubation with NM21-1480 induced a robust increase in IL-2 release as measured by ELISA ()). Maximal IL-2 release was more than twice that induced by any combination of αPD-L1/αPD-1 mAb (avelumab, nivolumab, or pembrolizumab) with an agonist α4-1BB mAb (urelumab or utomilumab) (p < .001). In the absence of SEA pre-stimulation, none of the treatments elicited measurable IL-2 secretion. The lack of cytokine release from quiescent PBMCs was further tested in a series of experiments using PBMCs from 6 healthy human donors. PBMCs were treated with either positive controls (muromonab-CD3 [OKT3], phytohemagglutinin [PHA], Rituxan), negative controls (infliximab) or one of three concentrations of NM21-1480 (1, 10, 100 μg/ml). No significant increase in cytokine (TNF-α, IFN-γ, IL-10, IL-6, or IL-2) release was observed for any concentration of NM21-1480 ()) In the same set of experiments shown in , we analyzed the effects of NM21-1480 on the viability of SEA pre-stimulated CD4+ and CD8+ T cells, antigen-presenting cells (CD11c+/CD123+), and monocytes (CD14+/CD16+). All cell classes retained viability 96 hours after treatment with NM21-1480, whereas avelumab treatment was associated with a reduction in the viability of antigen-presenting cells (APCs; )).
Dendritic cells (DCs) express PD-L1 and upregulate its expression after antigen uptake.Citation48 Recent studies have shown that PD-L1-expressing DCs play a uniquely important role in limiting T-cell responses in cancer and dampen the tumor response to immune checkpoint inhibitors.Citation49–51 Therefore, we studied the effects of NM21-1480 and the combination of avelumab + urelumab in co-cultures of T cells and monocyte-derived DCs. NM21-1480 increased markers of T-cell activation (TNFα, IL2, and IFNγ) and markers of DC activation (IL12p40, CD86, HLA-DR) significantly more than avelumab + urelumab ()). These results suggest that NM21-1480―by activating both T cells and DCs―may be uniquely suited to stimulating anti-tumor immune responses in the tumor microenvironment.
Anti-tumor activity of NM21-1480 analogues in xenograft models
Antitumor effects of NM21-1480 in xenograft-bearing mice employed closely related analogues of NM21-1480, in part to incorporate an anti-mouse serum albumin domain in place of the anti-HSA domain of NM21-1480. These analogues are described in Supplementary Table 1. The analogues were studied in severely immunodeficient (NOG) mice engrafted with human PBMCs. Human, PD-L1–positive non-small cell lung cancer (NSCLC) cell lines were used for xenografts. In an HCC-827 NSCLC xenograft model, mice with established tumors were treated with NM21-1186 and had significantly smaller increases in relative tumor volume and significantly greater tumor infiltration of human T cells versus control or comparator monotherapies (–c)). A combination therapy that included an αPD-L1 IgG and an agonist α4-1BB IgG yielded comparable results in terms of tumor volume, but exhibited significantly less tumor infiltration of T cells () and (c)). In another NSCLC xenograft model (H292 cells in NOG mice), NM21-1601 reduced the rate of tumor growth ()). Combined therapy using NM21-1601 with a bispecific T-cell engager targeting a tumor-associated antigen and CD3, eradicated tumors in all animals (n = 10). Taken together, studies in xenograft models indicate that NM21-1480 exerts potent anti-tumor activity and synergistic anti-tumor activity with bispecific T cell engagers.
Preclinical pharmacology and safety of NM21-1480
The biodistribution of 125I-labeled NM21-1601 was studied after intravenous administration to BalbC nude mice bearing a PD-L1-positive human breast cancer cell line xenograft (MDA-MB-231). Radioactivity in spleen, small intestine, liver, and kidney were highest at the first timepoint after administration (1 hour) and declined without marked evidence of tissue retention. In contrast, radioactivity in the tumor tissue peaked 24 hours after administration and exhibited evidence of tumor retention ()), suggesting successful tumor localization of NM21-1480.
Characteristics of NM21-1480 binding to human and cynomolgus monkey 4–1BB, PD-L1, and serum albumin are shown in Supplemental Table 2. The binding characteristics in humans and cynomolgus monkey were nearly identical, supporting the use of cynomolgus monkey to estimate human pharmacokinetics. Results of a single-dose pharmacokinetic study at three dose levels (0.2, 2, and 20 mg/kg) in cynomolgus monkey are shown in ). At the lowest dose level (0.2 mg/kg), the half-life of NM21-1480 was <20 hours, but at higher doses (≥20 mg/kg) the half-life was approximately 4.5 days. Shorter half-life at lower doses has been observed with other αPD-L1 mAbs,Citation52,Citation53 and is likely due to target-mediated drug disposition. On the basis of these results, we estimate a serum half-life in humans of approximately 2 weeks, potentially suitable for dosing every 3–4 weeks.
NM21-1480 stimulation of IL-2 release from SEA pre-stimulated PBMCs showed nearly identical concentration-response characteristics in cynomolgus monkey (EC50, 176 ± 87 pM) and humans (EC50, 339 ± 147 pM; Supplementary Figure S4), supporting cynomolgus monkey as an in vivo model of human pharmacodynamics and toxicity.
In a GLP toxicology study, three dosage cohorts [20 mg/kg (n = 6), 60 mg/kg (n = 6), and 140 mg/kg (n = 10)] and a control cohort (n = 10) of cynomolgus monkeys received five doses of NM21-1480 at weekly intervals. No changes in serum levels of liver transaminases were observed during the study ()), and no treatment-related changes were observed in organ weights or macroscopic characteristics. Furthermore, histopathologic examination of liver tissue sections, performed immediately after the last dose and at 4 weeks after the last dose, found no evidence of liver inflammation in any of the dose groups ()). No changes in levels of effector or memory T cells were observed at days 8 or 15 ()). Furthermore, no significant changes were observed in populations of cytotoxic T cells, helper T cells, activated cytotoxic T cells (CD8+/CD25+ and CD8+/CD69+), activated helper T cells (CD4+/CD5+ and CD4+/Cd69+), B cells, and NK cells compared with pre-treatment levels. No increases in serum levels of GM-CSF, IL-18, IL-1B, IFNγ, IL-2, IL-4, or IL-5 were observed following the administration of NM21-1480. IL-6, MCP-1, and IL-10 levels were increased in some animals in the 20 and 60 mg/kg cohorts, consistent with the emergence of anti-drug antibodies. Thus, we found no evidence of widespread systemic immune cell activation or liver toxicity during treatment with NM21-1480. No changes were observed in clinical chemistry, hematology, urinalysis, or cardiac parameters (PR interval, QRS duration, QT interval, QTc interval, heart rate, or systolic, diastolic or mean arterial blood pressure). In conclusion, weekly IV infusion of NM21-1480 at a dose level of 140 mg/kg was well tolerated with no signs of toxicity in cynomolgus monkeys, and was therefore considered to be the no-observed-adverse-effect-level. Plasma levels of NM21-1480 were proportional to dose during the study ()), except in animals that developed anti-drug antibodies. Analysis of blood samples collected at different time points during the study revealed that circulating levels of NM21-1480 saturated PD-L1 binding sites on circulating NK cells and B cells ()). High levels of PD-L1 occupancy were observed within 4 hours of the first dose and were maintained during the inter-dose interval.
Discussion
The goal of this drug development program was to engineer an immuno-oncology drug candidate that 1) effectively stimulates 4–1BB signaling in antigen-experienced T-cells, 2) inhibits the PD-L1/PD-1 checkpoint pathway, 3) effects these actions across a range of clinically useful concentrations and in a manner restricted to the tumor microenvironment, and 4) has an advantageous serum half-life. We used rational molecular design strategies coupled with empirical testing to refine and confirm our assumptions, and these efforts led to the development and preclinical validation of NM21-1480 as a tri-specific, anti-tumor drug candidate incorporating 4–1BB co-stimulation and PD-L1 blockade.
NM21-1480 has a number of molecular features supporting its function. First, its 4–1BB binding domain interacts with the membrane-distal epitope of 4–1BB, yielding better pathway stimulation than the membrane-proximal epitope, possibly by being more permissive of 4–1BB clustering and cross-linking.Citation19 Second, NM21-1480 incorporates a number of features important for reducing off-target stimulation of 4–1BB-expressing cells: it is monovalent for 4–1BB, it does not possess an Fc-domain, and it is engineered to stimulate 4–1BB only during concomitant binding of PD-L1. The lack of liver toxicity in cynomolgus monkey supports the success of these design strategies. Third, a systematic molecular engineering strategy was used to identify and deploy an optimal balance of binding affinities for the monovalent PD-L1– and 4–1BB–binding domains, providing for maximal and overlapping 4–1BB stimulation and PD-L1 inhibition across a broad dose range. Indeed, in the presence of PD-L1-expressing cells, NM21-1480 stimulated the 4–1BB pathway in antigen-experienced T cells as potently as bivalent agonist α4-1BB mAbs. A notable feature of 4–1BB stimulation by NM21-1480 is its bell-shaped dose–response curve. We believe this characteristic to be a consequence of saturation of both PD-L1 and 4–1BB binding sites at high drug concentrations, whereby drug molecules bound to 4–1BB encounter no available PD-L1 binding sites to function as crosslinking anchors, resulting in ineffective stimulation of 4–1BB signaling.
Another key molecular feature of NM21-1480 is its ultra-high affinity, monovalent binding to PD-L1. The results confirmed that this design achieved its goals. First, NM21-1480 blocks PD-L1/PD-1 signaling with high potency and efficacy. Second, the αPD-L1 domain localizes NM21-1480 to tissues expressing PD-L1 (eg, the tumor microenvironment), further reducing opportunities for off-target 4–1BB signaling. Third, the αPD-L1 domain serves as an anchor for the agonist α4-1BB domain, mimicking the membrane anchor of endogenous TNFSF ligands; such anchoring is known to improve the effectiveness of 4–1BB clustering and signaling.Citation20,Citation26,Citation45 This combination of desirable features has not been replicated in studies employing two mAbs to simultaneously target PD-L1 and 4–1BB.
Monovalency is a key feature of the αPD-L1 domain of NM21-1480. Studies characterizing the binding properties of monovalent versus bivalent mAbs have shown that the affinity-based binding of monovalent mAbs is predictable and independent of target density. In contrast, the avidity-based binding of bivalent mAbs leads to binding that is dependent on target density, and therefore highly variable.Citation32,Citation35,Citation36 Because one of our design goals was to have consistent and predictable dosing that was independent of (non-zero) PD-L1 density in the tumor, we chose to deploy monovalent PD-L1 binding. The results shown in & b) confirmed that the EC50 for 4–1BB stimulation was largely independent of PD-L1 target density, suggesting that clinical dosing of NM21-1480 should be independent of tumor PD-L1 density. However, the requirement for at least some tumor expression of PD-L1 was preserved (as shown in )), and this feature may partly account for the lack of NM21-1480 liver toxicity in cynomolgus monkey. Employing a monovalent αPD-L1 domain to 1) achieve highly effective checkpoint inhibition and 2) achieve overlapping dose–response curves for the αPD-L1 and α4-1BB domains rested on the ultra-high affinity of the αPD-L1 domain. Specifically, in order to inhibit PD-L1/PD-1 signaling at doses that also maximally stimulated 4–1BB signaling across a broad dose range required an ultra-high affinity αPD-L1 domain ()). In theory, this dose-response overlap could be achieved using bivalent αPD-L1 domains, but the resulting avidity-based binding would lead to 4–1BB stimulation that was dependent on PD-L1 target density, making clinical dosing challenging and inconsistent.
In contrast to standard IgGs, NM21-1480 contains no Fcγ region. One rationale for that design choice was to reduce the potential for on-target but off-tumor toxicity. Recent studies have provided evidence that the Fcγ portion of urelumab, by interacting with FcγR in resident liver cells, performed the crosslinking function essential for stimulation of 4–1BB signaling, contributing to urelumab liver toxicity. Fcγ-FcγR interaction may also activate resident or infiltrating immune cells, such as NK cells. Several groups have developed 4–1BB agonists in which the crosslinking function is performed by an alternative binding domain such as αEGFR, αHER2, αFAP, or PD-L1.Citation29–31,Citation54–58 These agonists have exhibited greatly reduced or absent liver toxicity. Our results showing lack of liver toxicity of NM21-1480 are consistent with those findings. A second rationale for eliminating the Fcγ domain was to replace it with an αHSA domain to confer a prolonged serum half-life. The estimated 2-week serum half-life in humans (based on cynomolgus monkey results), suggests that was successful.
The choice of targeting PD-L1 to combine with 4–1BB agonism implies that NM21-1480 is likely to be targeted to tumors expressing PD-L1 themselves or in their microenvironment. Thus, we anticipate that PD-L1 expression is likely to be a predictive biomarker of response to NM21-1480. However, with the ultra-high affinity of NM21-1480, it is possible that very low levels of PD-L1 expression may be sufficient for tumor targeting. It is not yet clear whether current PD-L1 assays are sensitive enough to detect such low levels. And it is not clear whether PD-L1 expression is required in the tumor itself, in the tumor microenvironment, or both. One theoretical possibility is the potential for NM21-1480 to trigger a positive feedback loop wherein initial 4–1BB stimulation triggers cytokine release that in turn increases PD-L1 expression, potentially providing the substrate for increased binding of NM21-1480. For example, studies using mixed lymphocyte cultures showed that NM21-1480 stimulated the release of IFN-γ ()), which is known to stimulate PD-L1 expression. Studies of PD-L1 expression in the tumor microenvironment will be needed to resolve these questions. Studies of other 4–1BB-stimulating molecules suggest that other biomarkers may be useful predictors of activity or tumor response.Citation31Citation59,Citation60
One feature of NM21-1480 that could present clinical challenges is its bell-shaped dose–response curve for 4–1BB stimulation. Ultimately, clinical studies will be needed to evaluate the magnitude of that problem. However, the success of NM21-1480 across a broad dose range in xenograft models suggests that the problem can be addressed. Because both of the main effector functions of NM21-1480 (PD-L1/PD-1 blockade and 4–1BB stimulation) are expected to increase the activity of tumor-exposed T cells, it may be difficult to know which effector function is responsible for tumor responses. This issue is likely to be shared by many multi-targeted agents; ultimately, efficacy will rest on clinical outcomes. Initial clinical studies of NM21-1480 are enrolling patients with advanced solid tumors who failed prior therapies, including those targeting the PD-L1/PD-1 pathway. That trial cohort is consistent with the initial design goal of “rescuing” anti-tumor T-cell activity.
Supplemental Material
Download ()Acknowledgments
Scientific writing support was provided by Ken Scholz, PhD. We thank the protein crystallization center (PCC, University of Zurich) for assistance in crystallization and Expose GmbH for assistance in data collection and structure solution.
Disclosure statement
All authors are employees of Numab Therapeutics AG. Research and manuscript preparation supported by Numab Therapeutics AG.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):1–17. doi:10.1038/s12276-018-0191-1.
- Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021;14(1):10. doi:10.1186/s13045-020-01027-5.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349.
- Abe BT, Macian F. Uncovering the mechanisms that regulate tumor-induced T-cell anergy. Oncoimmunology. 2013;2(2):e22679. doi:10.4161/onci.22679.
- Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood. 2018;131(1):49–57. doi:10.1182/blood-2017-06-741041.
- Vinay DS, Kwon BS. Immunotherapy of cancer with 4-1BB. Mol Cancer Ther. 2012;11(5):1062–1070. doi:10.1158/1535-7163.Mct-11-0677.
- Bremer E. Targeting of the tumor necrosis factor receptor superfamily for cancer immunotherapy. ISRN Oncol. 2013;2013:371854. doi:10.1155/2013/371854.
- Choi BK, Lee SC, Lee MJ, Kim YH, Kim YW, Ryu KW, Lee JH, Shin SM, Lee SH, Suzuki S, et al. 4-1BB-based isolation and expansion of CD8+ T cells specific for self-tumor and non-self-tumor antigens for adoptive T-cell therapy. J Immunother. 2014;37(4):225–236. doi:10.1097/cji.0000000000000027.
- Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, Greenberg PD. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110(1):201–210. doi:10.1182/blood-2006-11-056168.
- Wilcox RA, Tamada K, Flies DB, Zhu G, Chapoval AI, Blazar BR, Kast WM, Chen L. Ligation of CD137 receptor prevents and reverses established energy of CD8+ cytolytic T lymphocytes in vivo. Blood. 2004;103(1):177–184. doi:10.1182/blood-2003-06-2184.
- Weigelin B, Bolaños E, Teijeira A, Martinez-Forero I, Labiano S, Azpilikueta A, Morales-Kastresana A, Quetglas JI, Wagena E, Sánchez-Paulete AR, et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc Natl Acad Sci U S A. 2015;112(24):7551–7556. doi:10.1073/pnas.1506357112.
- Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, Gajewski TF. The EGR2 targets LAG-3 and 4-1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J Exp Med. 2017;214(2):381–400. doi:10.1084/jem.20160485.
- Chen S, Lee L-F, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3(2):149–160. doi:10.1158/2326-6066.Cir-14-0118.
- Dai M, Yip YY, Hellstrom I, Hellstrom KE. Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies. Clin Cancer Res. 2015;21(5):1127–1138. doi:10.1158/1078-0432.Ccr-14-1339.
- Wei H, Zhao L, Hellstrom I, Hellstrom KE, Guo Y. Dual targeting of CD137 co-stimulatory and PD-1 co-inhibitory molecules for ovarian cancer immunotherapy. Oncoimmunology. 2014;3(4):e28248. doi:10.4161/onci.28248.
- Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, Schmidt H, Robert C, Chiarion-Sileni V, Ascierto PA, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res. 2017;23(8):1929–1936. doi:10.1158/1078-0432.Ccr-16-1272.
- Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, Cesari R, Chen Y, Davis CB, Huang B, et al. Phase I study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin Cancer Res. 2018;24(8):1816–1823. doi:10.1158/1078-0432.CCR-17-1922.
- Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, Di Gravio D, Huang B, Gambhire D, Chen Y, et al. Phase Ib study of utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin Cancer Res. 2017;23(18):5349–5357. doi:10.1158/1078-0432.Ccr-17-1243.
- Chin SM, Kimberlin CR, Roe-Zurz Z, Zhang P, Xu A, Liao-Chan S, Sen D, Nager AR, Oakdale NS, Brown C, et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun. 2018;9(1):4679. doi:10.1038/s41467-018-07136-7.
- Vanamee ÉS, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signal. 2018;11(511):511. doi:10.1126/scisignal.aao4910.
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17(3):197–223. doi:10.1038/nrd.2017.227.
- Rabu C, Quéméner A, Jacques Y, Echasserieau K, Vusio P, Lang F. Production of recombinant human trimeric CD137L (4-1BBL). Cross-linking is essential to its T cell co-stimulation activity. J Biol Chem. 2005;280(50):41472–41481. doi:10.1074/jbc.M506881200.
- Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, and Brogdon JL. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210(9):1685–1693. doi:10.1084/jem.20130573.
- Fellermeier S, Beha N, Meyer JE, Ring S, Bader S, Kontermann RE, Müller D. Advancing targeted co-stimulation with antibody-fusion proteins by introducing TNF superfamily members in a single-chain format. Oncoimmunology. 2016;5(11):e1238540. doi:10.1080/2162402x.2016.1238540.
- Bartkowiak T, Jaiswal AR, Ager CR, Chin R, Chen CH, Budhani P, Ai M, Reilley MJ, Sebastian MM, Hong DS, et al. Activation of 4-1BB on liver myeloid cells triggers hepatitis via an interleukin-27-dependent pathway. Clin Cancer Res. 2018;24(5):1138–1151. doi:10.1158/1078-0432.Ccr-17-1847.
- Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, Yang X. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nat Commun. 2019;10(1):2141. doi:10.1038/s41467-019-10088-1.
- Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, Albrecht R, Herter S, Schlenker R, Hüsser T, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. 2019;11(496):496. doi:10.1126/scitranslmed.aav5989.
- Junttila TT, Li J, Johnston J, Hristopoulos M, Clark R, Ellerman D, Wang B-E, Li Y, Mathieu M, Li G, et al. Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res. 2014;74(19):5561–5571. doi:10.1158/0008-5472.Can-13-3622-t.
- Hinner MJ, Aiba RSB, Jaquin TJ, Berger S, Dürr MC, Schlosser C, Allersdorfer A, Wiedenmann A, Matschiner G, Schüler J, et al. Tumor-localized costimulatory T-cell engagement by the 4-1BB/HER2 bispecific antibody-anticalin fusion PRS-343. Clin Cancer Res. 2019;25(19):5878–5889. doi:10.1158/1078-0432.Ccr-18-3654.
- Compte M, Harwood SL, Muñoz IG, Navarro R, Zonca M, Perez-Chacon G, Erce-Llamazares A, Merino N, Tapia-Galisteo A, Cuesta AM, et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun. 2018;9(1):4809. doi:10.1038/s41467-018-07195-w.
- Piha-Paul S, Bendell J, Tolcher A, Hurvitz S, Patnaik A, Shroff R, Pohlmann P, Zettl M, Hahn N, Krishnamurthy A, et al. Phase I dose escalation study of PRS-343, a HER2/4-1BB bispecific molecule, in patients with HER2-positive malignancies. J Immunother Cancer.2020;8(Suppl 1) doi:10.1136/LBA2019.2.
- Slaga D, Ellerman D, Lombana TN, Vij R, Li J, Hristopoulos M, Clark R, Johnston J, Shelton A, Mai E, et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci Transl Med. 2018;10:463. doi:10.1126/scitranslmed.aat5775.
- Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, Brinkmann U, Kettenberger H, Regula JT, and Schaefer W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. MAbs. 2012;4(6):653–663. doi:10.4161/mabs.21379.
- Pacchiana G, Chiriaco C, Stella MC, Petronzelli F, De Santis R, Galluzzo M, Carminati P, Comoglio PM, Michieli P, Vigna E, et al. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J Biol Chem. 2010;285(46):36149–36157. doi:10.1074/jbc.M110.134031.
- Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs. 2015;7(2):377–389. doi:10.1080/19420862.2015.1007816.
- Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, Wu H, Dall’Acqua WF. Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs. 2015;7(3):461–469. doi:10.1080/19420862.2015.1022695.
- Egan TJ, Diem D, Weldon R, Neumann T, Meyer S, Urech DM. Novel multispecific heterodimeric antibody format allowing modular assembly of variable domain fragments. MAbs. 2017;9(1):68–84. doi:10.1080/19420862.2016.1248012.
- Novarra S, Grinberg L, Rickert KW, Barnes A, Wilson S, Baca M. A hingeless Fc fusion system for site-specific cleavage by IdeS. MAbs. 2016;8(6):1118–1125. doi:10.1080/19420862.2016.1186321.
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt2):125–132. doi:10.1107/s0907444909047337.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi:10.1107/s0021889807021206.
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi:10.1107/s0907444996012255.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi:10.1107/s0907444910007493.
- Eskiocak U, Guzman W, Wolf B, Cummings C, Milling L, Hj W, Ophir M, Lambden C, Bakhru P, Gilmore DC, et al. Differentiated agonistic antibody targeting CD137 eradicates large tumors without hepatotoxicity. JCI Insight. 2020;5:5. doi:10.1172/jci.insight.133647.
- Tan S, Liu K, Chai Y, Zhang CW, Gao S, Gao GF, Qi J. Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell. 2018;9(1):135–139. doi:10.1007/s13238-017-0412-8.
- Wyzgol A, Müller N, Fick A, Munkel S, Grigoleit GU, Pfizenmaier K, Wajant H. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol. 2009;183(3):1851–1861. doi:10.4049/jimmunol.0802597.
- Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol. 1997;15(2):159–163. doi:10.1038/nbt0297-159.
- Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. doi:10.1016/j.immuni.2007.05.016.
- Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45–56. doi:10.1038/nri3790.
- Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, Jost C, Fransen MF, Buser RB, Kowanetz M, et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med. 2020;12(534):534. doi:10.1126/scitranslmed.aav7431.
- Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nature Cancer. 2020;1(7):681–691. doi:10.1038/s43018-020-0075-x.
- Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, Guo J, Peng H, Chen M, Fu Y-X, et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun. 2020;11(1):4835. doi:10.1038/s41467-020-18570-x.
- Heery CR, O’Sullivan-Coyne G, Madan RA, Cordes L, Rajan A, Rauckhorst M, Lamping E, Oyelakin I, Marté JL, Lepone LM, et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): a phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 2017;18(5):587–598. doi:10.1016/s1470-2045(17)30239-5.
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi:10.1038/nature14011.
- Lakins MA, Koers A, Giambalvo R, Munoz-Olaya J, Hughes R, Goodman E, Marshall S, Wollerton F, Batey S, Gliddon D, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res. 2020;26(15):4154–4167. doi:10.1158/1078-0432.Ccr-19-2958.
- Garralda E, Geva R, Ben-Ami E, Maurice-Dror C, Calvo E, LoRusso P, Türeci Ö, Niewood M, Şahin U, Jure-Kunkel M, et al. First-in-human phase I/IIA trial to evaluate the safety and initial clinical activity of duobody®-PD-L1×4–1BB (GEN1046) in patients with advanced solid tumors. J Immunother Cancer. 2020;8(Suppl 3):A437–A437. SITC Abstract 412. doi:10.1136/jitc-2020-SITC2020.0412.
- Bol K, Marissen J, Elassiass-Schaap J, Tacken P, Engels S, Wang L-C, Mondal A, Throsby M, Roberts A, Mayes P, et al. MCLA −145 (CD137xPD 145 (CD137xPD -L1): a potent CD137 agonist and immune checkpoint inhibitor that does not show signs of peripheral toxicity in preclinical models. J Immunother Cancer. 2020. SITC 2020 Poster P814. doi:10.1136/jitc-2020-SITC2020.0814.
- Zhai T, Wang C, Xu Y, Huang W, Yuan Z, Wang T, Dai S, Peng S, Pang T, Jiang W, et al. Generation of a safe and efficacious llama single-domain antibody fragment (vHH) targeting the membrane-proximal region of 4-1BB for engineering therapeutic bispecific antibodies for cancer. J Immunother Cancer. 2021;9(6):6. doi:10.1136/jitc-2020-002131.
- Jeong S, Park E, Kim HD, Sung E, Kim H, Jeon J, Kim Y, Jung U-J, Son Y-G, Hong Y. Novel anti-4-1BB×PD-L1 bispecific antibody augments anti-tumor immunity through tumor-directed T-cell activation and checkpoint blockade. J Immunother Cancer. 2021;9:7. doi:10.1136/jitc-2021-002428.
- Garralda E, Geva R, Ben-Ami E, Maurice-Dror C, Calvo E, LoRusso P, Tureci O, Niewood M, Sahin U, Jure-Kinkel M, et al. First-in-human phase I/IIA trial to evaluate the safety and initial clinical activity of duobody-PD-L1x4-1BB (GEN1046) in patients with advanced solid tumors. J Immunother Cancer. 2020;8(Suppl 3):Abstract 412. doi:10.1136/jitc-2020-SITC2020.0412.
- Bol K, Marissen W, Elassiass-Schaap J, Tacken P, Engels S, Wang L-C, Mondal A, Throsby M, Roberts A, Mayes P, et al. MCLA-145 (CD137xPD-L1): a potent CD137 agonist and immune checkpoint inhibitor that that does not show signs of peripheral toxicity. J Immunother Cancer. 2020;8(Suppl 3):Abstract 814. doi:10.1136/jitc-2020-SITC2020.0814.