
ABSTRACT
Bladder cancer (BC) and melanoma are amenable to immune checkpoint blockade (ICB) therapy, yet most patients with advanced/metastatic disease do not respond. CD122-targeted interleukin (IL)-2 can improve ICB efficacy, but mechanisms are unclear. We tested αPD-L1 and CD122-directed immunotherapy with IL-2/αIL-2 complexes (IL-2c) in primary and metastatic bladder and melanoma tumors. IL-2c treatment of orthotopic MB49 and MBT-2 BC generated NK cell antitumor immunity through enhanced activation, reduced exhaustion, and promotion of a mature, effector NK cell phenotype. By comparison, subcutaneous B16-F10 melanoma, which is IL-2c sensitive, requires CD8+ T and not NK cells, yet we found αPD-L1 efficacy requires both CD8+ T and NK cells. We then explored αPD-L1 and IL-2c mechanisms at distinct metastatic sites and found intraperitoneal B16-F10 metastases were sensitive to αPD-L1 and IL-2c, with IL-2c but not αPD-L1, increasing CD122+ mature NK cell function, confirming conserved IL-2c effects in distinct cancer types and anatomic compartments. αPD-L1 failed to control tumor growth and prolong survival in B16-F10 lung metastases, yet IL-2c treated B16-F10 lung metastases effectively even in T cell and adaptive immunity deficient mice, which was abrogated by NK cell depletion in wild-type mice. Flow cytometric analyses of NK cells in B16-F10 lung metastases suggest that IL-2c directly boosts NK cell activation and effector function. Thus, αPD-L1 and IL-2c mediate nonredundant, immune microenvironment-specific treatment mechanisms involving CD8+ T and NK cells in primary and metastatic BC and melanoma. Mechanistic differences suggest effective treatment combinations including in other tumors or sites, warranting further studies.
Background
Bladder cancer (BC) and melanoma are among the most common cancers in the United States, collectively accounting for an estimated 190,000 new cancer diagnoses and 25,000 deaths annually.Citation1 Nearly all cancer-related mortality is from metastasesCitation2–4 and 5-year survival rates for patients with metastatic BC and melanoma are poor at 6% and 30%, respectively.Citation5 BC and melanoma share common metastatic sites, including regional lymph nodes, bones, and lung.Citation6,Citation7 Melanoma is also one of the most common extra-abdominal malignancies to develop intraperitoneal metastasis, which is uniformly associated with a poor prognosis.Citation8
Immune checkpoint blockade (ICB) antibodies targeting programmed death receptor 1 (PD-1) or one of its ligands (programmed death ligand 1, PD-L1), and cytotoxic T-lymphocyte-associate protein 4 (CTLA-4) have provided durable clinical efficacy in patients with advanced or metastatic melanoma, yet more than 50% do not respond to ICB.Citation9–12 BC, like melanoma, is thought to be among the most ICB-responsive cancers due to high tumor mutational burden, yet only 10%–30% of BC patients respond to ICB.Citation13
High dose interleukin-2 (IL-2) became a United States Food and Drug Administration-approved metastatic melanoma immunotherapy in 1998, yet is highly toxic with low overall efficacy, in part because of its promotion of regulatory T cells (Tregs).Citation14,Citation15 IL-2 signals by binding to the IL-2 receptor (IL-2R), which exists as either a trimer of IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2 Rγ (γc, CD132) subunits or as a dimeric βγ IL-2 R.Citation16,Citation17 Activated T cells and CD4+ Foxp3+ Tregs express trimeric receptors,Citation18 whereas memory-phenotype CD8+ CD44+ T cells and NK cells highly express dimeric βγ IL-2 Rs and can mediate anti-tumor immunity.Citation17 Selective dimeric βγ IL-2R stimulation with CD122-directed IL-2/αIL-2 complexes (IL-2c) boosts antitumor immunity in murine cancer models by augmenting CD122+ effector T cell functions while avoiding the deleterious Treg-promoting effects of IL-2 monotherapy.Citation19
However, specific effector populations activated by IL-2c are highly dependent on tumor type and the tumor immune microenvironment, which also differentially affects αPD-L1 efficacy despite potentially targeting similar immune populations.Citation20 Here, we studied IL-2c and αPD-L1 immunotherapy treatment mechanisms in orthotopic mouse models of BC (MB49 and MBT-2) and melanoma (B16-F10) versus mechanisms in peritoneal and lung metastatic sites. We show that IL-2c promotes NK cell maturation and effector function in distinct primary (orthotopic bladder) and metastatic (peritoneum and lung) sites in a T cell-independent manner. NK cells were indispensable for efficacious treatment of lung metastatic BC and melanoma. Notably, IL-2c effects on CD8+ T cells are complementary with αPD-L1 in metastatic melanoma models and treatment effects show tumor and anatomic compartment-dependence. These data shed light on conserved mechanisms of selective IL-2 receptor targeting on NK cell functions and help define treatment response mechanisms, in addition to potentially responsive patient populations, as CD122-directed immunotherapy and ICB combinations are currently in human trials.Citation21
Methods
Mice
Wild-type C57BL/6J (BL6) and C3H/HeJ (C3H) mice and B6.129P2-Tcrdtm1Mom/J (TCRδKO), B6.129P2-Tcrbtm1Mom/J (TCRβKO), and B6.129S7-Rag1tm1Mom/J (RAG1KO) genetic knock out mice all on the BL6 background were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were bred in our animal facility, given ad libitum water and food, and housed under specific pathogen-free conditions. All mice were at least 8 weeks old and age- and sex-matched when used for each experiment.
Tumor cell lines and cell culture
Mouse MB49 and MBT-2 BC cell lines were engineered to express luciferase as we described.Citation19 Mouse B16-F10 (herein B16) was purchased from ATCC (Manassas, VA). We developed Luciferase-expressing B16 using CMV-Firefly luciferase-IRES-Puro lentivirus from Cellomics Technology (Halethrope, MD), following the manufacturer’s protocol. All cell lines are PD-L1+Citation22–24 and were passaged <5 times prior to tumor challenge. Cells were maintained in 5% fetal bovine serum (FBS)-containing DMEM (Dulbecco’s Modified Eagle Medium, MB49, and B16) or RPMI-1640 (Roswell Park Memorial Institute, MBT-2), supplemented with 1/100 dilutions of penicillin/streptomycin, L-glutamate, and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES).
In vivo tumor challenges
Orthotopic (intravesical) BC challenge with MB49 and MBT-2 used 8 × 104 or 1 × 106 cells, respectively, in 50 μL Dulbecco’s phosphate buffered saline (DPBS, Sigma Aldrich), via indwelling urinary catheter in female mice under isoflurane anesthesia, as described.Citation25 Orthotopic (subcutaneous) B16 challenge used 5 × 105 cells in 200 μL DPBS into each flank of male mice (2 tumors/mouse). Subcutaneous B16 tumor growth was measured every 2 days by calipers and volume was calculated as (length x width2)/2. Intraperitoneal B16 challenge used 4 × 105 cells injected into the peritoneum. Lung BC tumors were generated via intravenous tail vein injection of 7 × 105 MB49 or 2.5 × 105 MBT-2 cells in 200 μL DPBS into male mice.Citation20 Lung B16 tumors were generated via similar methods using 3 × 105 B16 cells. Bladder tumor and peritoneal B16 tumor growth was assessed by tumor weight at sacrifice. Lung B16 tumor growth was measured in vivo by tumor bioluminescence every 4 days, starting on day 7 post challenge. Survival was defined as spontaneous death, moribundity, tumor volume ≥2000 mm3 (subcutaneous B16), or significant weight loss from baseline (≥15% for bladder and ≥20% for lung tumors). To ensure uniform tumor size across all treatment groups, mice were occasionally excluded upon first tumor measurement before randomization, if determined to be an outlier by Grubbs’ test.
In vivo bioluminescence monitoring
Bioluminescent imaging (IVIS Lumina Imaging System; Perkin Elmer; Waltham, MA) was used for treatment randomization and in vivo tumor monitoring as described.Citation19 In brief, mice were imaged 15 minutes after intraperitoneal injection of 3 mg PBS-dissolved d-luciferin K+ (Gold Biotechnology; St. Louis, MO) with one-minute exposure, small binning, and F/stop = 1. Identical regions of interest were drawn over each mouse and average radiance (photons/sec/cm2/sr) was quantified using Living Image software version 4.2.
In vivo treatments
αPD-L1 (clone 10 F.9G2), αIL-2 (clone JES6-5H4), and isotype control antibodies (clone LTF-2 rat IgG2b and polyclonal Armenian hamster IgG) were purchased from BioXCell (Lebanon, NH). Carrier-free recombinant mouse IL-2 was purchased from Biolegend (San Diego, CA). IL-2c is 1.5 µg/mouse IL-2 complexed with 7.5 µg/mouse αIL-2 at a 1:2 molar ratio defined as optimal in PBS at 37°C for 15–30 min,Citation26 before intraperitoneal administration in 100 μL DPBS. IL-2c treatment was every other day for four doses. Treatment began on day 6 or 7 post challenge for orthotopic BC, subcutaneous melanoma, or peritoneal melanoma challenge, and day 7 or 8 for intravenous lung tumor challenge, unless noted otherwise. 100 μg/mouse αPD-L1 was given intraperitoneally every five days for three doses starting on days 7–9 post tumor challenge, with all experiments using relevant isotype controls.
In vivo cell depletions
α-asialo ganglioside GM1 (asGM1, clone Rabbit) was purchased from FUJIFILM Wako Chemicals U.S.A. Corporation (Richmond, VA) and prepared according to manufacturer’s instructions using 1 mL of deionized water. αNK1.1 (clone PK136), αCD8 (clone 2.43), and isotype control antibodies were purchased from BioXCell. Antibodies were given intraperitoneally to deplete relevant cells concurrently with the tumor treatment regimen per figure legends. Doses/mouse: 200 µL α-asGM1, 250 µg αNK1.1, and 250 µg αCD8.
Flow cytometry
Tumors were harvested for flow cytometry after mice were sacrificed via cervical dislocation and induction of deep isoflurane anesthesia. Bladder tumor dissection was as previously described.Citation20 Subcutaneous and peritoneal B16 tumors were dissected and placed in a 6-well plate filled with serum-free RPMI-1640 and manually dissociated with the back of a syringe. Cells were incubated for 45–60 min in 3 mL serum-free RPMI-1640 with 0.25 mg/mL DNAse I and 1.65 mg/mL collagenase Type IV (both Sigma Aldrich; St. Louis, MO) and passed through a 70 µm filter to generate single-cell suspensions. 3–5 × 106 cells were then transferred to 96-well plates and samples with <5 × 106 cells from the same group were pooled to ensure uniform cell counts for all samples.
Single-cell suspension from lung was prepared as previously reported.Citation27 Briefly, lungs bearing B16 tumors were excised from mice and lung tissue was dissociated via razor blades into fragments of ~1 mm in size. Then, tissue fragments were transferred into a 24-well plate containing 2 mL/lung of digestion medium (complete RPMI containing 2 mg/mL collagenase IV + 0.02 mg/mL DNase I) and incubated at 37°C for 30 min. Cells were then passed through a 70 µm filter and a single-cell suspension was generated similar to subcutaneous and peritoneal B16 tumors.
Dead cells were excluded using either LIVE/DEAD Fixable Blue Dead Cell Stain Kit for ultraviolet excitation (Thermo Fisher Scientific; Waltham, Massachusetts, USA) or Ghost Dye Violet 510 (Tonbo biosciences; San Diego, California, USA). Fc blocking to prevent nonspecific labeling was done using 1:100 dilution anti-CD16/32 (clone 93, Biolegend). A list of used antibodies, including surface and intracellular staining, is provided in . Cells were stained for surface antigens by incubating at 4°C for 30–45 min with antibodies against: CD45 (30-F11), CD3 (17A2/145-2C11), CD8 (53–6.7), B220 (RA3-6B2), PD-1 (J43), CD27 (LG.3A10), CD11b (M1/70), CD69 (H1.2F3), CD49b (HMα2), CD122 (TM-β1), KLRG1 (2F1), NK1.1 (PK136), DNAM-1 (10E5), NKG2A (16a11), and gp100 [EP4863(2)], all at manufacturer recommended dilutions.
Table 1. Antibodies and fluorescent dyes used for flow cytometry
For intracellular staining, cells were fixed and permeabilized with FoxP3/transcription factor buffer (eBioscience; San Diego, CA) according to manufacturer instructions, and incubated at 4°C for 45 min. To assess effector molecule production, cells were stimulated with Cell Activator Cocktail (Biolegend) containing phorbol 12‐myristate 13‐acetate, ionomycin, and brefeldin A at 2 µL cocktail/mL CR10 medium (RPMI-1640 with 10% FBS, L-glutamine, sodium pyruvate, non-essential amino acids, penicillin/streptomycin, and HEPES buffer) for 6 hours in a 37°C incubator. Following stimulation, intracellular staining was performed by incubating cells at 4°C for 30–45 min with antibodies against: Eomes (W17001A), Perforin (S16009A), Granzyme B (NGZB/GB11), Interferon (IFN)-γ (XMG1.2), all at manufacturer recommended dilutions in FoxP3 permeabilization buffer. Brilliant Stain Buffer Plus (BD) was added to surface and intracellular staining at the manufacturer recommended dilution. Absolute cell numbers were determined by multiplying the cell ratio per live, singlet cell in each flow sample by total cell numbers in the sample. Flow data were acquired on a Cytek Aurora flow cytometer (Cytek Biosciences; Freemont, CA) and analyzed using FlowJo software (BD) version 10.7.1. Representative flow cytometry gating strategies and relevant immune phenotype comparisons across tumor models are shown in Supplementary Data and , respectively.
Table 2. Flow cytometric assessment of NK and CD8+ T cell phenotypes across tumor models
Statistical and data analyses
Data were analyzed and graphed with GraphPad Prism 9.1.2. Data with error bars are mean ± SEM. To compare two means, we used an unpaired t test. Three or more means were compared with one-way ANOVA and post hoc Sidak’s test. Tumor growth curves were compared by two-way ANOVA, analyzed for overall treatment effect, followed by post hoc Sidak’s test of discrete time points. Log-rank test was used to compare Kaplan-Meier survival curves. Occasionally, data sets with suspected outliers were identified by Grubbs’ test (used only once for a given data set) and removed from analysis. For all analyses, significance was based on a multiplicity-corrected α of 0.05.
Results
IL-2c enhances NK cell activation and maturation in orthotopic BC
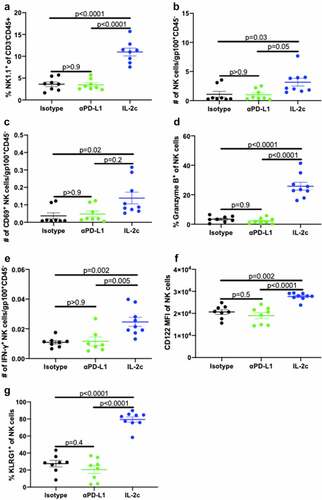
We previously reported that CD122-targeting IL-2c activated γδ T cells but not CD8+ T cells to treat orthotopic MB49 BC.Citation20 However, effects on other CD122+ immune cell populations in bladder are unreported. Like MB49, we observed reduced tumor growth and tumor weight in IL-2c treated mice bearing orthotopic MBT-2 (Fig. S1A,B), a syngeneic mouse BC line on a distinct genetic background (C3H/He) from MB49 (BL6). To assess NK cell effects in IL-2c treatment, we performed flow cytometric analysis of bladders from mice bearing orthotopic MB49 and MBT-2 tumors (Fig. S2). Compared to isotype treatment, IL-2c increased intratumoral NK cell frequency (), reduced PD-1 expression () and increased CD69 expression (), corresponding with reduced exhaustion and enhanced activation, respectively. We also observed a trend toward decreased expression of the NK cell inhibitory receptor (NKG2A) in IL-2c treated bladders, yet other NK activating receptors (e.g., DNAM-1) were unaffected by IL-2c treatment (Fig. S3A).
Figure 1. IL-2c enhances NK activation and maturation to treat orthotopic BC. Wild-type female mice were challenged orthotopically with 8 × 104 MB49 and treated with IL-2c or an isotype control every other day starting on day 7 for a total of 4 doses. (a-e) Mice were sacrificed on day 16 post challenge. Flow cytometric analysis of intratumoral bladder NK cell frequency (a), exhaustion (PD-1+) (b), activation (CD69+) (c), maturation (CD27−) (d), and CD69+ Eomes+ expression by CD27− NK cells (e). N = 6–8 pooled bladders/group. p, unpaired t test. NK cells in MB49 defined as CD45+ CD3− NK1.1+ cells. (f) Mouse survival in orthotopic MB49 challenge treated with IL-2c (as above) ± 200 µL α-asialo GM1 (asGM1). N = 7–9 mice/group. p, log-rank. Note: We previously reported data showing survival of isotype-treated mice bearing orthotopic MB49.Citation20 Eomes, Eomesodermin. IL-2c, interleukin-2 complex. NK, Natural Killer. PD-1, Programmed Death Receptor 1
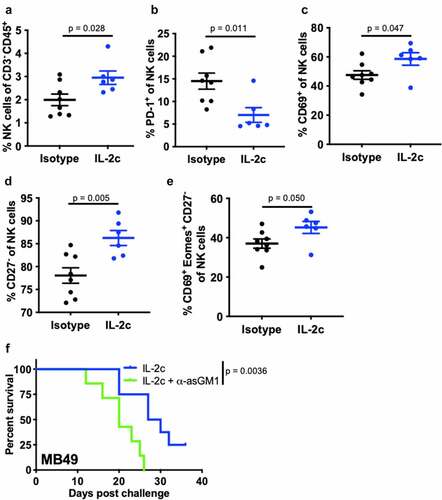
NK cell maturation describes the process of terminal differentiation and acquisition of optimal effector function and can be defined by the loss of CD27 expression in nonlymphoid tissues.Citation28 We noted a significant increase in the frequency of CD27− NK cells () in MB49 tumors, suggesting increased NK cell maturation after IL-2c treatment. Moreover, an increase in frequency of CD69+ Eomes+ CD27− NK cells () in the context of efficacious IL-2c treatment suggests that these activated, mature NK cells may be important mediators of the IL-2c driven antitumor immune response. We also observed a trend for increased perforin-producing NK cells after IL-2c treatment (Fig. S3B), further suggesting IL-2c driven improvement in NK cell effector function. Data in the MBT-2 model corroborated these results, with IL-2c also increasing CD27− NK cell frequency (Fig. S3C) in this model. CD27− NK cells in MBT-2 tumors also highly expressed KLRG1 (Fig. S3D), a marker only present in the final stage of NK cell maturation.Citation29 To test NK contributions to IL-2c efficacy directly, we treated wild-type mice with IL-2c ± NK cell depletion using α-asialoGM1,Citation30 which significantly reduced treatment efficacy in orthotopic MB49 (). Collectively, these data implicate NK cells as critical mediators of IL-2c antitumor efficacy in orthotopic bladder tumors through promotion of NK cell maturation and effector function.
IL-2c promotes NK cell maturation in a γδ T cell-independent manner
We previously reported that antibody-mediated or genetic depletion of γδ T cells significantly abrogated IL-2c efficacy in orthotopic MB49 challenge,Citation20 similar to NK cell depletion shown here (). Thus, we hypothesized that IL-2c treatment efficacy is either the summation of independent γδ T and NK cell effects or the result of their coordinated interactions. To test interactions, we performed flow cytometric analysis of intratumoral bladder NK cell content in orthotopic MB49 challenged TCRδKO mice (lacking all γδ T cells) ± IL-2c treatment. As in wild-type mice (), IL-2c NK cell effects included increased intratumoral bladder NK cell frequency (), reduced PD-1 expression (), and increased frequency of perforin-producing NK cells (). We then evaluated if the NK cell maturation phenotype observed in IL-2c treated wild-type mice was also present in TCRδKO mice. As in wild-type mice, we found a robust increase in the frequency and number of NK cells co-expressing KLRG1 and Eomes (), both of which are essential for NK cell maturation and function.Citation29,Citation31 These data suggest that IL-2c driven NK cell maturation in this model appears to be γδ T cell independent, suggesting potential benefits of IL-2c in other contexts.
Figure 2. IL-2c promotes NK cell maturation in a γδ T cell independent manner. TCRδKO female mice were challenged orthotopically with 8 × 104 MB49 cells and treated with IL-2c on days 9, 11, 13, and 15 or isotype control. Mice were sacrificed on day 17 for flow cytometric analysis of NK cell frequency (a), exhaustion (i.e., PD-1+) (b), perforin production (c), and KLRG1+ Eomes+ expression by CD27− NK cells (d,e). NK cells in MB49 defined as CD45+ CD3− NK1.1+ cells. N = 9–11 mice/group. Tumors were pooled to achieve 5 × 106 live cells per sample. p, unpaired t test. Eomes, Eomesodermin. IL-2c, interleukin-2 complex. KLRG1, Killer Cell Lectin Like Receptor G1. NK, Natural Killer. PD-1, Programmed Death Receptor 1
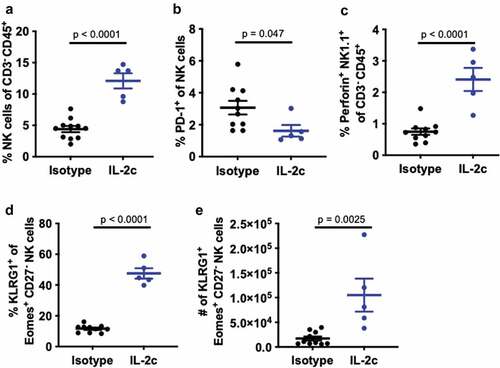
We then performed more detailed analyses of NK cell maturation in TCRδKO mice bearing orthotopic MB49 by examining CD49b+ NK cells, as CD49b is a marker acquired at the onset of NK cell maturation in BL6 mice.Citation29 Using differential expression of CD11b and CD27 to evaluate the final three stages of NK cell maturation,Citation32 we found that IL-2c significantly increased the frequency of terminally differentiated, mature CD11b+ CD27− NK cells (Fig. S4A,B), with striking increases in KLRG1+ NK cell frequency (Fig. S4C), as observed in wild-type hosts (). IL-2c also increased the number of Perforin+ Eomes+ KLRG1+ mature NK cells (Fig. S4D) suggesting enhanced effector function.
αPD-L1 requires CD8+ T and NK cells to treat subcutaneous B16 melanoma
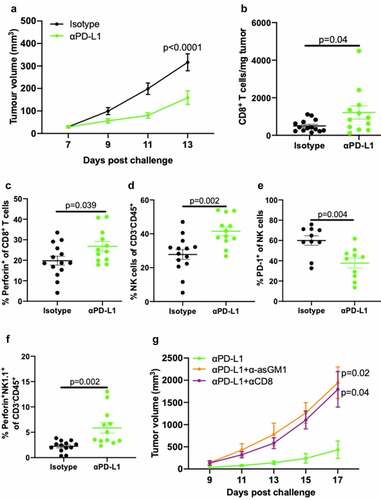
IL-2 activates downstream STAT5 signaling via CD122 in NK cells and facilitates NK cell cytotoxicity,Citation33 yet prior studies in subcutaneous B16 melanoma showed that CD8+ T cells but not NK cells, are required for IL-2c treatment efficacy.Citation34 To determine if lack of NK cell requirements for IL-2c treatment efficacy in orthotopic B16 is tissue microenvironment-specific, we studied αPD-L1 which has established effects on CD8+ T and NK cells in several cancers.Citation35 We confirmed αPD-L1 efficacy in B16 in our mice (), consistent with prior reports.Citation36,Citation37 Flow cytometric analysis of subcutaneous B16 tumors (Fig. S5) showed that αPD-L1 improved CD8+ T cell number () and effector function detected as increased perforin expression (), consistent with enhanced T cell cytotoxicity. However, αPD-L1 also increased intratumoral NK cell frequency (), reduced PD-1 expression (), and increased prevalence of perforin-producing NK cells (), showing the potential for beneficial αPD-L1 effects on NK cells in skin. We then depleted CD8+ T cells with αCD8 (Fig. S6) or NK cells with α-asialoGM1 during αPD-L1 treatment and found that both depletions significantly abrogated αPD-L1 mediated tumor control of subcutaneous B16 melanoma (). Together, these data confirm distinct tumor microenvironment-dependent effects of IL-2c and αPD-L1 in treatment of orthotopic (primary) B16 and provide an important baseline for comparison with mechanisms of these agents at metastatic sites.
Figure 3. αPD-L1 requires NK and CD8+ T cells to treat subcutaneous B16 melanoma. Wild-type male mice were challenged subcutaneously with 5 × 105 B16 cells on both flanks and treated with 100 μg αPD-L1 on days 7, 12, 17 or an isotype control (a-f). (a) Tumor growth of mice bearing subcutaneous B16. N = 7 mice/group. p, two-way ANOVA of day 13 tumor volume. (b-e) Mice were sacrificed on day 14 for flow cytometric analysis of CD8+ T cell number (b) and perforin production (c), and NK cell prevalence (d), exhaustion (i.e., PD-1+) (e), and perforin production (f). N = 6–7 mice/group. p, unpaired t test. (g) Wild-type male mice were challenged subcutaneously with 5 × 105 B16 cells on a single flank and treated with 100 μg αPD-L1 on days 7, 10, and 13 ± 200 µL α-asGM1, 250 ug αCD8, or an isotype control every three days starting the day before the challenge until the end of αPD-L1 treatment. N = 5 mice/group. p, two-way ANOVA of day 17 tumor volume. ANOVA, analysis of variance. asGM1, asialo GM1. NK, Natural Killer. PD-L1, Programmed Death Ligand 1. PD-1, Programmed Death Receptor 1
IL-2c or αPD-L1 alone treat peritoneal B16 melanoma
We have extensively studied orthotopic (peritoneal) ID8agg mouse ovarian cancer, which is refractory to αPD-L1 but sensitive to IL-2c.Citation19 To compare IL-2c and αPD-L1 mechanisms in orthotopic (subcutaneous) B16 and ID8agg, we used a clinically relevant peritoneal metastatic melanoma modelCitation8 via intraperitoneal injection of B16 into wild-type mice.Citation38 Peritoneal tumors from αPD-L1 treated mice were marginally smaller than isotype controls, yet IL-2c treatment led to a dramatic reduction in tumor weight versus isotype and αPD-L1 treated mice (Fig. S7A). Both agents equally improved mouse survival (Fig. S7B).
IL-2c promotes NK cell maturation in peritoneal B16, whereas αPD-L1 does not
Flow cytometric analyses of peritoneal B16 tumors (Fig. S8) showed that IL-2c increased total intratumoral CD45+ immune cell content () compared to isotype control and αPD-L1 treated mice. αPD-L1 and IL-2c shared many beneficial effects on CD8+ T cells including increasing their prevalence () and production of effector molecules including perforin, IFN-γ, and granzyme B (), yet IL-2c effects were superior to αPD-L1 in these regards (). IL-2c also had NK cell activating effects not elicited by αPD-L1, including increased NK cell concentration (Fig. S9A) and CD69 expression (Fig. S9B), though both agents reduced NK cell PD-1 expression versus isotype controls (Fig. S9C). IL-2c-mediated NK activation also increased effector molecule production (), frequency of CD69+ Eomes+ CD27− activated mature NK cells (), and increased NK cell CD122 expression (), effects not observed with αPD-L1 ().
Figure 4. IL-2c activates of CD8+ T cells and promotes NK cell maturation in peritoneal B16. Wild-type male mice were challenged peritoneally with 4 × 105 B16 cells and treated with 100 μg αPD-L1 on days 7, 12, 17 or IL-2c on days 7, 9, 11, 13. Mice were sacrificed on day 15 for flow cytometric analysis of immune (CD45+) cell number normalized to tumor weight (a). (b-e) Intratumoral CD8+ T cell frequency (b) and effector molecule production (c-e). (f-i) Intratumoral NK cell perforin (f) and granzyme B (g) production, CD69+ Eomes+ expression by CD27− NK cells (h), and CD122 expression (i). N = 11–13 mice/group. p value, one-way ANOVA. (j) Survival of wild-type male mice challenged peritoneally with 4 × 105 B16 cells and treated with αPD-L1 (as above) ± 250 ug αNK1.1 or 250 ug αCD8 on days 6, 9, 12, 15, 18. N = 8–14 mice/group. p value, log-rank. (k) Survival of wild-type male mice challenged peritoneally with 4 × 105 B16 cells and treated with IL-2c (as above) ± 250 ug αNK1.1 or 250 ug αCD8 on days 6, 9, 12, 15. N = 8–9 mice/group. p value, log-rank. Note: Survival of mice in Figure 4j,k can be compared to survival of isotype-treated mice with peritoneal B16 in Fig. S7B. ANOVA, analysis of variance. Eomes, Eomesodermin. IFN-γ, interferon-gamma. IL-2, interleukin-2 complex. MFI, Mean Fluorescence Intensity. NK, Natural Killer. PD-L1, Programmed Death Ligand 1
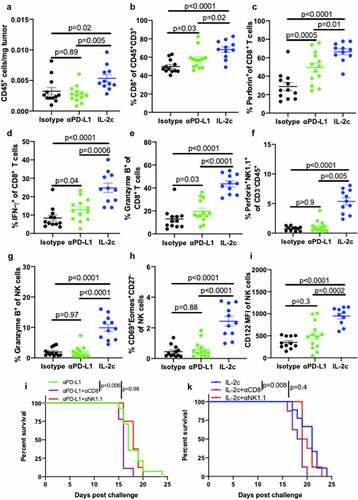
To assess CD8+ T and NK cell contributions to αPD-L1 and IL-2c treatment mechanisms, we administered αPD-L1 ± CD8+ T and NK cell depleting antibodies to peritoneal B16 tumor-bearing mice. Consistent with our flow cytometric analyses that showed αPD-L1 effects primarily via CD8+ T cells and not NK cells in this model, CD8+ T cell depletion significantly reduced survival of mice treated with αPD-L1, whereas NK cell depletion did not impact survival (). As CD8+ T cell depletion did not fully abrogate treatment efficacy, other immune cells likely also contribute. IL-2c treatment of peritoneal B16 had similar CD8+ T cell requirements, yet surprisingly, NK cell depletion did not significantly reduce survival (). This may be due to the strength of IL-2c CD8+ T cell effects in this context compared to NK cells, as CD8+ T cells expressed higher levels of CD122 (the IL-2c target) both at baseline and during IL-2c treatment (Fig. S9D). These data show that in addition to many shared effects on CD8+ T cells with αPD-L1, IL-2c-mediated promotion of NK cell maturation and function extends across tumor types and anatomic locations, though specific contributions of pleiotropic IL-2c immune effects to immunotherapy efficacy is tumor microenvironment-specific.
IL-2c treats B16 lung metastases, whereas αPD-L1 is ineffective
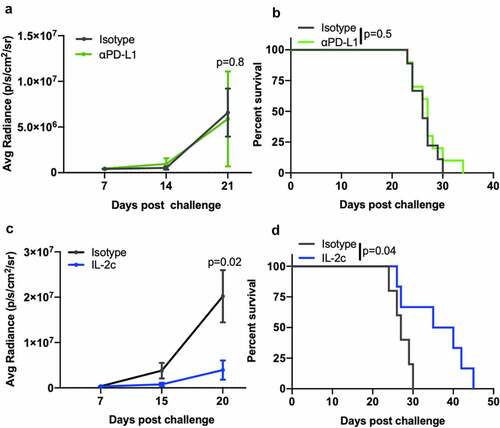
The lung is the most common site of visceral metastasis in melanomaCitation39 and approximately one-third of BC metastases are to the lungs.Citation7,Citation40 We previously showed that IL-2c and αPD-L1 fail as single-agent treatments in lung metastatic MB49 or MBT-2 BC, but their combination was effective and required CD8+ T cells.Citation20 To test if treatment mechanisms were cancer-specific, we studied a model of melanoma lung metastasis via intravenous B16 cell injection. As in BC lung metastasis, αPD-L1 was unable to slow tumor growth () or prolong mouse survival () in lung metastatic B16 melanoma despite its efficacy in subcutaneous () and peritoneal (Fig. S7) metastases. However, IL-2c significantly slowed B16 lung metastatic tumor growth () and improved survival as a single agent versus isotype treated controls (), mirroring outcomes in orthotopic (peritoneal) ID8agg ovarian cancer which is also IL-2c sensitive but αPD-L1 resistant.Citation19
Figure 5. IL-2c, but not αPD-L1, treats B16 lung metastases. Wild-type male mice were challenged intravenously with 3 × 105 B16 cells and treated with 100 μg αPD-L1 (a, b) on days 8, 13, 18, or IL-2c (c, d) on days 8, 10, 12, 14, or isotype control (a-c). (a,c) Tumor bioluminescence corresponding with pre-treatment, mid-treatment, and post treatment timepoints. N = 6–10 mice/group. p, two-way ANOVA of day 21 (a) or 20 (c) signal. (b,d) Mouse survival of mice in (a) and (c). N = 6–10 mice/group. p, log-rank. ANOVA, analysis of variance. IL-2c, interleukin-2 complex. PD-L1, Programmed Death Ligand 1
IL-2c treatment efficacy in lung metastasis is NK cell-dependent
Next, we tested if treatment mechanisms for IL-2c treatment efficacy in B16 lung metastases were similar to subcutaneous or peritoneal B16. Using TCRβKO mice that lack CD4+ and CD8+ T cells, we found that IL-2c still reduced B16 lung tumor growth () and improved mouse survival versus isotype control treatment (). IL-2c acts through γδ T cells in orthotopic bladder cancer.Citation20 To assess possible γδ T cell effects in B16 lung metastases, we challenged RAG1KO mice, which additionally lack γδ T cells and B cells compared to TCRβKO mice. IL-2c still improved mouse survival versus controls (), supporting the concept that IL-2c treats lung metastatic B16 independent of adaptive immunity or γδ T cells.
Figure 6. NK cells are essential for IL-2c efficacy in lung metastasis. TCRβKO (a, b), RAG1KO (c), or wild-type (d,e) male mice were challenged intravenously with 3 × 105 B16. (a-e) Mice received IL-2c on days 8, 10, 12, 14, an isotype control (a-c), or 250 ug αNK1.1 on days 7, 10, 13, 16 (d, e). (a) Tumor bioluminescence in intravenous B16 challenge of TCRβKO mice. N = 9–10 mice/group. p, two-way ANOVA of day 20 signal. (b) TCRβKO mouse survival following intravenous B16 challenge. N = 9–10 mice/group. p, log-rank. (C) RAG1KO mouse survival following intravenous B16 challenge. N = 9 mice/group. p, log-rank. (d) Tumor bioluminescence in intravenous B16 challenge of wild-type mice. N = 11–12 mice/group. p, two-way ANOVA of day 22 signal. (e) Wild-type mice survival following intravenous B16 challenge. Note: Survival of isotype-treated mice bearing B16 lung metastasis is in . N = 11–12 mice/group. p, log-rank. ANOVA, analysis of variance. RAG1, Recombinase Activating Gene 1. TCR, T Cell Receptor
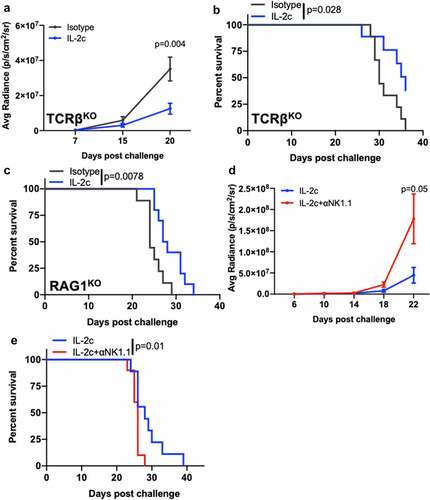
As IL-2c significantly promoted NK cell activity in both orthotopic BC () and metastatic melanoma models (), we tested NK cell-driven antitumor immunity as a mediator of IL-2c efficacy in metastatic B16 lung tumors. We depleted NK cells using αNK1.1 (Fig. S10) in mice bearing lung metastatic B16 melanoma treated with IL-2c, which reduced IL-2c-mediated control of tumor growth () and survival (), supporting a critical role for NK cells in IL-2c efficacy here.
In lung metastatic MB49 and MBT-2 BC, IL-2c, or αPD-L1 alone are ineffective, but their combination effectively treats both models.Citation20 To test NK cell contributions to treatment of lung metastases in BC, we challenged mice with either lung metastatic MB49 or MBT-2 and treated with αPD-L1 + IL-2c with or without NK cell depletion (Fig. S11). In support of significant NK cell contributions, NK cell depletion significantly reduced mouse survival in both models (Fig. S12A,B). Together, these data show that IL-2c effects on NK cells are disparate from CD8+ T cell effects and contribute independently to immunotherapy efficacy across multiple primary (orthotopic) and metastatic models of BC and melanoma on distinct genetic backgrounds.
IL-2c activates lung NK cells to treat B16 lung metastases
We then performed flow cytometric analyses of B16 lung tumors (Fig. S13) to explore possible mechanisms for NK cell-mediated IL-2c efficacy in lung. We used gp100+ (melanoma-associated antigenCitation41) cell number to normalize tumor-infiltrating NK and CD8+ T cell numbers and found that IL-2c but not aPD-L1, significantly increased the frequency () and total numbers () of lung tumor-infiltrating NK cells, as well as numbers of activated NK cells (). IL-2c also improved NK cell production of anti-tumor effector molecules granzyme B and IFN-γ (), and increased CD122 expression (), with αPD-L1 having no observable NK cell effects (). Similar to orthotopic MB49 BC, IL-2c treatment also significantly increased expression of the NK cell maturation marker KLRG1 (), consistent with terminal differentiation and enhanced effector function of lung NK cells.
Figure 7. IL-2c promotes CD122 expression and NK cell maturation in B16 lung metastases. Wild-type male mice were challenged intravenously with 3 × 105 B16 cells and treated with 100 μg αPD-L1 on days 14, 19, or IL-2c on days 14, 16, 18, or isotype control, and sacrificed on day 20. Total lung NK cell frequency (a) and tumor-normalized NK cell number (b), CD69 expression (c), granzyme B production (d), IFN-γ production (e), CD122 expression (f), and KLRG1 expression (g). Ν = 5–9 mice/group. p value, one-way ANOVA. ANOVA, analysis of variance. IFN-γ, interferon-gamma. IL-2c, interleukin-2 complex. KLRG1, Killer Cell Lectin Like Receptor G1. MFI, Mean Fluorescence Intensity. NK, Natural Killer. PD-L1, Programmed Death Ligand 1
In contrast, neither IL-2c nor αPD-L1 treatment affected CD8+ T cell prevalence (Fig. S14A), total numbers (Fig. S14B), activation (Fig. S14 C), or effector molecule production (Fig. S14D). These results support our findings in T cell deficient mice () that CD8+ T cells are dispensable for IL-2c efficacy in B16 lung metastasis and further emphasize the role of NK cells in mediating the IL-2c efficacy in this model.
Discussion
ICB with αPD-1 or αPD-L1 is FDA-approved to treat nearly 20 cancer subtypesCitation42 yet even in the most ICB-sensitive cancers, many patients do not respond,Citation43 necessitating improved treatment approaches. Here, we studied IL-2c and αPD-L1 treatment mechanism differences in primary (orthotopic) and metastatic BC and melanoma, heading toward our goal of identifying conserved immune cell intrinsic mechanisms of these two agents that persist in varied tumor immune microenvironments, which could help define rational treatment combinations for improved treatment efficacy.
We previously reported that γδ T cells were required for optimal IL-2c treatment efficacy in orthotopic bladder cancerCitation20 and recently showed that γδ T cells augment antigen-specific immunity in the treatment of MB49 BC with Bacillus Calmette-Guérin (BCG) immune therapy.Citation44 Here, we found that NK cells also contribute to IL-2c efficacy in orthotopic bladder cancer in a γδ T cell-independent manner, suggesting that both cells could contribute individually, rather than serially, to treatment efficacy. Thus, strategies to boost function of either cell type could improve IL-2c efficacy, such as ultra-low dose rapamycin, which we demonstrated to be a safe and tolerable γδ T cell adjuvant in early phase BC trials when combined with BCG immune therapy.Citation45
To understand tissue-selective effects and assess differences from αPD-L1, we found that αPD-L1 required both CD8+ T cells and NK cells to treat orthotopic B16 melanoma, whereas IL-2c only required CD8+ T cells.Citation34 These data show that IL-2c has differential mechanisms of action against the same tumor in distinct anatomic compartments, and that αPD-L1 effects in the same environment and tumor can differ from IL-2c. Our prior study showed that CD8+ T cells are dispensable for IL-2c treatment of orthotopic bladder cancer (where γδ T cellsCitation20 and NK cells (this report) are required), whereas αPD-L1 requires CD8+ T cells to treat orthotopic bladder cancer, suggesting potential benefit of combining both agents. We also found that NK cells from IL-2c treated mice consistently expressed lower PD-1, which could also impact ICB therapy. Further studies evaluating the impact of CD122-directed IL-2 therapy on ICB targets (e.g., PD-1, PD-L1) are warranted to gain better mechanistic insights that can facilitate optimized combination therapies.
We considered that metastatic tumors represent an additional immune challenge to hosts, whereby the tumor appears in distinct anatomic compartments. To understand IL-2c and αPD-L1 effects in metastatic tumors, we evaluated peritoneal B16 melanoma, a large unmet clinical need, as patients with peritoneal metastatic melanoma have a median survival of only two months.Citation8 We found that either IL-2c or αPD-L1 alone effectively treated, but did not cure, peritoneal B16. By contrast, we previously showed that IL-2c very effectively treated peritoneal (orthotopic) ID8agg ovarian cancer, where αPD-L1 is entirely ineffective.Citation19 Thus, these outcomes do not reflect the generic efficacy of αPD-L1 in the peritoneal compartment. In peritoneal B16, IL-2c activated CD8+ T cells more effectively versus NK cells, which could be due to greater CD122 expression on local CD8+ T cells versus NK cells that we show here. Cell depletion data were consistent with flow data for αPD-L1 efficacy mechanisms, but not for IL-2c. IL-2c greatly improved NK cell numbers and functions over αPD-L1, yet NK cell depletion very surprisingly did not affect IL-2c treatment efficacy in peritoneal B16. There are several potential explanations that require further investigations. Among these, the NK cell effects required for treatment efficacy might not be reflected in flow analyses, specific suppressive factors could impede NK cell efficacy after IL-2c but not after αPD-L1, and NK cell/tumor co-colocalization could differ based on IL-2c versus αPD-L1 treatments. These data underscore the fact that flow cytometric analyses in this model might not fully reflect all mechanistic details. Further, as CD8+ T cell depletion did not fully reverse IL-2c treatment efficacy in peritoneal metastases, additional mechanisms might exist, requiring further studies.
In the clinically relevant setting of B16 melanoma lung metastases, IL-2c outperformed αPD-L1 in tumor control and host survival. As αPD-L1 treats subcutaneous B16, these data show that the local immune microenvironment can significantly affect treatment efficacy. In accord with much other data here, IL-2c efficacy in lung metastases depended on NK cells. Further, lung metastatic IL-2c efficacy was independent of CD8+ T cells (or tumor-specific immunity), as we found that IL-2c efficacy was preserved in RAGKO mice lacking any tumor-specific immunity. Flow cytometry analyses of B16 lung tumors showed that IL-2c preferentially boosts lung NK cell activation and effector function but not lung CD8+ T cells, confirming the necessity of NK cells in IL-2c treatment of B16 lung metastasis and providing a striking contrast to peritoneal B16 tumors where IL-2c activated both CD8+ T and NK cells. We also identified an increase in lung KLRG1+ NK cells following IL-2c treatment, which denotes a terminally mature NK cell population with a protective effect against pulmonary metastasis.Citation46,Citation47 The robust effects on this population in both bladder and lung following IL-2c treatment warrants further investigation, as IL-2c-driven NK cell maturation may occur via a CD122-Eomes axis,Citation48 with expansion of KLRG1+ NK cells serving as a possible biomarker for responsiveness to CD122-directed immunotherapies. In this regard, our NK cell immunophenotyping generally agrees with prior reports.Citation29 However, as tissue-specific NK cell differences are known,Citation49 detailed NK cell studies in specific anatomic compartments with tumor metastases will help further understanding of immunotherapy effects. Thus, detailed understandings of the immune landscape in distinct anatomic compartments and effects of specific treatment agents therein will help define more effective treatment regimens.
All together, we establish differential responsiveness of the same tumor to the same agent in distinct (metastatic) compartments, and IL-2c mechanisms of efficacy appear largely to depend on NK cells, whereas αPD-L1 effects depend to a greater extent on CD8+ T cells. These data suggest that combining αPD-L1 with IL-2c could be more effective against a variety of cancers, including in metastatic settings where there is a great unmet medical need. Further, other CD122-selective treatment approaches under evaluation, such as pegylated IL-2 (bempegaldesleukin) which shows efficacy in metastatic melanoma trials in combination with the αPD-1 agent, nivolumab,Citation50 should be similarly tested. It remains to be seen whether distinct CD122-targeted IL-2 constructs have differing capacities to activate CD8+ T cells versus NK cells or γδ T cells, but such data will greatly help define optimal combinations for further clinical evaluations.
Abbreviations
Analysis of variance (ANOVA), asialo GM1 (asGM1), Bacillus Calmette-Guérin (BCG), bladder cancer (BC), cytotoxic T-lymphocyte-associate protein 4 (CTLA-4), Eomes (Eomesodermin), IL-2 complex (IL-2c), IL-2 receptor (IL-2R), immune checkpoint blockade (ICB), interleukin (IL), interferon-gamma (IFN-γ), killer cell lectin-like receptor G1 (KLRG1), mean fluorescence intensity (MFI), milligram (mg), natural killer (NK), programmed death receptor 1 (PD-1), programmed death-ligand 1 (PD-L1), recombination activating gene 1 (RAG1), T regulatory cells (Tregs), T cell receptor (TCR)
Consent for publication
All authors of this manuscript consent to its submission and publication.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Authors’ contributions (CRediT – Contributor Roles Taxonomy)
RMR: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, validation, visualization, writing – original draft, writing – review and editing CZ: data curation, formal analysis, investigation, methodology, visualization, writing – original draft, and writing – review and editing. YD: data curation, formal analysis, investigation, methodology, visualization, and writing – review and editing. NJ: investigation, methodology, resources. NM: data curation, methodology, resources. ASP: investigation, resources. CAC: investigation. RSS: conceptualization, data curation, formal analysis, funding acquisition, resources, supervision, and writing – review and editing. TJC: conceptualization, data curation, formal analysis, funding acquisition, methodology, project administration, resources, software, supervision, writing – original draft, writing – review and editing.
Supplemental Material
Download ()Acknowledgments
This work was supported by the Flow Cytometry Shared Resource Facility, which is supported by UL1 TR001120.
Disclosure statement
Ethics approval and consent to participate: All animal studies were approved by the UT Health San Antonio Institutional Animal Care and Use Committee and each experiment was conducted in accordance with the standards required by the UT Health San Antonio Department of Laboratory Animal Resources.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–15. doi:10.3322/caac.21590.
- Suhail Y, Cain MP, Vanaja K, Kurywchak PA, Levchenko A, Kalluri R. Kshitiz: systems biology of cancer metastasis. Cell Syst. 2019;9(2):109–127. doi:10.1016/j.cels.2019.07.003.
- Riggi N, Aguet M, Stamenkovic I. Cancer metastasis: a reappraisal of its underlying mechanisms and their relevance to treatment. Annu Rev Pathol. 2018;13(1):117–140. doi:10.1146/annurev-pathol-020117-044127.
- Ganesh K, Massagué J. Targeting metastatic cancer. Nat Med. 2021;27(1):34–44. doi:10.1038/s41591-020-01195-4.
- Chensue SW, Warmington KS, Ruth JH, Sanghi PS, Lincoln P, Kunkel SL. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen-induced granuloma formation: relationship to local inflammation, Th cell expression, and IL-12 production. J Immunol. 1996;157:4602–4608.
- Tas F. Metastatic behavior in melanoma: timing, pattern, survival, and influencing factors. J Oncol. 2012;2012:1–9. doi:10.1155/2012/647684.
- Shinagare AB, Ramaiya NH, Jagannathan JP, Fennessy FM, Taplin M-E, Van Den Abbeele AD. Metastatic pattern of bladder cancer: correlation with the characteristics of the primary tumor. Am J Roentgenol. 2011;196(1):117–122. doi:10.2214/AJR.10.5036.
- Flanagan M, Solon J, Chang KH, Deady S, Moran B, Cahill R, Shields C, Mulsow J. Peritoneal metastases from extra-abdominal cancer - A population-based study. Eur J Surg Oncol. 2018;44(11):1811–1817. doi:10.1016/j.ejso.2018.07.049.
- Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17–35. doi:10.1016/j.immuni.2019.12.011.
- Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P, Demidov L, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London, England). 2020;395(10240):1835–1844. doi:10.1016/S0140-6736(20)30934-X.
- Hamid O, Molinero L, Bolen CR, Sosman JA, Muñoz-Couselo E, Kluger HM, McDermott DF, Powderly JD, Sarkar I, Ballinger M, et al. Safety, clinical activity, and biological correlates of response in patients with metastatic melanoma: results from a phase I trial of atezolizumab. Clin Cancer Res. 2019;25(20):6061–6072. doi:10.1158/1078-0432.CCR-18-3488.
- Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA, et al. Combined nivolumab and Ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379(8):722–730. doi:10.1056/NEJMoa1805453.
- Darvin P, Toor SM, Nair VS, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):1–11. doi:10.1038/s12276-018-0191-1.
- Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, Matory YL, Skibber JM, Shiloni E, Vetto JT, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313(23):1485–1492. doi:10.1056/NEJM198512053132327.
- Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A. 2010;107(26):11906–11911. doi:10.1073/pnas.1002569107.
- Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol. 2015;36(12):763–777. doi:10.1016/j.it.2015.10.003.
- Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26(1):453–479. doi:10.1146/annurev.immunol.26.021607.090357.
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164.
- Drerup JM, Deng Y, Pandeswara SL, Padrón ÁS, Reyes RM, Zhang X, Mendez J, Liu A, Clark CA, Chen W, et al. CD122-selective IL2 complexes reduce immunosuppression, promote treg fragility, and sensitize tumor response to PD-L1 blockade. Cancer Res. 2020;80(22):5063–5075. doi:10.1158/0008-5472.CAN-20-0002.
- Reyes RM, Deng Y, Zhang D, Ji N, Mukherjee N, Wheeler K, Gupta HB, Padron AS, Kancharla A, Zhang C, et al. CD122-directed interleukin-2 treatment mechanisms in bladder cancer differ from αPD-L1 and include tissue-selective γδ T cell activation. J Immunother Cancer. 2021;9(4):e002051. doi:10.1136/jitc-2020-002051.
- Diab A, Tannir NM, Bentebibel S-E, Hwu P, Papadimitrakopoulou V, Haymaker C, Kluger HM, Gettinger SN, Sznol M, Tykodi SS. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 2020;10(8):1158–1173. doi:10.1158/2159-8290.CD-19-1510.
- Zhang D, Reyes RM, Osta E, Kari S, Gupta HB, Padron AS, Kornepati AV, Kancharla A, Sun X, Deng Y. Bladder cancer cell‐intrinsic PD‐L1 signals promote mTOR and autophagy activation that can be inhibited to improve cytotoxic chemotherapy. Cancer Med. 2021;10(6):2137–2152. doi:10.1002/cam4.3739.
- Lyon A, Fallon J, Boyerinas B, Schmitz R, Hance KW, Lan Y, Sabzevari H, Tsang K, Schlom J, Greiner J. Anti-tumor effects of anti-PD-L1 therapy in an orthotopic bladder tumor model. J ImmunoTher Cancer. 2014;2(S3): P101. doi:10.1186/2051-1426-2-S3-P101.
- Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, Drerup JM, Padron A, Conejo-Garcia J, Murthy K. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 2016;76(23):6964–6974. doi:10.1158/0008-5472.CAN-16-0258.
- Svatek RS, Zhao XR, Morales EE, Jha MK, Tseng TY, Hugen CM, Hurez V, Hernandez J, Curiel TJ. Sequential intravesical mitomycin plus bacillus calmette–guérin for non–muscle-invasive urothelial bladder carcinoma: translational and phase I clinical trial. Clin Cancer Res. 2015;21(2):303–311. doi:10.1158/1078-0432.CCR-14-1781.
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311(5769):1924–1927. doi:10.1126/science.1122927.
- Tait Wojno ED, Beamer CA. Isolation and identification of innate lymphoid cells (ILCs) for immunotoxicity testing. Methods Mol Biol. 2018;1803:353–370.
- Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006;176(3):1517–1524. doi:10.4049/jimmunol.176.3.1517.
- Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. 2018;9:1869. doi:10.3389/fimmu.2018.01869.
- Nishikado H, Mukai K, Kawano Y, Minegishi Y, Karasuyama H. NK cell-depleting anti-asialo GM1 antibody exhibits a lethal off-target effect on basophils in vivo. J Immunol. 2011;186(10):5766–5771. doi:10.4049/jimmunol.1100370.
- Simonetta F, Pradier A, Roosnek E. T-bet and eomesodermin in NK cell development, maturation, and function. Front Immunol. 2016;7:241. doi:10.3389/fimmu.2016.00241.
- Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood J Am Soc Hematol. 2009;113:5488–5496.
- Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, Rudensky AY. IL-2–dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med. 2013;210(6):1167–1178. doi:10.1084/jem.20122462.
- Arenas-Ramirez N, Zou C, Popp S, Zingg D, Brannetti B, Wirth E, Calzascia T, Kovarik J, Sommer L, Zenke G. Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci Transl Med. 2016;8(367):367ra166–367ra166. doi:10.1126/scitranslmed.aag3187.
- Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. 2018;7(11):e1509819. doi:10.1080/2162402X.2018.1509819.
- Padrón Á, Hurez V, Gupta HB, Clark CA, Pandeswara SL, Yuan B, Svatek RS, Turk MJ, Drerup JM, Li R, et al. Age effects of distinct immune checkpoint blockade treatments in a mouse melanoma model. Exp Gerontol. 2018;105:146–154. doi:10.1016/j.exger.2017.12.025.
- Ji S, Lee J, Lee ES, Kim DH, and Sin JI. B16 melanoma control by anti-PD-L1 requires CD8+ T cells and NK cells: application of anti-PD-L1 Abs and Trp2 peptide vaccines. Hum Vaccin Immunother. 2021;17(7): 1910–1922. PMID: 33522416
- Gómez-Cuadrado L, Tracey N, Ma R, Qian B, Brunton VG. Mouse models of metastasis: progress and prospects. Dis Model Mech. 2017;10(9):1061–1074. doi:10.1242/dmm.030403.
- Damsky WE, Rosenbaum LE, Bosenberg M. Decoding melanoma metastasis. Cancers. 2010;3(1):126–163. doi:10.3390/cancers3010126.
- Babaian RJ, Johnson DE, Llamas L, Ayala AG. Metastases from transitional cell carcinoma of urinary bladder. Urology. 1980;16(2):142–144. doi:10.1016/0090-4295(80)90067-9.
- Gelbart Y, Frankenburg S, Pinchasov Y, Krispel S, Eliahu D, Drize O, Morag E, Bartfeld D, Lotem M, Peretz T, et al. Production and purification of melanoma gp100 antigen and polyclonal antibodies. Protein Expr Purif. 2004;34(2):183–189. doi:10.1016/j.pep.2003.12.006.
- Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers. 2020;12(3):738. doi:10.3390/cancers12030738.
- Sun J-Y, Zhang D, Wu S, Xu M, Zhou X, Lu X-J, Ji J. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms, predictive factors, and future perspectives. Biomarker Res. 2020;8(1):1–10. doi:10.1186/s40364-020-00212-5.
- Ji N, Mukherjee N, Shu Z-J, Reyes RM, Meeks JJ, McConkey DJ, Gelfond JA, Curiel TJ, Svatek RS. γδ T cells support antigen-specific αβ T cell-mediated antitumor responses during BCG treatment for bladder cancer. Cancer Immunol Res. 2021. doi:10.1158/2326-6066.CIR-21-0285.
- Ji N, Mukherjee N, Reyes RM, Gelfond J, Javors M, Meeks JJ, McConkey DJ, Shu Z-J, Ramamurthy C, Dennett R. Rapamycin enhances BCG-specific γδ T cells during intravesical BCG therapy for non-muscle invasive bladder cancer: a randomized, double-blind study. J ImmunoTher Cancer. 2021;9(3):e001941. doi:10.1136/jitc-2020-001941.
- Malaisé M, Rovira J, Renner P, Eggenhofer E, Sabet-Baktach M, Lantow M, Lang SA, Koehl GE, Farkas SA, Loss M, et al. KLRG1+ NK cells protect T-bet-deficient mice from pulmonary metastatic colorectal carcinoma. J Immunol. 2014;192(4):1954–1961. doi:10.4049/jimmunol.1300876.
- Huntington ND, Tabarias H, Fairfax K, Brady J, Hayakawa Y, Degli-Esposti MA, Smyth MJ, Tarlinton DM, Nutt SL. NK cell maturation and peripheral homeostasis is associated with KLRG1 up-regulation. J Immunol. 2007;178(8):4764–4770. doi:10.4049/jimmunol.178.8.4764.
- Zhang J, Marotel M, Fauteux‐Daniel S, Mathieu AL, Viel S, Marçais A, Walzer T. T‐bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur J Immunol. 2018;48(5):738–750. doi:10.1002/eji.201747299.
- Goh W, Huntington ND. Regulation of murine natural killer cell development. Front Immunol. 2017;8:130. doi:10.3389/fimmu.2017.00130.
- Hurwitz M, Cho D, Balar A, Curti B, Siefker-Radtke A, Sznol M, Kluger H, Bernatchez C, Fanton C, Iacucci E, et al. Baseline tumor immune signatures associated with response to bempegaldesleukin (NKTR-214) and nivolumab. J Clin Oncol. 2019;37(15_suppl): Abstract 2623. doi:10.1200/JCO.2019.37.15_suppl.2623.