
ABSTRACT
Antibody–drug conjugates (ADCs) are used to target cancer cells by means of antibodies directed to tumor-associated antigens, causing the incorporation of a cytotoxic payload into target cells. Here, we characterized the mode of action of ADC costing of a TWEAKR-specific monoclonal antibody conjugated to a small molecule kinesin spindle protein (KSP) inhibitor (KSPi). These TWEAKR-KSPi-ADCs showed strong efficacy in a TWEAKR expressing CT26 colon cancer model in mice. TWEAKR-KSPi-ADCs controlled the growth of CT26 colon cancers in immunodeficient as well as in immunocompetent mice. However, when treated with suboptimal doses, TWEAKR-KSPi-ADCs were still active in immunocompetent but not in immunodeficient mice, indicating that TWEAKR-KSPi-ADCs act – in addition to the cytotoxic mode of action – through an immunological mechanism. Indeed, in vitro experiments performed with a cell-permeable small molecule KSPi closely related to the active payload released from the TWEAKR-KSPi-ADCs revealed that KSPi was capable of stimulating several hallmarks of immunogenic cell death (ICD) on three different human cancer cell lines: cellular release of adenosine triphosphate (ATP) and high mobility group B1 protein (HMGB1), exposure of calreticulin on the cell surface as well as a transcriptional type-I interferon response. Further, in vivo experiments confirmed that treatment with TWEAKR-KSPi-ADCs activated immune responses via enhancing the infiltration of CD4+ and CD8+ T lymphocytes in tumors and the local production of interferon-γ, interleukin-2, and tumor necrosis factor-α. In conclusion, the antineoplastic effects of TWEAKR-KSPi-ADCs can partly be attributed to its ICD-stimulatory properties.
Introduction
It is well established that anticancer agents mediate their effects in vivo through a dual mode of action, namely, direct cytostatic or cytotoxic effects on malignant cells and indirect effects mediated by the immune system.1,Citation2 Such indirect effects can be explained by the capacity of some antineoplastic agents to kill tumor cells in a fashion that makes them recognizable to the immune system. This “immunogenic cell death” (ICD) stimulates an anticancer immune response that allows the host to control residual tumor cells.Citation3–5 Cancer cell death triggered by “immunogenic chemotherapeutics” comprises a specific combination of pre-mortem stress responses and postmortem alterations that together is used to characterize ICD.Citation6,Citation7
ICD is typically preceded by endoplasmic reticulum (ER) stress and autophagy. The process occurs as a spatiotemporal sequence of events with pre-mortem ER stress underlying the exposure of the usually ER sessile protein calreticulin (CALR) on the cell surface and pre-mortem autophagy facilitating the release of adenosine triphosphate (ATP) from the cells.Citation8–13 ATP is actively secreted from the intracellular pool to the extracellular space where it acts on purinergic receptors expressed by immune cells. Immunogenic cell death is further characterized by the release of high mobility group box 1 protein (HMGB1) from the nucleus into the extracellular space as the plasma membrane becomes permeable.Citation14–16 Furthermore, type-I interferon (IFN) signaling has been identified as a requirement for ICD. Thus, cancer cells respond to an ICD inducer by expressing and releasing type-I IFNs, activating autocrine/paracrine type-I IFN signaling, and secreting chemokine (C-X-C motif) ligand 10 (CXCL10) and additional proteins for local immune stimulation.Citation17,Citation18 These alterations together constitute the aforementioned ICD characteristics and can be measured in vitro to identify anticancer agents with immunostimulatory properties.Citation19,Citation20
Only a few FDA approved chemotherapeutics are known that are able to induce immunogenic cell death (ICD).Citation21 These chemotherapeutics (like anthracyclines, oxaliplatin and a variety of microtubule poisons) are of particular interest for combination treatments with immune checkpoint inhibitors.Citation1,Citation6,Citation22,Citation23 Cancer cells that are undergoing ICD can vaccinate immunocompetent mice against a subsequent challenge with living cells of the same kind.Citation24–26
One strategy to reduce systemic toxicity of cytotoxic drugs consists in coupling them to monoclonal antibodies recognizing tumor-associated antigens expressed on the surface of malignant cells. If appropriately engineered, such antibody–drug conjugates (ADCs) will release the cytotoxic payload upon their internalization, thus causing the selective death of the target population while sparing nonmalignant cells including immune effectors.Citation27 In this context, it appears important to choose a payload that has ICD-inducing properties. Indeed, some ADC payloads such as maytansinoids and auristatins, which are microtubule inhibitors, and pyrrolobenzodiazepines, which are DNA-alkylating agents as well as the anthracycline derivate T-PNU have been described to induce ICD.Citation28–30 Moreover, recently, the FDA approved belantamab mafodotin, an ADC that targets B-cell maturation antigen (BCMA) on multiple myeloma cells using monomethyl auristatin F (MMAF) as a payload, based on its capacity to trigger ICD.Citation31,Citation32
Here, we characterized TWEAKR-KSPi-ADC1 and -ADC2 that consist of an antibody specific for fibroblast growth factor-inducible 14 (Fn14, best known as the receptor for tumor necrosis factor-like weak inducer of apoptosis, TWEAKR/ TNFRSF12A)Citation33 and a small molecule inhibitor of kinesin family member 11 (KIF11, kinesin-5, BimC, Eg5, best known as KSP) and just differ in the way how they are linked to the antibody. Targeting of TWEAKR for cancer therapy either by anti-TWEAKR antibodiesCitation34 or anti-TWEAKR immunoconjugatesCitation33 has been explored in preclinical studies.Citation35,Citation36 KSP inhibitors induce monoaster formationCitation37 and several distinct chemical classes of KSP inhibitors have entered clinical trials.Citation38,Citation39 However, side effects such a neutropenia and mucositis most probably precluded escalation of these pure small molecules to efficacious doses in the clinical trials in monotherapy.
Several TWEAKR-KSPi-ADCs have been built on the rationale that TWEAKR is overexpressed on multiple human cancers including glioblastomaCitation40 as well as breast,Citation41,Citation42 lung,Citation43 ovarian,Citation44 endometrial,Citation45 and colorectal cancerCitation46 (https://www.proteinatlas.org) and KSPi may specifically inhibit proliferating as well as hyperploid cells.Citation47
We here assessed the immunological effects of TWEAKR-KSPi-ADCs in suitable mouse models and characterized the induction of immunogenic cell death by a small molecule KSPi in vitro.
Results and discussion
Design of TWEAKR-KSPi-ADCs
We previously explored a pyrrole-based, potent subclass KSPi as a novel payload class in ADCsCitation35 and have shown strong efficacy of KSPi-ADCs addressing different targets with both, cleavable and non-cleavable linkers.Citation35,Citation36,Citation48 From a variety of ADCs with KSPi payloads, we selected the TWEAKR-KSPi-ADC1 shown in with the agonistic, mouse-cross-reactive TWEAKR antibody BAY-356,Citation49 previously described as ADC 6c*Citation36 to investigate potential ICD properties. Furthermore, we here also investigated TWEAKR-KSPi-ADC2 shown in , differing from ADC1 solely in the linkage to the antibody. Upon legumain-mediated cleavage in the lysosomal compartment, the same active metabolite () is formed.Citation36 This metabolite had been optimized for low cellular permeability to achieve an accumulation inside the tumor cells upon release from the ADC. Due to its poor membrane permeability which hampers cell-based in vitro studies, we selected a closely related, cell-permeable small molecule KSPi for investigation of ICD in in vitro studies (). TWEAKR-KSPi-ADC1 and ADC2 have been shown to be equipotent in inducing cell death in cell culture models, namely, human colon carcinoma LoVo, human pancreatic cancer BxPC-3 and human lung cancer NCI-H292 cells (Supplemental Figure S1A-C, Supplemental Table S1).
Figure 1. Scheme of TWEAKR-KSPi-ADCs, its metabolite and the small molecule KSPi.
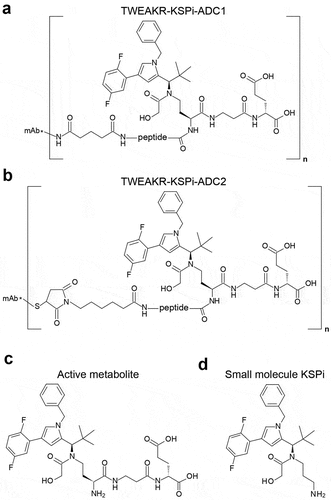
Immune cell-dependent effects of TWEAKR-KSPi-ADC
As it has been shown by us and others that signaling via an agonistic anti-TWEAKR antibody can induce recruitment of immune effector cells and immune-modulation via secretion of cytokine such as IL-8,Citation49,Citation50 anti-tumor efficacy studies were performed in immunocompetent mice bearing CT26 tumors in order to analyze at which dose agonistic effects of the anti-TWEAKR antibody BAY-356 can be observed. These studies demonstrated an efficacy of the unconjugated TWEAKR antibody at 10 mg/kg biweekly but not at 5 mg/kg biweekly (data not shown). To exclude anti-tumor effects based on the agonistic activity of the anti-TWEAKR antibody, a maximum dose of 5 mg/kg biweekly was chosen for investigating the anti-tumor activity of TWEAKR-KSPi-ADC1 in mice bearing CT26 tumors. In a second model, using MC38 tumor bearing immunocompetent mice, the anti-TWEAKR antibody BAY-356 did not show any anti-tumor activity at 10 mg/kg biweekly (data not shown). Regardless of this, also for the MC38 model, a maximum dose of 5 mg/kg biweekly of the TWEAKR-KSPi-ADC1 was chosen.
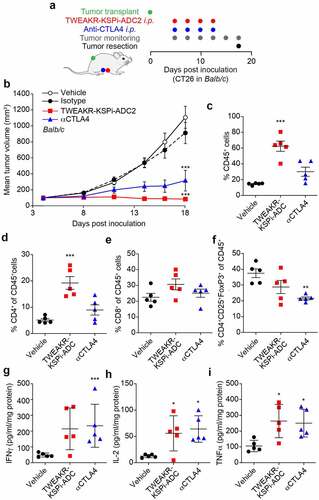
The immunomodulatory effects of the TWEAKR-KSPi-ADC1 and TWEAKR-KSPi-ADC2 were investigated in immunocompetent BALB/c or C57Bl/6 mice bearing subcutaneous TWEAKR positive CT26 or MC38 colon cancers as well as TWEAKR positive LLC lung cancer (; Supplemental Figure S2A,B and S3A), respectively, and in immunodeficient NOD.Cg-Prkdcscid/J (best known as NOD-SCID) mice bearing the same tumor. After establishment of palpable tumors, the mice received biweekly intraperitoneal (i.p.) injections of vehicle, TWEAKR-KSPi-ADC or a monoclonal antibody (mAb) blocking CTLA-4.
Figure 2. TWEAKR-KSPi-ADC controls tumor growth in an immune-dependent fashion in TWEAKR expressing CT26.
At a dose level of 5 mg/kg biweekly, the TWEAKR-KSPi-ADC2 was equally efficacious in controlling tumor growth in immunocompetent and immunodeficient mice bearing MC38 tumors (Supplemental Figure S2C-E; ), demonstrating the cytotoxic component of the ADC activity. As expected, CTLA-4 blockade was not efficacious in immunodeficient mice (Supplemental Figure S2E).
Table 1. In vivo treatment schedule and outcome
However, when reducing the dose of the TWEAKR-KSPi-ADC1 to 2.5 mg/kg applied as a single dose, TWEAKR-KSPi-ADC1 and CTLA-4 blockade were equally efficacious in controlling tumor growth in immunocompetent mice (, ), but at this low single dose of 2.5 mg/kg TWEAKR-KSPi-ADC1 did not show any anti-tumor activity in immunodeficient mice (). In conclusion, it appears that the antineoplastic effect of TWEAKR-KSPi-ADC1 induces ICD in vivo and partly relies on the immune system.
KSPi induces immunogenic cell death
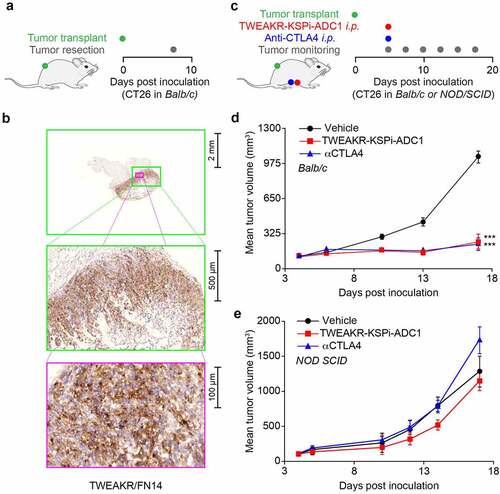
Since the active metabolite () released from the TWEAKR-KSPi-ADC1 and -ADC2 () is not cell-permeable, we investigated a cell-permeable variant, the small molecule KSP inhibitor () for induction of ICD. We took advantage of a predictive artificial intelligence tool to calculate ICD scores for each compound from the NCI60 collection and to compare their distribution with the one of the small molecule KSPi. In this approach, the small molecule KSPi had a predictive ICD score similar to the one of the prototype ICD inducer MTX () spurring our interest in further characterizing its potential in vitro. We thus validated the prediction by means of a phenotypic in vitro screening platform that combines the use of biosensors for the detection of ICD hallmarks with a fluorescence-based data acquisition operating in a high-throughput format.Citation19 The biosensors that we used included quinacrine, a small molecule that accesses the intracellular space and interacts with ATP in an acidic environment (mostly lysosomes) to emit a green fluorescence, meaning that the decrease of the intracellular fluorescent signal can be interpreted as a surrogate marker of ATP release (). We also stably transfected cells with a green fluorescent protein (GFP) under the control of the type-I interferon-induced GTP-binding protein MX1, meaning that such cells constitute biosensors of type-I interferon responses (). Alternatively, cancer cells were engineered to express an HMGB1-GFP fusion protein, allowing to monitor the release of HMGB1 from the nucleus (). Finally, cells were equipped with a CALR-GFP fusion protein to follow the translocation of CALR from the ER to the cell periphery (). These biosensor experiments were calibrated in a way that mitoxantrone (MTX), an established ICD inducer, would induce all hallmarks of ICD (). Of note, we found that the small molecule KSPi was as efficient as MTX in inducing the four ICD hallmarks in a time- and dose-dependent fashion in HT29 colorectal adenocarcinoma biosensor cells (, Supplemental Fig. S4) and two additional cell lines different in origin, namely, NCI-H929 myeloma and HCC70 mammary carcinoma (Supplemental Fig. S5–7). In conclusion, the small molecule KSPi can efficiently induce ICD.
Figure 3. Prediction of the potential of KSPi to induce immunogenic cell death.
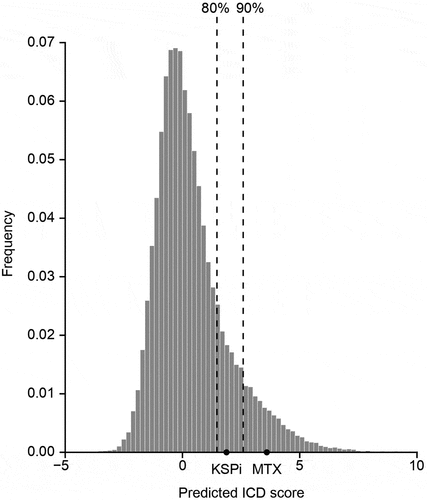
Figure 4. Biosensor cells for the immunogenic cell death fingerprinting (a-c) Human colorectal adenocarcinomas HT29 cells were either stained with quinacrine to assess ATP release, or were genetically modified to express CALR-GFP as surrogate marker for CALR exposure, HMGB1-GFP to measure nuclear HMGB1 exodus or GFP expression under the control of the type-I interferon-induced GTP-binding protein MX1 promoter to measure type-I interferon responses in a high throughput fashion. A schematic representation is depicted in (a) and representative images of HT29 cells untreated (CTRL) or treated with the KSPi or mitoxantrone are shown in (b). Size bar equals 10 µm. (c) To assess ICD fingerprints, HT29 cells were treated with the indicated concentrations of KSPi. Mitoxantrone and recombinant IFNα at the indicated concentrations were used as positive controls. The data is plotted together with solvent controls (vehicle) in form of a heatmap. White boxes indicate: “data not available”.
Figure 5. TWEAKR-KSPi-ADC triggers anticancer immune responses.
TWEAKR-KSPi-ADCs trigger an anticancer immune response
In the final step of our analysis, we compared the local immune effects of TWEAKR-KSPi-ADC2 (B) and CTLA-4 blockade on CT26 tumors evolving on immunocompetent hosts (; ). While TWEAKR-KSPi-ADC2 was more efficacious than CTLA-4-targeting immunotherapy in inducing an increase in tumor-infiltrating CD45+ leukocytes () and more specifically CD4+ and CD8+ T lymphocytes (), while only a minor effect on FoxP3+ regulatory T cells was observed (). Moreover, TWEAKR-KSPi-ADC2 and CTLA-4 blockade were equally potent in enhancing the local production of interferon-γ (), interleukin-2 (), and tumor necrosis factor-α (). Altogether, it appears that TWEAKR-KSPi-ADC2 mobilizes T cell effectors against the tumors.
We were interested in understanding the anti-tumor activity of TWEAKR-targeted ADCs with the KSP inhibitor as payload and performed anti-tumor activity experiments in immunocompetent mouse models. For the in vivo studies in immunocompetent mouse models, we used the anti-TWEAKR antibody BAY-356 cross-reactive with murine TWEAKR. The naked antibody BAY-356 had no anti-tumor activity at doses and in models used in this study. TWEAKR-KSPi-ADC1 applied as single dose of 2.5 mg/kg was equally efficacious to CTLA-4 blockade in controlling tumor growth in immunocompetent mice. In contrast, a single dose of 2.5 mg/kg TWEAKR-KSPi-ADC1 did not show any anti-tumor activity in immunodeficient mice. In conclusion, it appears that at least part of the antitumor efficacy of TWEAKR-KSPi-ADC1 relies on a contribution of the immune system. This was further supported by studies using closely related TWEAKR-KSPi-ADC2. This ADC showed monotherapy efficacy in immunocompetent mice and induced an increase in tumor-infiltrating CD45+ leukocytes as well as an increase of the pro-inflammatory cytokines IFNγ, IL-2, and TNFα. In summary, the KSPi payload for which induction of ICD has been demonstrated may contribute to the induction of therapy-relevant anti-tumor activity of TWEAKR-KSPi-ADCs.
The results provided in this paper indicate that the KSP inhibitor payloads of specific ADCs designed to target various TWEAKR-overexpressing cancers, may act by inducing immunogenic cell death and enhancing T cell infiltration and cytokine secretion in addition to their cytotoxic mode of action. To our knowledge, this is the first time that a KSP inhibitor was investigated in the context of ICD. Besides the induction of ICD, the KSPi-induced apoptosis of KSPi-ADCs may enhance the release of tumor-associated antigens such further potentiating an adaptive immune response.Citation48
These results have been obtained on human cell lines as well as in mouse models, meaning that they require future confirmation in clinical trials. Based on preclinical results, we firmly recommend that immune-monitoring should be incorporated into the trial design of KSPi-ADCs as one of the secondary endpoints. Indeed, signs of a specific immune response may be considered as surrogate markers of therapeutic efficacy and may guide toward the design of therapeutic schedules combining TWEAKR-KSPi-ADCs with established immune checkpoint inhibitors targeting CTLA-4 or PD-1/PD-L1. Several recent preclinical and clinical studies point to the possibility of advantageously combining ICD inducers with such checkpoint blockers.Citation52,Citation53
The pro-ICD activity of the payload of the TWEAKR-KSPi-ADCs requires future mechanistic exploration. TWEAKR is a member of the TNF receptor family, and prior work supports the idea that direct activation of the extrinsic pathway of apoptosis by the activation of such receptors is sufficient to stimulate caspase-8-dependent ICD.Citation54,Citation55 Ligation of TWEAKR by its ligand TWEAK can activate this pathway, and some TWEAKR-specific antibodies have similar effects,Citation56–58 spurring the launch of clinical trials using anti-TWEAKR antibodies without a payload.Citation46,Citation50 KSPi acts as a mitotic inhibitor and as such might perturb cellular physiology in a similar fashion as do microtubule inhibitors.Citation47,Citation59 However, this conjecture needs further exploration. Altogether, efficient ICD induction by TWEAKR-KSPi-ADCs might involve the antibody moiety, the payload or the combination of both. Irrespective of this incognita, it appears obvious that TWEAKR-KSPi-ADCs deserve further clinical characterization.
Materials and methods
Cell culture
Mitoxantrone was purchased from Sigma-Aldrich (St. Louis, MO, USA). Culture media were from Gibco-Invitrogen (Carlsbad, CA, USA). Human colon carcinoma HT-29, human multiple myeloma NCI-H929, human breast cancer HCC70, human colon carcinoma LoVo, human pancreatic cancer BxPC-3 and human lung cancer NCI-H292 and murine colon carcinoma CT26 cells were obtained from the American Type Culture Collection (ATCC), and murine colon carcinoma MC38 cells were obtained from NCI. Cell lines were cultured under standard cell culture conditions (5% CO2, 37°C) in a water saturated atmosphere in a cell culture incubator (HeraCell, Heraeus, Germany). HT-29 were cultured in McCoy’s 5A media supplemented with 10% FCS, Lovo were cultured in F-12 K media supplement with 10% FCS and BxPC-3, NCI-H292, HCC70, and NCI-H929 as well as murine colon carcinoma CT26 and MC38 cells were cultured in RPMI 1640 GlutaMax supplemented with 10% FCS. Media for culture of LoVo, NCI-H292, and NCI-H929 were supplemented with 50 µM beta-mercaptoethanol.
Generation of recombinant reporter cell lines
Mycoplasma-free human colon carcinoma HT-29, human breast carcinoma HCC70, and human multiple myeloma NCI-H929 cells obtained from the ATCC were stably transfected with cDNA expression vectors that code for CALR-green fluorescent protein (GFP) or HMGB1-GFP or GFP under the control of the promoter of the type-I interferon response gene MX1, respectively. Subsequently, stable expressing cells were selected by means of appropriate selection antibiotics and clones were obtained by single cell sorting using a FACS DIVA (Becton Dickinson). Monoclonal reporter cell lines were propagated in the presence of respective selection antibiotics.
Automated image acquisition and analysis
One day prior to the experiment, 5 × 103 cancer cells were seeded in the tissue culture treated 96-well µClear imaging plates (Greiner BioOne) and incubated under standard tissue culture conditions at 37°C 5% CO2 in a water saturated atmosphere in a cell culture incubator (HeraCell). The following day, the cells were treated with the indicated compounds. The treatment duration for each cell line was adapted to the course of cell death. Thus, after incubation for 8, 16, 32, and 48 h for NCI-H929; 16, 32, 72, and 96 h for HCC70; and 8, 16, 32, and 72 h for HT-29, respectively, the cells were fixed with 3.7% formaldehyde solution containing 2 µg/ml Hoechst 33342 for 20 min at RT. The fixative was changed to PBS and the plates were subjected to automated image analysis. MX1 expression in NCI-H929 and HCC70 in response to type-I interferon was detected by immunofluorescence utilizing an MX1-specific antibody (Origene; TA308496) and automated image analysis. For the detection of ATP containing vesicles, the cells were labeled after 4, 8, 16, or 32 hours of incubation with quinacrine. In short, cells were labeled with 5 µm quinacrine and 2 µg/ml Hoechst 33342 in Krebs-Ringer solution (125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0,7 mM KH2PO4, 2 mM CaCl2, 6 mM glucose, and 25 mM Hepes, pH 7.4) for 30 minutes at 37°C. Thereafter, cells were rinsed with Krebs-Ringer and living cells were microscopically examined. Automated fluorescence microscopy was conducted with a robot-assisted Molecular Devices IXM XL BioImager (Molecular Devices, Sunnyvale, CA, USA) equipped with Sola light sources (Lumencor, Beaverton, OR, USA), adequate excitation and emission filters (Semrock, Rochester, NY, USA) and a 16-bit monochromes sCMOS PCO.edge 5.5 camera (PCO Kelheim, Germany) and a 20× PlanAPO objective (Nikon, Tokyo, Japan) was used to acquire at least six fields, followed by image processing with the custom module editor within the MetaXpress software (Molecular Devices). Dependent on the utilized biosensor cell line, the primary region of interest (ROI) was defined by a polygon mask around the nucleus allowing for the enumeration of cells, the detection of morphological alterations of the nucleus and nuclear fluorescence intensity. Cellular debris was excluded from the analysis and secondary cytoplasmic ROIs were used for the quantification of CALR-GFP or quinacrine containing vesicles. For the latter, the images were segmented and analyzed for GFP granularity by comparing the standard deviation of the mean fluorescence intensity of groups of adjacent pixels (coefficient of variation) within the cytoplasm of each cell to the mean fluorescence intensity in the same ROI using the MetaXpress software (Molecular Devices).
In vivo treatment in mice
Murine colon carcinoma cell lines CT26 and MC38 cells expressing the TWEAK receptor (TWEAKR) were subcutaneously inoculated in the flank of immunocompetent female Balb/c (for CT26) or C57Bl/6 (for MC38) mice provided by Envigo Netherlands or Charles River, Germany, respectively. For comparison with immunodeficient mice, female NOD-SCID mice provided by Charles River, Germany were used. First, 5 × 105 cells were detached from the cell culture flasks, centrifuged and re-suspended in a mix of medium and Matrigel (1:1). This cell suspension was then injected subcutaneously and tumors established within 3–5 days. Treatment started at a mean tumor volume of 100 mm3 by intraperitoneal injection of the ADCs or antibodies formulated in PBS in a volume of 5 ml/kg. Each group consisted of 8–10 mice. The mice were treated either with a single dose or twice per week and tumor area was measured by means of a caliper (length, width) three times per week followed by calculation of the tumor volume (Tvol = 0.5 x length x width2). T/Cvol served as parameter describing the ratio of mean tumor volumes of treated mice compared to the volume of vehicle control treated mice. All animal studies were performed in the facilities of Bayer AG, Berlin, Germany and conducted according to the Animal Welfare Guidelines under the approved regulation A0089/17.
Ex vivo immune infiltrate analysis
At the end of the study, dissected tumors were freshly dissociated using a gentleMACS Octo instrument (Miltenyi) and single-cell suspensions were investigated for immune-antigen expression by flow cytometry (FACS Canto II, Becton Dickinson) after Fc-blocking. For the antibody Set #1, cells were permeabilized to investigate intracellular FoxP3.
Additional tumor samples (used either as fresh tissue or snap-frozen) were analyzed for cytokines or chemokines by a sandwich ELISA on the Mesoscale (MSD) platform. The following murine cytokines and chemokines were investigated: IL-1β, IL-12p70, IFN-γ, IL-2, IP-10, IL-17A, VEGF, MCP-1, IL-33, and TNF-α (mouse U-Plex Biomarker Group 1 Assay Kit).
Anti-TWEAKR IHC of murine colon cancer models CT26 and MC38
Cryosections of CT26 and MC38 murine colon tumors from vehicle control groups were subjected to acetone fixation, dried, washed with TBS-T wash buffer (tris buffered saline buffer from ProTaqs (biocyc GmbH &Co. KG, Luckenwalde, Germany) with 0.05% Tween-80), followed by peroxidase block (#S2023, Dako), protein block (#Y0909, Dako) and incubated for 65 min with recombinant rabbit anti-TWEAKR monoclonal antibody (EPR3179, #ab109365, abcam) or rabbit immunoglobulin fraction (solid phase absorbed) as control (#X0936, rabbit IgG, Dako) at 0,5 µg/ml. After TBS-T wash, the sections were incubated with horseradish peroxidase-coupled rabbit EnVision (#K4011, Dako) for 35 min and stained for 10 min with the DAB peroxidase detection kit (#K3953, Dako) and counterstained with hematoxylin (#53275, Sigma). After dehydration with increasing ethanol concentrations, isopropanol, and xylol, the sections were mounted using Cytoseal Xyl medium (Thermo Scientific #8312-4).
Statistical procedures
Unless otherwise specified, in vitro experiments were performed in quadruplicate instances. Data were analyzed with the freely available software R (https://www.r-project.org) or GraphPad PRISM. Significances were calculated using a Welch’s student t-test, followed by p values adjustment using Bonferroni correction. Thresholds for each assay were applied according to the cell-based distribution of positive and negative controls. In vivo experiments were conducted on 8–10 mice per group. Statistics were performed by One-Way ANOVA using log-transformed final tumor volumes followed by a Dunnett’s multiple comparisons test. P-values equal *0,05; **0,01; ***0,001.
Abbreviations
Supplemental Material
Download Zip (1.1 MB)Acknowledgments
GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancellerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM). We are grateful for the help of Anna-Maria DiBetta, Beate Koenig and Dirk Wolter for synthesis of ADCs; Henryk Bubik, Sebastian Deitz, Daniel Grab, Simone Greven and Christoph Mahlert for analytics of ADCs; Manuela Brand and Sven Golfier for the TWEAKR IHC, Norman Dittmar, Simone Zolchow and Jessica Moellmann for in vivo and ex vivo studies, and Rukiye Tamm for the visualization of data.
Disclosure statement
GK and OK are cofounders of Samsara Therapeutics. GK received funding from Bayer AG during the conduct of this study. HGL, SB, DM and AS are shareholders of Bayer AG.
Supplementary materials
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Kepp O, Zitvogel L, Kroemer G. Clinical evidence that immunogenic cell death sensitizes to PD-1/PD-L1 blockade. Oncoimmunology. 2019;8(10):e1637188. doi:10.1080/2162402X.2019.1637188.
- Vandenabeele P, Vandecasteele K, Bachert C, Krysko O, Krysko DV. Immunogenic apoptotic cell death and anticancer immunity. Adv Exp Med Biol. 2016;930:133–11. doi:10.1007/978-3-319-39406-0_6.
- Garg AD, Dudek-Peric AM, Romano E, Agostinis P. Immunogenic cell death. Int J Dev Biol. 2015;59(1–3):131–140. doi:10.1387/ijdb.150061pa.
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31(1):51–72. doi:10.1146/annurev-immunol-032712-100008.
- Zhou J, Wang G, Chen Y, Wang H, Hua Y, Cai Z. Immunogenic cell death in cancer therapy: present and emerging inducers. J Cell Mol Med. 2019;23(8):4854–4865. doi:10.1111/jcmm.14356.
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi:10.1038/nri.2016.107.
- Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8(1):e000337. doi:10.1136/jitc-2019-000337.
- Kasikova L, Hensler M, Truxova I, Skapa P, Laco J, Belicova L, Praznovec I, Vosahlikova S, Halaska MJ, Brtnicky T, et al. Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J Immunother Cancer. 2019;7(1):312. doi:10.1186/s40425-019-0781-z.
- Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, Vosahlikova S, Klouckova J, Church SE, Cremer I, et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood. 2016;128(26):3113–3124. doi:10.1182/blood-2016-08-731737.
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi:10.1038/nm1523.
- Wang Y, et al. Autophagy induction by thiostrepton improves the efficacy of immunogenic chemotherapy. J Immunother Cancer. 2020;8. doi:10.1136/jitc-2019-000462.
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–1178. doi:10.1038/nm.2028.
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–1577. doi:10.1126/science.1208347.
- Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira J-P, Delaloge S, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220(1):47–59. doi:10.1111/j.1600-065X.2007.00573.x.
- Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29(4):482–491. doi:10.1038/onc.2009.356.
- Liu P, Zhao L, Loos F, Iribarren K, Kepp O, Kroemer G. Epigenetic anticancer agents cause HMGB1 release in vivo. Oncoimmunology. 2018;7(6):e1431090. doi:10.1080/2162402X.2018.1431090.
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–1309. doi:10.1038/nm.3708.
- Le Naour J, Liu P, Zhao L, Adjemian S, Sztupinszki Z, Taieb J, Mulot C, Silvin A, Dutertre C-A, Ginhoux F, et al. A TLR3 ligand reestablishes chemotherapeutic responses in the context of FPR1 deficiency. Cancer Discov. 2021;11(2):408–423. doi:10.1158/2159-8290.CD-20-0465.
- Kepp O, Sauvat A, Leduc M, Forveille S, Liu P, Zhao L, Bezu L, Xie W, Zitvogel L, and Kroemer G, . A fluorescent biosensor-based platform for the discovery of immunogenic cancer cell death inducers. Oncoimmunology. 2019;8(8):1606665. doi:10.1080/2162402X.2019.1606665.
- Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, Engblom C, Rickelt S, Yamazaki T, Iribarren K, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. 2019;10(1):1486. doi:10.1038/s41467-019-09415-3.
- Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4(143):143ra199. doi:10.1126/scitranslmed.3003807.
- Xie W, Forveille S, Iribarren K, Sauvat A, Senovilla L, Wang Y, Humeau J, Perez-Lanzon M, Zhou H, Martínez-Leal JF, et al. Lurbinectedin synergizes with immune checkpoint blockade to generate anticancer immunity. Oncoimmunology. 2019;8(11):e1656502. doi:10.1080/2162402X.2019.1656502.
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012;337(6102):1678–1684. doi:10.1126/science.1224922.
- Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1691–1701. doi:10.1084/jem.20050915.
- Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res. 2011;71(14):4809–4820. doi:10.1158/0008-5472.CAN-11-0753.
- Liu P, Zhao L, Kepp O, Kroemer G. Crizotinib - a tyrosine kinase inhibitor that stimulates immunogenic cell death. Oncoimmunology. 2019;8(7):1596652. doi:10.1080/2162402X.2019.1596652.
- Gerber HP, Sapra P, Loganzo F, May C. Combining antibody-drug conjugates and immune-mediated cancer therapy: what to expect? Biochem Pharmacol. 2016;102:1–6. doi:10.1016/j.bcp.2015.12.008.
- Bauzon M, Drake PM, Barfield RM, Cornali BM, Rupniewski I, Rabuka D. Maytansine-bearing antibody-drug conjugates induce in vitro hallmarks of immunogenic cell death selectively in antigen-positive target cells. Oncoimmunology. 2019;8(4):e1565859. doi:10.1080/2162402X.2019.1565859.
- Rios-Doria J, Harper J, Rothstein R, Wetzel L, Chesebrough J, Marrero A, Chen C, Strout P, Mulgrew K, McGlinchey K, et al. Antibody-drug conjugates bearing pyrrolobenzodiazepine or tubulysin payloads are immunomodulatory and synergize with multiple immunotherapies. Cancer Res. 2017;77(10):2686–2698. doi:10.1158/0008-5472.CAN-16-2854.
- D’Amico L, Menzel U, Prummer M, Müller P, Buchi M, Kashyap A, Haessler U, Yermanos A, Gébleux R, Briendl M, et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J Immunother Cancer. 2019;7(1):16. doi:10.1186/s40425-018-0464-1.
- Montes de Oca R, Alavi AS, Vitali N, Bhattacharya S, Blackwell C, Patel K, Seestaller-Wehr L, Kaczynski H, Shi H, Dobrzynski E, et al. Belantamab Mafodotin (GSK2857916) drives immunogenic cell death and immune-mediated antitumor responses in vivo. Mol Cancer Ther. 2021;20(10):1941–1955. doi:10.1158/1535-7163.MCT-21-0035.
- Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, Abdallah A-O, Callander N, Lendvai N, Sborov D, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21(2):207–221. doi:10.1016/S1470-2045(19)30788-0.
- Zhou H, Marks JW, Hittelman WN, Yagita H, Cheung LH, Rosenblum MG, Winkles JA. Development and characterization of a potent immunoconjugate targeting the Fn14 receptor on solid tumor cells. Mol Cancer Ther. 2011;10(7):1276–1288. doi:10.1158/1535-7163.MCT-11-0161.
- Michaelson JS, Kelly R, Yang L, Zhang X, Wortham K, Joseph IBJK. The anti-Fn14 antibody BIIB036 inhibits tumor growth in xenografts and patient derived primary tumor models and enhances efficacy of chemotherapeutic agents in multiple xenograft models. Cancer Biol Ther. 2012;13(9):812–821. doi:10.4161/cbt.20564.
- Lerchen HG, Wittrock S, Stelte‐Ludwig B, Sommer A, Berndt S, Griebenow N, Rebstock A-S, Johannes S, Cancho‐Grande Y, Mahlert C, et al. Antibody-drug conjugates with pyrrole-based KSP inhibitors as the payload class. Angew Chem Int Ed Engl. 2018;57(46):15243–15247. doi:10.1002/anie.201807619.
- Lerchen HG, Stelte-Ludwig B, Sommer A, Berndt S, Rebstock A-S, Johannes S, Mahlert C, Greven S, Dietz L, and Jörißen H, . Tailored linker chemistries for the efficient and selective activation of ADCs with KSPi payloads. Bioconjug Chem. 2020;31(8):1893–1898. doi:10.1021/acs.bioconjchem.0c00357.
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286(5441):971–974. doi:10.1126/science.286.5441.971.
- Myers SM, Collins I. Recent findings and future directions for interpolar mitotic kinesin inhibitors in cancer therapy. Future Med Chem. 2016;8(4):463–489. doi:10.4155/fmc.16.5.
- Wakui H, Yamamoto N, Kitazono S, Mizugaki H, Nakamichi S, Fujiwara Y, Nokihara H, Yamada Y, Suzuki K, Kanda H, et al. A phase 1 and dose-finding study of LY2523355 (litronesib), an Eg5 inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74(1):15–23. doi:10.1007/s00280-014-2467-z.
- Hersh DS, Harder BG, Roos A, Peng S, Heath JE, Legesse T, Kim AJ, Woodworth GF, Tran NL, and Winkles JA, . The TNF receptor family member Fn14 is highly expressed in recurrent glioblastoma and in GBM patient-derived xenografts with acquired temozolomide resistance. Neuro Oncol. 2018;20(10):1321–1330. doi:10.1093/neuonc/noy063.
- Chao DT, Su M, Tanlimco S, Sho M, Choi D, Fox M, Ye S, Hsi ED, Durkin L, Yin J, et al. Expression of TweakR in breast cancer and preclinical activity of enavatuzumab, a humanized anti-TweakR mAb. J Cancer Res Clin Oncol. 2013;139(2):315–325. doi:10.1007/s00432-012-1332-x.
- Zhou H, Mohamedali KA, Gonzalez-Angulo AM, Cao Y, Migliorini M, Cheung LH, LoBello J, Lei X, Qi Y, Hittelman WN, et al. Development of human serine protease-based therapeutics targeting Fn14 and identification of Fn14 as a new target overexpressed in TNBC. Mol Cancer Ther. 2014;13(11):2688–2705. doi:10.1158/1535-7163.MCT-14-0346.
- Whitsett TG, Cheng E, Inge L, Asrani K, Jameson NM, Hostetter G, Weiss GJ, Kingsley CB, Loftus JC, Bremner R, et al. Elevated expression of Fn14 in non-small cell lung cancer correlates with activated EGFR and promotes tumor cell migration and invasion. Am J Pathol. 2012;181(1):111–120. doi:10.1016/j.ajpath.2012.03.026.
- Gu L, Dai L, Cao C, Zhu J, Ding C, Xu H-B, Qiu L, Di W. Functional expression of TWEAK and the receptor Fn14 in human malignant ovarian tumors: possible implication for ovarian tumor intervention. PLoS One. 2013;8(3):e57436. doi:10.1371/journal.pone.0057436.
- Wang D, Fung JN, Tuo Y, Hu L, Chen C. TWEAK/Fn14 promotes apoptosis of human endometrial cancer cells via caspase pathway. Cancer Lett. 2010;294(1):91–100. doi:10.1016/j.canlet.2010.01.027.
- Culp PA, Choi D, Zhang Y, Yin J, Seto P, Ybarra SE, Su M, Sho M, Steinle R, Wong MHL, et al. Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin Cancer Res. 2010;16(2):497–508. doi:10.1158/1078-0432.CCR-09-1929.
- Rello-Varona S, Vitale I, Kepp O, Senovilla L, Jemaá M, Métivier D, Castedo M, Kroemer G. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell Cycle. 2009;8(7):1030–1035. doi:10.4161/cc.8.7.7950.
- Kirchhoff D, Stelte-Ludwig B, Lerchen H-G, Wengner AM, Ahsen OV, Buchmann P, Märsch S, Mahlert C, Greven S, Dietz L, et al. IL3RA-targeting antibody-drug conjugate BAY-943 with a kinesin spindle protein inhibitor payload shows efficacy in preclinical models of hematologic malignancies. Cancers (Basel). 2020;12(11):3464. doi:10.3390/cancers12113464.
- Berndt S, et al. Abstract 1210: preclinical pharmacology and repeated dose toxicity of the novel agonistic TWEAK receptor binding antibody BAY-356. Cancer Res. 2016;76:1210. doi:10.1158/1538-7445.Am2016-1210.
- Ye S, Fox MI, Belmar NA, Sho M, Chao DT, Choi D, Fang Y, Zhao V, Keller SF, Starling GC, et al. Enavatuzumab, a humanized anti-TWEAK receptor monoclonal antibody, exerts antitumor activity through attracting and activating innate immune effector cells. J Immunol Res. 2017;2017:5737159. doi:10.1155/2017/5737159.
- Humeau J, Sauvat A, Cerrato G, Xie W, Loos F, Iannantuoni F, Bezu L, Lévesque S, Paillet J, Pol J, et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol Med. 2020;12(5):e11622. doi:10.15252/emmm.201911622.
- Heath EI, Rosenberg JE. The biology and rationale of targeting nectin-4 in urothelial carcinoma. Nat Rev Urol. 2021;18(2):93–103. doi:10.1038/s41585-020-00394-5.
- Bailly C, Thuru X, Quesnel B. Combined cytotoxic chemotherapy and immunotherapy of cancer: modern times. NAR Cancer. 2020;2(1):zcaa002. doi:10.1093/narcan/zcaa002.
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund A-C, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578–590. doi:10.1038/emboj.2009.1.
- Orzechowska BU, Kukowska-Latallo JF, Coulter AD, Szabo Z, Gamian A, Myc A. Nanoemulsion-based mucosal adjuvant induces apoptosis in human epithelial cells. Vaccine. 2015;33(19):2289–2296. doi:10.1016/j.vaccine.2015.03.002.
- Wilson CA, Browning JL. Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Differ. 2002;9(12):1321–1333. doi:10.1038/sj.cdd.4401107.
- Schneider P, Schwenzer R, Haas E, Mühlenbeck F, Schubert G, Scheurich P, Tschopp J, Wajant H. TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur J Immunol. 1999;29(6):1785–1792. doi:10.1002/(SICI)1521-4141(199906)29:06<1785::AID-IMMU1785>3.0.CO;2-U.
- Nakayama M, Ishidoh K, Kayagaki N, Kojima Y, Yamaguchi N, Nakano H, Kominami E, Okumura K, Yagita H. Multiple pathways of TWEAK-induced cell death. J Immunol. 2002;168(2):734–743. doi:10.4049/jimmunol.168.2.734.
- Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68(9):3269–3276. doi:10.1158/0008-5472.CAN-07-6699.