
ABSTRACT
Myeloid-derived suppressor cells (MDSCs) are a population of immune suppressive cells that are involved in tumor-associated immunosuppression, and dominate tumor progression and metastasis. In this study, we report that the leukocyte immunoglobulin-like receptor subfamily B member 4 (LILRB4, murine ortholog gp49B) orchestrates the polarization of MDSCs to exhibit pro-tumor phenotypes. We found that gp49B deficiency inhibited tumor metastases of cancer cells, and reduced tumor-infiltration of monocytic MDSCs (M-MDSCs) in tumor-bearing mice. Gp49B−/− MDSCs inhibited pro-tumor immune responses, such as activation of Treg cells, promotion of cancer cell migration, and stimulation of tumor angiogenesis. Treatment of wild-type tumor-bearing mice with gp49B−/− M-MDSCs reduced cancer metastasis. Furthermore, gp49B knockout affected plasma exosome composition in terms of increased miR-1 family microRNAs (miRNAs) expression, which correlates with the upregulation of gp49B−/− MDSC-derived anti-tumor miRNAs. Collectively, our findings reveal that LILRB4/gp49B promotes MDSC-mediated tumor metastasis by regulating the M2-polarization of MDSCs and suppressing the secretion of miR-1 family miRNAs, which facilitate tumor migration and invasion.
Abbreviations
CTLA-4: cytotoxic T-lymphocyte-associated protein-4; FBS: fetal bovine serum; G-MDSCs: granulocytic-MDSCs; GP49B: glycoprotein 49B; HE: hematoxylin-eosin; ICI: immune checkpoint inhibitor; ITIM: immunoreceptor tyrosine-based inhibition motif; LILRB4: leukocyte immunoglobulin-like receptor B4; M-CSF: macrophage colony stimulating factor; MDSC: myeloid-derived suppressor cell; M-MDSC: monocytic MDSC; MMP-9: metallopeptidase-9; mAb: monoclonal antibody; PBS: phosphate-buffered saline; PCR: polymerase chain reaction; PD-1: programmed death-1; PD-L1: programmed death ligand-1; PMN-MDSC: polymorphonuclear-MDSC; qRT-PCR: quantitative reverse transcription PCR; TAM: tumor associated macrophage; TME: tumor microenvironment; TMM: trimmed mean of M value; VEGFA: vascular endothelial growth factor A
Introduction
Tumor metastasis, the spread of malignant cells to distant organs, is the main cause of death in cancer patients. The ability of tumor cells to modulate immune response and evade immune recognition is necessary for successful metastasis.Citation1,Citation2 In the tumor environment, myeloid cell-induced immunosuppression that inhibits anti-tumor immune responses is a pivotal requirement for cancer cells to grow and metastasize.Citation3
Myeloid-derived suppressor cells (MDSCs), a population of immune suppressive cells, strongly inhibit anti-tumor immune reactions mediated by T cells and enhance angiogenesis for metastatic formation.Citation4 MDSCs consist of two major subsets in human and mice, monocytic-MDSC (M-MDSC) and polymorphonuclear MDSC (PMN-MDSC), also known as granulocytic MDSC (G-MDSC).Citation5 Both MDSC subsets are found in the bone marrow, spleen, lung, peripheral blood, and tumor tissue.Citation6 MDSCs are capable of polarizing to a classically activated (M1) or an alternatively activated (M2) phenotype. M1 MDSCs exhibit anti-tumor activities by secreting tumor necrosis factor (TNF-α) and nitric oxide (NO); whereas M2-polarized MDSCs suppress effector T cells (Teff) but activate regulatory T cells (Treg) through the expression of interleukin-10 (IL-10), transforming growth factor (TGF-β) and arginase-1 (ARG1).Citation7,Citation8 Several studies have shown that MDSCs infiltrated in various cancers, both in the primary tumor and metastatic sites.Citation9 MDSC-mediated immunosuppression limits the potency of cancer immunotherapy drugs, such as anti-programmed death receptor-1 (PD-1), anti-programmed death-ligand 1 (PD-L1) and anti-cytotoxic T lymphocyte antigen 4 (CTLA4);Citation10,Citation11 therefore, targeting tumor-infiltrating MDSCs is an important issue in cancer therapy. Interestingly, the leukocyte immunoglobulin-like receptor subfamily B member 4 (LILRB4) is expressed on MDSCs and correlates with survival in human lung cancer patients.Citation12
LILRB4, also known as ILT3, CD85k, LIR-5, or gp49B (mouse), is expressed on monocytes, dendritic cells, macrophages, plasma cells, activated T cells, NK cells, and osteoclasts.Citation13,Citation14 LILRB4 contains two extracellular immunoglobulin domains, a transmembrane domain, and three immunoreceptor tyrosine-based inhibitory motifs (ITIMs).Citation15 Upon ligand engagement, the ITIMs of LILRB4 are phosphorylated by tyrosine kinases of the Src family, thereby recruiting phosphotyrosine phosphatases (SHP-1/2) or the inositol-phosphatase SHIP to inhibit the activation of molecules that contribute in cell signaling.Citation16–18 LILRB4 signal attenuates FcγRI-mediated activation and endocytosis/phagocytosis of monocyte by SHP-1-dependent phosphorylation of multiple kinases.Citation19,Citation20 In addition, LILRB4 shows constitutive phosphorylation and association with SHP-1, which down-regulates osteoclast differentiation from monocyte.Citation21 Previous studies reported that activated leukocyte cell adhesion molecule (ALCAM/CD166/MEMD) and apolipoprotein E (APOE) are ligands of LILRB4, which mediate the growth of tumor cells and the development of acute myeloid leukemia, respectively.Citation22,Citation23
Herein, we hypothesized that LILRB4/gp49B may be involved in tumor metastasis, and that LILRB4/gp49B signaling may contribute to MDSC-mediated immunosuppression in the tumor environment. We found that metastases of Lewis lung carcinoma (LLC) and B16F10 melanoma cells were reduced in gp49B-deficient mice. Blocking of gp49B using anti-gp49 mAb also decreased tumor metastases in tumor-bearing mice. The tumor-infiltrated MDSCs from gp49B−/− mice exhibited an anti-tumor phenotype, as shown by decreased secretion of pro-tumor cytokines, reduced immunosuppression of T cells, and decreased angiogenesis. Particularly, gp49B knockout increased the expression of anti-tumor microRNAs (miRNAs), including miR-1a-3p, miR-133a-3p, and miR-206-3p in MDSCs and plasma exosomes. Our results suggest that LILRB4/gp49B facilitates MDSC-mediated tumor metastasis by inducing M-MDSCs to differentiate into tumor-promoting M2 phenotype and inhibiting the expression of anti-tumor miRNAs that inhibit cancer cell migration and invasion.
Materials and methods
Animals
C57BL/6 (B6, WT) mice were purchased from CLEA Japan (Tokyo, Japan). The generation of gp49B−/− mice with B6 background was described previously.Citation24 All animals were bred and conducted in accordance with the animal guidelines of the Laboratory for Animal Resources of the Institute of Development, Aging and Cancer, Tohoku University. All protocols of animal experiments were approved by the Animal Studies Committee of Tohoku University. Male mice between 8 and 10 weeks of age were used.
Cell lines
Murine melanoma cells (B16F10, TKG 0348), Lewis lung cancer cells (LLC, TKG 0153), and human monocytic THP-1 cells (TKG 0267) were obtained from the Cell Resource Center for Biomedical Research of Tohoku University (Sendai, Japan). The LILRB4−/− THP-1 cells were established using CRISPR/Cas9 system as described previously.Citation25 To generate luciferase-expressing LLC (LLC-Luc2) cells, LLC cells were transfected with pcDNA3.1(+)/Luc2 = tdT and selected with geneticin (G418 sulfate, 400 μg/ml) for one month. The stable LLC-Luc2 cells were confirmed by flow cytometry (FACSAriaIII, BD bioscience) and using the luciferase reporter assay (Promega). B16F10 and LLC cell lines were, respectively, maintained in DMEM medium (Sigma-Aldrich) and RPMI-1640 medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Biowest, Nuaillé, France) and 1× antibiotic-antimycotic (ThermoFisher Scientific, Waltham, MA, USA). THP-1 and LILRB4−/− THP-1 cells were maintained in RPMI-1640 medium supplemented with 10% FBS, 1x antibiotic-antimycotic, and 50 μM 2-mercaptoethanol (Sigma-Aldrich). Human vascular endothelial cells (HUVECs, CRL-1730) were cultured in endothelia cell growth medium (PromoCell, Heidelberg, Germany; c-22110).
Plasmids and H1.1-mIgG1 chimeric antibody production
pcDNA3.1(+)/Luc2 = tdT was a gift from Christopher Contag (Addgene plasmid #32904). Plasmid extraction kit (PureLink HiPure Plasmid Midiprep Kit) and Lipofectamine® 2000 for cell transfection were purchased from Invitrogen (Thermo Fisher Scientific).
Regarding H1.1-mIgG1 antibody production, the construction of the heavy chain and the light chain cDNAs of H1.1-chimeric antibody (H1.1γ1-mouse chimera and H1.1κ-mouse chimera) by chimerization of the variable regions of the heavy chain and light chain from Armenian hamster anti-mouse LILRB4/gp49B hybridoma (H1.1) with the constant regions of mouse IgG1 heavy chain and κ light chain separately into the HindIII/NotI site of pUC19/+Not I/ΔLacZ vector was performed by Evec Inc. (Sapporo, Japan). The HindIII/NotI-digested cDNA fragments of H1.1γ1-mouse chimera and H1.1Κ-mouse chimera were inserted into the HindIII/NotI site of pEHX1.1 vector and pELX2.2 vector (TOYOBO, Osaka Japan), respectively. Plasmids pEHX/H1.1mγ1 and pELX/H1.1mκ were digested with BglII/EcoRI and ligated to form a single plasmid pEHX-ELX/H1.1mγ1-mκ. The resultant plasmid was linearized with AseI digestion and used for the transfection of CHO-K1 cells with Lipofectamine® 2000 (Thermo Fischer Scientific). A H1.1-mIgG1 chimeric antibody stable-producing CHO-K1 clone was selected by the limiting dilution method. The H1.1-mIgG1 chimeric antibody was purified from the culture supernatant by using HiTrap™ Protein G HP (Cytiva, Sheffield, UK) according to the manufacture’s protocol.
Adoptive transfer experiment
For bone marrow transplantation (BMT), B6 mice were irradiated with 8.5 Gy total body irradiation (MBR-1520 R-4, Hitachi Power Solutions Co., Ltd.). At 1-day post-irradiation, mice were transplanted with bone marrow cells from WT or gp49B−/− donor mice via tail vein injection (5 × 106 cells/mouse). Following adoptive transfer, mice were treated with gentamycin (1.2 mg/ml) for one month, and the total blood donor reconstitution was evaluated by flow cytometry at four weeks post-transplantation. For MDSC adoptive transfer, splenic MDSCs were isolated from WT or gp49B−/− tumor-bearing mice by flow sorting, and isolated MDSCs (2 × 105 cells/mouse) were then co-injected with LLC cells in a 1:1 ratio into WT mice on day 0. The adoptive transfer of splenic MDSCs was repeated 1 week later.
Mouse model of tumor metastases and therapeutic protocols
Regarding tumor metastases, LLC (1.5 × 106 cells/mouse) and B16F10 (5 × 105 cells/mouse) cells were inoculated into B6 and gp49B−/− mice by intravenous injection. After inoculation, LLC- and B16F10-bearing mice were sacrificed on day 30 and day 21, respectively, and lung and liver tissues from tumor-bearing mice were harvested for analysis of tumor metastasis by hematoxylin–eosin (H&E) staining or counting of surface nodules. For antibody treatment, the B6 mice were inoculated intravenously at a low dose of 5 × 105 LLC-Luc2 cells or 5 × 104 B16F10 cells. At 3 days post-tumor inoculation, mice were treated with anti-PD-1 (RPM1-14, Bio X Cell; 200 μg/mouse), anti-gp49 (H1.1-Amenian hamster IgG from hybridoma cellsCitation26 or H1.1-mIgG1, 200 μg/mouse), or isotype matched control antibody (2A3 (Bio X cell), HTK888 (Biolegend) or MOPC-21 (Bio X Cell); 200 μg/mouse) by intraperitoneal injection six times at 3-day intervals. LLC-Luc2 tumor-bearing mice with antibody treatment were imaged using the IVIS Spectrum In Vivo Imaging System (Perkin Elmer) at three weeks after tumor inoculation, and sacrificed at five weeks after tumor injection for detection of tumor metastases in lung and liver by H&E staining.
Cell isolation
For isolation of MDSCs, splenocytes and bone marrow cells harvested from WT and gp49B−/− mice were fractionated with Percoll (GE Healthcare) density solutions of 40%, 50%, 60%, 70% and 100% as previously described.Citation27 Cells banding at gradient of 50%–60% (monocyte fraction) were isolated for MDSC positive-selection by PE-anti-CD115 and anti-PE microbeads (Miltenyi Biotec). For the adoptive transfer of MDSCs, the monocyte fraction was subjected to isolate CD11b+Gr1 (Ly6G/Ly6C)+ MDSCs with a purity greater than 99% by flow sorting using a FACSAria III (BD Biosciences). For the detection of tumor-infiltrated MDSC, minced tumors were dissociated with mouse tumor dissociation kit (Miltenyi Biotec), while lung tissues were dissociated with mouse lung dissociation kit (Miltenyi Biotec) according to the manufacturer’s instructions.
Treg cell expansion by MDSCs
Splenocytes (2 × 106 cells/well) from OT-II mice were labeled with CFSE (Thermo Fischer Scientific) according to the manufacturer’s instructions, and subsequently co-cultured with MDSCs (5 × 105 cells/well) from tumor-bearing mice in the presence of OVA peptides (1 μg/ml) for five days. The co-cultured cells were stained FITC-conjugated anti-CD4, APC-conjugated anti-CD25, and PE-conjugated anti-Foxp3 antibodies for flow cytometry analysis.
Flow cytometry
For surface marker staining, cells (1 × 106) were washed with 1 × PBS and incubated with fluorochrome-conjugated antibodies in blocking buffer (1% FBS in PBS) for 15 min at 4°C. For intracellular Foxp3 staining, cells (1 × 106) were permeabilized with Foxp3/transcription factor staining buffer set (eBioscience) and then incubated with fluorochrome-conjugated anti-Foxp3 antibody for 30 min at room temperature. After washing with blocking buffer, stained cells were resuspended with PBS and analyzed using a FACSAria III (BD Biosciences).
ELISA
Murine TNF-α, TGF-β, and IL-10 in cell culture supernatants were measured with the ELISA Max standard TNF-α, TGF-β, and IL-10 sets (BioLegend) according to the manufacturer’s protocol. Absorbance was read at 450 nm on a Model 680 microplate reader (Bio-Rad).
Transwell migration assay
LLC cells (1 × 104 cells/well) were seeded in the upper chamber of a 24-well 8-μm-pore transwell plate (Corning, NY, USA) with serum-free RPMI-1640 medium for 24 h. Subsequently, 1-day starved LLC cells were co-cultured with 1.2 × 105 of WT or gp49B−/− bone marrow-derived MDSCs (BM-MDSCs) from tumor-bearing mice plated in the lower chamber for another 72 h. In PC9 cell migration assay, WT or LILRB4−/− THP-1 cells (4 × 105 cells/well) were seeded on a 24-well culture plate for PMA (50 ng/ml) stimulation. At 48 h post-stimulation, the activated THP1 cells were co-cultured with 1 × 104 of starved PC9 cells plated on the upper chamber (8-μm-pore) for 36 h. Non-migrated cells on the upper side of the transwell membrane were removed with a cotton swab, and the migrated cells on the underside of the transwell membrane were stained with 0.5% crystal violet solution. Images of the migrated cells were captured with a Keyence BZ-9000 microscope (Keyence Corporation, Osaka, Japan). Cell number of migrated cells in 20 random fields was analyzed using ImageJ.
Wound healing assay
LLC or PC9 cells (105 cells/well) were plated into a 24-well plate and incubated in RPMI-1640 medium supplemented with 10% FBS for 24 h. Monolayer cells were then incubated in serum-free medium supplemented with 5 μg exosomes for another 24 h. Wounds were created by scratching the cell monolayer with a 1000–μl pipette tip, and non-adherent cells were washed off with medium twice. Cells were then cultured in RPMI-1640 containing 5% FBS for another 18 h and cell images were captured using a microscope Keyence BZ-9000 microscope. Cell migration was determined by the rate of wound closure analyzed using ImageJ.
Exosome isolation
Murine whole blood samples from tumor-bearing mice were collected with 4 mM EDTA, pH 8.0 (Invitrogen Inc., Carlsbad, CA, USA), and subjected to plasma separation by centrifugation at 3500 × g for 5 min at room temperature. Plasma exosomes were isolated using the ExoQuick exosome precipitation solution (SBI, System Biosciences, CA, USA) according to the manufacturer’s protocol. The extracted exosomes were resuspended in 1 × PBS and sterilized by Ultrafree-MC Centrifugal Filter Devices (0.22 μm, Millipore Co.), and stored at – 80°C for subsequent experiments.
Exosomal miRNA extraction and sequencing
Total exosomal RNA (including miRNA) was isolated using the miRNAeasy Mini kit (Qiagen Company, Hilden, Germany) according to the manufacturer’s instructions. The integrity of miRNA was examined on Agilent 2100 Bioanalyzer using the RNA 6000 Pico Kit and the Small RNA Kit (Agilent Technologies Inc., Santa Clara, CA, USA) and subjected to library preparation using QIAseq miRNA Library Kit (Qiagen Company, Hilden, Germany). The quality control analysis of the library was assessed using the High Sensitivity DNA kit (Agilent Technologies Inc., Santa Clara, CA, USA). The miRNA library was sequenced using an Illumina NextSeq-500 Sequencing System (Illumina, San Diego, CA, USA).
miRNA-Seq analysis
Approximately 14–20 million reads were obtained per library. RNA-seq raw reads were quality checked by FastQC (version 0.11.9), and were then pre-processed by filtering out sequencing adapters, short-fragment reads (<10 nt), and other low-quality reads. Unique Molecular Identifier analysis was performed on GeneGlobe data analysis center (Qiagen), and the processed reads was mapped to the mouse [Mus musculus, GRCm38 (mm10) assembly] reference genome. The mapped reads were normalized using Strand NGS with the Trimmed Mean of M value (TMM) method. Normalized reads containing miRNA sequences were annotated compliantly with existing sequences in the miRBase database (http://www.mirbase.org/). The fold-change was calculated from the normalized reads, and an MA plot showing base-2 log fold-change (Log2[gp49B−/−]-Log2[WT]) along the y-axis and normalized average expression of each miRNA ({Log2[gp49B−/−] +Log2[WT]}/2) along the x-axis was generated using GraphPad Prism version 10 software. Additionally, base-2 log fold-change data from two independent miRNA sequencing were analyzed with a comparison analysis using Ingenuity Pathway Analysis tools (Qiagen).
Quantitative reverse transcription PCR (qRT-PCR)
For mRNA expression analysis, total RNA was isolated using the RNeasy Micro kit (Qiagen, Hilden, Germany), and was then reverse transcribed into cDNA using the ReverTra Ace qPCR RT Master Mix with a gDNA remover kit (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. The cDNA was then used as the template for the qPCR reaction, which was performed using the CFX Connect Real-Time PCR detection system (Bio-Rad, Hercules, CA, USA) with the SYBR Green Real-time PCR Master Mix Plus kit (Toyobo). For miRNA expression analysis, total miRNA was isolated using the miRNAeasy Mini kit (Qiagen), and was then reverse transcribed into cDNA using the Mir-X miRNA First-Strand Synthesis Kit (TaKaRa Holdings Inc., Kyoto, Japan) according to the manufacturer’s instructions. The qPCR was performed with the reagent of Mir-X miRNA qRT-PCR TB Green Kit (TaKaRa). All samples were analyzed in triplicate, and data were analyzed using the 2−ΔΔCt method. β-actin, and U6 snRNA were used as internal reference genes for mRNA and miRNA analysis, respectively. The primer sequences of the corresponding mRNAs and miRNAs are shown in Supplementary Table 1.
Analysis of publicly available genomic data
To evaluate the correlation between LILRB4 and PD-1 in human lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), genomic data obtained from The Cancer Genome Atlas (TCGA)-LUAD and TCGA-LUSC database were analyzed using USCS Xena (http://xena.ucsc.edu/).
Statistical analysis
Statistical analyses were performed using GraphPad Prism® 9 (Version 9.0; GraphPad Software, San Diego, CA, USA). Data are presented as means ± SEM. Data were compared for statistical differences by Pearson’s correlation coefficients with two-tailed test, two-tailed unpaired Student’s t-test, as stated in the figure legends. P < .05 was considered statistically significant.
Data availability
Raw and processed miRNA sequencing data have been deposited into the GEO online database (accession number GSE185118).
Results
Genetic deficiency of gp49B impairs tumor metastases in tumor-bearing mice
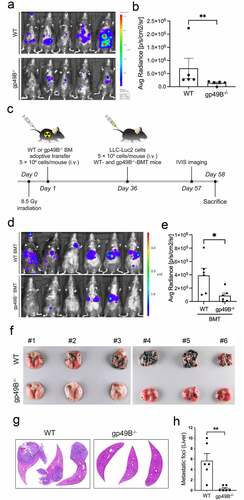
To determine whether LILRB4/gp49B contributes to tumor metastases, wild-type (WT) B6 mice and gp49B−/− mice were intravenously injected with luciferase expressing-Lewis lung carcinoma cells (LLC-Luc2) and examined for the metastases 21 days later. We found that the metastatic ability of LLC-Luc2 was significantly inhibited in gp49B−/− mice (, and ). In addition, the LLC-mediated lung and liver metastases were reduced in gp49B−/− tumor-bearing mice compared to those in WT tumor-bearing mice (Supplemental Figure 1). With adoptive bone marrow transplantation (BMT), the bone marrow cells of WT mice were reconstituted with that of gp49B−/− mice to confirm the gp49B-mediated tumor metastasis (). In the comparison of WT BMT and gp49B−/− BMT mice, LLC-Luc2 tumor growth monitored by a noninvasive IVIS bio-image system was reduced in gp49B−/− BMT-tumor-bearing mice (, and ). We also inspected whether gp49B knockout could reduce the metastatic ability of melanoma B16F10. The numbers of pulmonary metastatic nodule and liver metastatic foci in B16F10-injected gp49B−/− mice were significantly decreased compared to those in B16F10-injected WT mice (, -). Together, the above results indicated that LILRB4/gp49B is involved in the metastases of tumor cells.
Figure 1. Gp49B genetic deficiency decreases tumor metastases in tumor-bearing mice. (a) WT and gp49B−/− mice (n = 5 mice per group) were intravenously injected with LLC-Luc2 cells (5 × 105 cells per mouse) for 21 days. In vivo bioluminescence imaging of LLC-Luc2 tumor metastases in mice was detected using the IVIS Spectrum in vivo imaging system. (b) Graph showing bioluminescence measurements of IVIS image mice as average radiance (photons/s/cm2/steradian) corresponding to (a). (c) Schematic of adoptive transfer and tumor inoculation. Bone marrows cells from WT or gp49B−/− donor mice were adoptively transferred into irradiated B6 mice (n = 6 mice per group) by tail vein injection. After 36 days, mice were inoculated with LLC-Luc2 cells and monitored for tumor metastasis. (d) In vivo bioluminescence image of tumor metastasis in WT (above) and gp49B−/− (below) BMT mice (n = 6 mice per group) challenged with LLC-Luc2 cells for 21 days. (e) Quantification of average radiance (photons/s/cm2/steradian) from IVIS imaged mice as shown in (d) (f-h) Tumor nodules formed on lung surface (f) and H&E stained liver sections (g and h) from WT and gp49B−/− mice (n = 6 mice per group) challenged with B16F10 cells for 21 days. All numerical data are represented as mean ± SEM. *p < .05, **p < .005 calculated by unpaired t-test.
Gp49B blockade inhibits tumor metastases in tumor-bearing mice
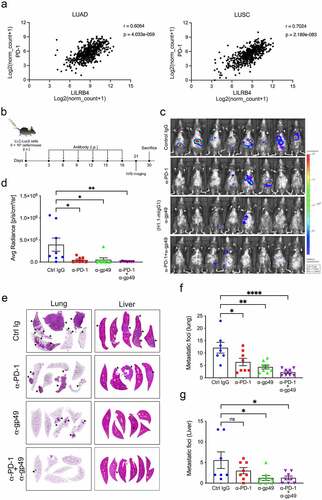
An analysis of RNA expression data from The Cancer Genome Atlas (TGCA) database via the Xena platformCitation28 revealed a strong correlation between LILRB4 and PD-1 mRNA expression in LUAD and LUSC samples (r = 0.6064 and 0.7024, respectively) (). Since deficiency of gp49B reduced the tumor metastasis, we then assessed the effect of gp49B blockade on tumor metastases using anti-gp49 (H1.1) monoclonal antibody (mAb) or a combination of anti-gp49 with anti-PD-1 (RMP1-14) mAb. LLC-Luc2 cells-challenged B6 mice were intraperitoneally injected with isotype control IgG, anti-PD-1 mAb, anti-gp49 mAb, or a combination of anti-PD-1 mAb and anti-gp49 mAb six times (). After antibody treatment, the metastatic growth by bioluminescence image was decreased in LLC-Luc2 tumor-bearing mice treated with anti-gp49 mAb or anti-PD-1 mAb (p = .0124 and .011, respectively) (, and ). In addition, a combination treatment of anti-PD-1 mAb and anti-gp49 mAb showed a modest synergistic effect on tumor metastatic growth in LLC-Luc2 tumor-bearing mice (p = .046) (, and ). We also observed the metastatic foci in mAb-treated tumor-bearing mice by histological analysis. Blockade of gp49, as well as blockade of both PD-1 and gp49, indeed impaired tumor metastases in the lung and liver of LLC-Luc2 tumor-bearing mice (, -). Furthermore, B6 mice were inoculated with B16F10 cells for monitoring tumor metastasis, and then, the efficacy of anti-gp49 and anti-PD-1 mAbs on tumor metastasis was evaluated. (Supplemental Figure 2). The pulmonary tumor nodule formation was significantly decreased in anti-gp49 mAb-treated tumor-bearing mice, similar to that in anti-PD-1-treated and combination-treated B16F10 tumor-bearing mice (Supplemental Figure 2B, left), and anti-gp49 mAb and/or anti-PD-1 mAb treatment also showed a trend toward reducing the liver metastasis in B16F10-bearing mice (Supplemental Figure 2B, right). These results suggested that LILRB4 blockade inhibits tumor metastasis and that LILRB4 may serve as a potential therapeutic target for combination therapy in cancer diseases.
Figure 2. Blockade of gp49B impaired tumor metastases in tumor-bearing mice. (a) Correlation between LILRB4 and PD-1 expression observed in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) tissues from the TCGA database. (b) Schematic representation of antibody treatment schedule. B6 mice were intravenously injected with LLC-Luc2 cells (n = 8 per each group) on day 0, and then treated with the indicated antibodies (200 μg per mouse) six times at 3-days intervals. (c) In vivo bioluminescence imaging of LLC-Luc2 tumor metastases in mice on day 21 post-tumor inoculation. (d) Bioluminescence measurements expressed as average radiance (photons/s/cm2/steradian) corresponding to (c). (e) Representative graph of tumor metastatic sites in the lung (left) and liver (right) of antibody treated LLC-Luc2 tumor-bearing mice on day 30 after tumor inoculation. (f and g) Quantification of metastatic sites in lung (f) and liver (g) from LLC tumor-bearing mice with antibody treatment corresponding to (e). Statistical results are represented as mean ± SEM. P values were calculated by Ordinary one-way ANOVA with Dunnett’s multiple comparisons test. *p < .05, **p < .005, ****p < .0001.
Knockout of gp49B impairs the infiltration of MDSCs in the tumor environment
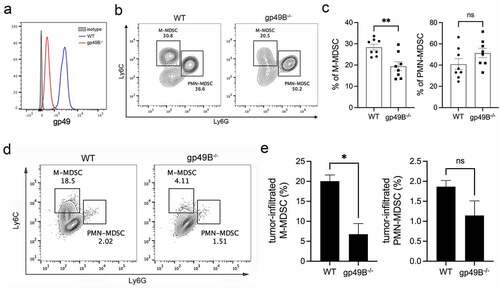
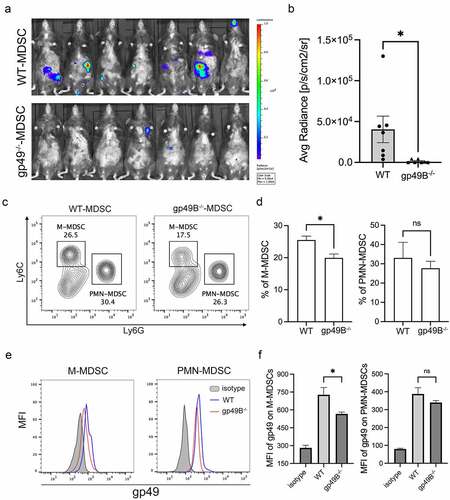
Since MDSCs play an important role in tumor-associated immunosuppression, we considered whether gp49B regulates MDSC infiltration in the tumor environment. To this end, we confirmed that gp49B is expressed on MDSCs in WT mice, but not in gp49B−/− mice (). To measure the population of tumor-infiltrated MDSCs, lung tissues from WT and gp49B−/− LLC-Luc2 tumor-bearing mice were dissociated for flow cytometry analysis. The result showed that lung-infiltrated monocytic (M)-MDSCs were reduced in gp49B−/− tumor-bearing mice, while lung-infiltrated polymorphonuclear (PMN)-MDSCs were comparable in WT and gp49B−/− tumor-bearing mice (, and ). With LLC subcutaneous inoculation, the population of tumor-infiltrating M-MDSCs were reduced in gp49B−/− tumor-bearing mice (, and ). Furthermore, the expression of MHC-II (M1 marker) and CD206 (M2 marker) on tumor-associated M-MDSCs was evaluated by flowcytometry. The result revealed that MHC-II expression was comparable in WT and gp49B−/− M-MDSCs, while CD206 expression was decreased on gp49B−/− M-MDSCs, indicating that M-MDSCs from gp49B−/− tumor-bearing mice exhibited the M1-phenotype (Supplemental Figure 3A). These results suggested that LILRB4/gp49B regulates M-MDSC differentiation and infiltration in the tumor environment.
Figure 3. Lung- and tumor-infiltrated M-MDSCs were decreased in gp49B−/− tumor-bearing mice. (a) The expression of gp49 on MDSCs of WT and gp49B−/− mice was measured by flow cytometry using anti-CD11b, anti-CD115, anti-Gr1 and anti-gp49 antibodies. (b and c) The percentage of M-MDSCs and PMN-MDSCs in the lung from WT and gp49B−/− tumor-bearing mice (n = 8 mice per group) was measured by flow cytometry using anti-CD11b, anti-Ly6C and anti-Ly6G antibodies. Graph showing percentage of M-MDSCs (Ly6C+ Ly6G−) and PMN-MDSC (Ly6C−Ly6G+) in CD11b+ lung cells. (d and e) Tumors from WT and gp49B−/− tumor-bearing mice were dissociated to detect tumor-infiltrated MDSCs by staining with anti-CD11b, anti-CD115, anti-Ly6C and, anti-Ly6G antibodies. Graph showing percentage of M-MDSCs (CD115+Ly6C+ Ly6G−) and PMN-MDSC (CD115− Ly6C−Ly6G+) in CD11b+ cells. All numerical data are represented as mean ± SEM. *p < .05, **p < .005, ns: not significant calculated by unpaired t-test.
Gp49B−/− MDSCs reduce immunosuppressive cytokine secretion, Treg activation, tumor cell migration, and angiogenesis
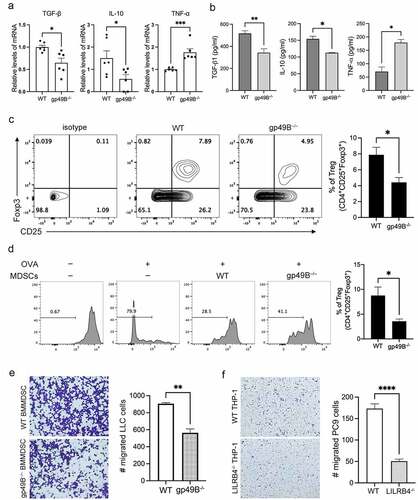
Immunosuppressive cytokines, such as IL-10 and TGF-β, are secreted by activated MDSCs to down-regulate IL-12 secretion by macrophages, block cytotoxic T lymphocyte activity, and activate Treg cells.Citation29 To determine the expression level of immunosuppressive genes in MDSCs from tumor-bearing mice, the splenic MDSCs from WT and gp49B−/− tumor-bearing mice were isolated for gene expression analysis by qRT-PCR. The expression of TGF-β, IL-10, and ARG-1 was downregulated, while the expression of TNF-α and iNOS was upregulated in gp49B−/− splenic MDSCs, suggesting that gp49B−/− MDSCs exhibit an M1 phenotype ( and Supplemental Figure 3B). In comparison with WT splenic MDSCs, gp49B−/− splenic MDSCs secreted low levels of TGF-β and IL-10 (). To verify whether gp49B could regulate the MDSC-mediated expansion and activation of Treg cells, MDSCs from WT or gp49B−/− tumor-bearing mice were co-cultured with OT-II splenocytes with OVA peptides stimulation. Upon co-culture with gp49B−/− MDSCs, the expansion of Treg cells decreased, whereas the proliferation of CD4 Teff increased (, and ), suggesting that the knockout of gp49B affects the immunosuppressive function of MDSCs. Furthermore, results of the in vitro T cell activation assay revealed that coculture with gp49B−/−MDSCs did not suppress the anti-CD3/anti-CD28 stimulated CD4 and CD8 T cell activation compared to the WT controls (Supplemental Figure 4). The above results suggested that gp49B knockout affects the immunosuppressive function of M-MDSCs.
Figure 4. MDSCs from tumor-bearinggp49B−/− mice exhibited the M1-like (anti-tumor) phenotype. (a) gene expression of splenic MDSCs from WT and gp49B−/− tumor-bearing mice was measured by qRT-PCR as described in Materials and Methods. (b) MDSCs from tumor-bearing mice were harvested for cytokine detection by ELISA. (c) Splenic MDSCs from WT or gp49B−/− tumor-bearing mice were co-cultured with splenocytes from OT-II mice in the presence of OVA peptide for 5 days. The population of Treg cells was measured by flow cytometry using anti-CD4, anti-Foxp3 and anti-CD25 antibodies. (d) Co-culture of CFSE-labeled OT-II splenocytes stimulated with OVA peptide with splenic MDSCs from WT and gp49B−/− tumor-bearing mice. T cell proliferation was measured by flow cytometry. (e) Bone marrow-derived MDSCs from WT or gp49B−/− tumor-bearing mice were plated in a 48-well plate and co-cultured with LLC cells for 48 h. The migration of LLC cells was determined using the transwell migration assay. (f) Representative graph of migrated human PC9 cells co-cultured with PMA-stimulated WT or LILRB4−/− THP-1 cells. The cell migration ability corresponding to (e and f) was calculated by counting migrated cells on the underside of the filters. Representative of three independent experiments with similar results. Data are represented as means ± SEM. *p < .05, **p < .005, ***p < .001 calculated by unpaired t-test.
Since MDSCs also contribute in the migration of tumor cells,Citation30,Citation31 we therefore examined the effect of gp49B knockout on MDSC-regulated tumor cell migration. In vitro transwell migration assay revealed a significantly decreased migration of murine LLC cells co-cultured with gp49B−/− BM-MDSCs as well as human PC9 cells co-cultured with activated LILRB4−/− THP-1 cells (, and ). MDSCs also directly promote tumor development by inducing angiogenesis and vasculogenesis via VEGFA and MMP-9 secretion.Citation32–34 To determine whether LILRB4/gp49B regulates MDSC-induced angiogenesis, we observed the expression of angiogenic biomarker CD31 in LLC tumors harvested from WT and gp49B−/− mice. The result of immunostaining showed that CD31 expression was reduced in LLC tumor from gp49B−/− mice compared to that from WT mice, indicating that tumor vasculogenesis was inhibited in gp49B−/− mice (Supplemental Figure 5A). We further confirmed that the expression of VEGFA and MMP-9 was downregulated in splenic MDSCs from gp49B−/− tumor-bearing mice (Supplemental Figure 5B). With stimulation of IL-4 ex-vivo to promote M2-phenotype differentiation of bone marrow (BM)-MDSCs, gp49B knockout decreased the expression of VEGFA and MMP-9 in M2-polarized BM-MDSCs compared to WT controls (Supplemental Figure 5C). Additionally, in vitro angiogenesis assay showed that tube formation of HUVECs was reduced in the co-culture with the supernatant from IL-4-stimulated gp49B−/− BM-MDSCs (Supplemental Figure 5, D and E). These results suggested that knockout of LILRB4/gp49B triggers MDSCs to preferentially differentiate into anti-tumor phenotype, and the secretome by gp49B−/− MDSC may contain lower pro-tumor factors and higher anti-tumor factors.
Treatment of tumor-bearing mice with gp49B−/− MDSCs suppresses tumor metastasis
In order to verify whether tumor-primed gp49B−/− MDSCs have anti-tumor activity, WT LLC-Luc2 tumor-bearing mice were intravenously injected with tumor-primed WT or gp49B−/− MDSCs twice, and the bioluminescence activity was monitored to assess tumor metastatic growth. Indeed, gp49B−/− MDSC treated tumor-bearing mice revealed a significant reduction of LLC-Luc2 metastasis compared to WT controls (, and ). With a lower expression level of gp49B, lung-infiltrated M-MDSCs were also reduced in gp49B−/− MDSC treated WT tumor-bearing mice as well as in gp49B−/− tumor-bearing mice. In contrast, gp49B−/− MDSC adoptive transfer did not affect the population frequency of PMN-MDSCs resident in lung tissue (, -). These data suggested that gp49B knockout in MDSCs triggers anti-tumor activity, and gp49B might be involved in M-MDSCs recruitment in the tumor environment.
Figure 5. Gp49B−/− MDSC treatment inhibited the metastasis of LLC-Luc2 cells in WT mice. (a) Splenic MDSCs from WT or gp49B−/− tumor-bearing mice were isolated and injected into WT LLC-Luc2 tumor-bearing mice twice at 1-week intervals as described in Materials and Methods. After 4 weeks of LLC-Luc2 cells inoculation, the tumor metastasis in WT mice was observed using the IVIS system. (b) Bioluminescence measurements expressed as average radiance (photons/s/cm2/steradian) corresponding to (a). (c) The percentage of M-MDSCs and PMN-MDSCs in lung from WT and gp49B−/− MDSC-adoptive transferred tumor-bearing mice was measured by flow cytometry using anti-CD11b, anti-Ly6C, and anti-Ly6G antibodies. (d) Graph showing percentage of M-MDSCs (left) and PMN-MDSCs (right) in CD11b+ lung cells corresponding to (c). (e) The gp49 expression on M-MDSCs and PMN-MDSCs in lung from tumor-bearing mice corresponding to (c). (f) Mean fluorescent intensity (MFI) analysis of gp49 expression corresponding to (e). Data are represented as means ± SEM. *p < 0.05, ns: not significant calculated by unpaired t-test.
Gp49B knockout increases anti-tumor miRNAs expression in MDSCs and plasma exosomes
Exosomes, with a size range of 30–150 nm in diameter, are an important part of the tumor microenvironment (TME).Citation35 MicroRNAs (miRNAs) carried in exosomes are taken up by neighboring or distant cells to regulate cancer progression.Citation36 To explore whether gp49B knockout affects the amount and composition of exosomal miRNAs, plasma exosomes from WT and gp49B−/− tumor-bearing mice were isolated for miRNA sequencing (miRNA-seq). An MA plot analysis revealed miRNAs that are differentially expressed between the WT miRNA-seq and gp49B−/− miRNA-seq (). By comparing the fold changes in gp49B−/− to that of WT samples, we observed that 4.46% miRNAs were upregulated while 3.80% miRNAs were downregulated in gp49B−/− plasma exosomes. Particularly, some of anti-tumor miRNAs showed higher fold changes in miRNA-seq data, including miR-1a-3p, miR-1b-5p, miR-133a-3p, and miR-206-3p (). We further validated that the expression levels of miR-1a-3p, miR-133a-3p, and miR-206-3p were increased in gp49B−/− plasma exosomes by qRT-PCR (). Moreover, the presence of gp49B−/− plasma exosomes from tumor-bearing mice indeed decreased the migration of LLC cells in vitro compared to the WT control (, and ). A comparative analysis revealed that the expression levels of miR-1a-3p and miR-133a-3p were significantly decreased in LUAD and LUSC tumor tissues compared to that in normal tissues (Supplemental Figure 6, A and B). The miR-206-3p expression was also downregulated in LUAD and LUSC samples, although with no statistical significance (Supplemental Figure 6C). These data suggested that LILRB4/gp49B contributes in the reduction of anti-tumor miRNAs in exosomes, which correlated with lung cancer diseases.
Figure 6. Anti-tumor miRNAs, including miR-1a-3p, miR-133a-3p, and miR-206-3p, were increased in plasma exosomes from gp49B−/− tumor-bearing mice. (a) MA plot reveals a set of exosomal miRNA candidates that changed in the plasma of gp49B−/− tumor-bearing mice. Analysis was performed on exosomal miRNA profiles generated using NextSeq 500 next-generation sequencing system. Two independent experiments were conducted. (b) The miRNA expression level in plasma exosomes and from WT and gp49B−/− tumor-bearing mice (n = 7 mice per group) was measured by qRT-PCR. (c) LLC cells (1 × 104 cells/well) were seeded in the upper chamber and cultured with serum-free medium supplemented with plasma exosomes from tumor-bearing mice for 16 h. Representative graph showed the migrated LLC cells with crystal violet staining. (d) The migratory ability of LLC cells cultured with plasma exosomes from WT or gp49B−/− tumor-bearing mice was determined using the wound healing assay as described in Materials and Methods. Graph showing the percentage of wound closure was represented as mean ± SEM. (e) The miRNA expression level in splenic MDSCs from WT and gp49B−/− tumor-bearing mice (n = 7 mice per group) was measured by qRT-PCR. (f and g) Exosomes from cell culture supernatants of M2-macrophage-polarized WT and LILRB4−/− THP1 cells were harvested for the detection of exosomal miR-1a-3p, miR-133a-30 and miR-206-3p expression. (f) or PC9 migration ability by wound healing assay (g). All numerical data are represented as means ± SEM. *p < .05, **p < .005, ***p < .001, ****p < .0001, ns: not significant calculated by unpaired t-test.
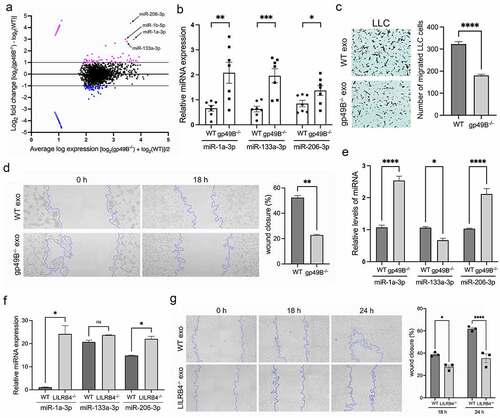
Recent studies have reported that MDSC-derived exosomes exert immunosuppressive and tumor-promoting effects in the tumor environment.Citation6,Citation37,Citation38 Therefore, we examined whether these anti-tumor miRNAs were indeed increased in splenic gp49B−/− MDSCs from tumor-bearing mice. There were higher levels of miR-1a-3p and miR-206-3p and lower levels of miR-133a-3p detected in gp49B−/− MDSCs compared to WT controls (). Since M-MDSCs swiftly differentiate to tumor associated macrophages (TAMs),Citation9 we then investigated whether LILRB4 also affects the content of anti-tumor miRNAs in exosomes derived from M2 macrophages. In comparison to WT controls, LILRB4−/− M2-polarized THP1 cell-derived exosomes that inhibited the migration ability of PC9 cells showed higher levels of miR-1a-3p and miR-206-3p (, and ). Together, the above results suggested that LILRB4/gp49B contributes to the suppression of anti-tumor miRNA expression in MDSC-derived exosomes, thereby promoting tumor progression.
Discussion
Herein, we demonstrate the tumor-promoting role of LILRB4/gp49B in LLC and B16F10 metastases using a murine tumor model with intravenous injection of cancer cells. Tumor metastases were inhibited in gp49B−/− tumor-bearing mice, similar to results in gp49-blocking tumor-bearing mice. We also demonstrated that the expression of gp49B on MDSCs plays a pivotal role in MDSC-mediated immunosuppression. An increase in plasma anti-tumor exosomal miRNAs from gp49B−/− mice was correlated with upregulation of gp49B−/− MDSC-derived anti-tumor miRNAs in vivo. LILRB4−/− M2-polarized macrophages also produced higher levels of anti-tumor exosomal miRNAs in vitro. Hence, our finding suggested that LILRB4/gp49B signaling inhibits the production of anti-tumor exosomal miRNAs to benefit tumor metastasis.
Several studies have reported that anti-LILRB4 antibody treatment inhibits tumor growth and prolongs survival in tumor-bearing mice.Citation22,Citation39,Citation40 Recently, Sharma et al. reported that anti-LILRB4 antibody treatment prolongs survival in B16F10 tumor-bearing mice.Citation41 Consistently, our results revealed that the anti-gp49 antibody treatment significantly inhibited pulmonary tumor nodule formation (p = .0057), and showed a trend to reduce liver metastasis in B16F10 tumor-bearing mice. Combination immunotherapies such as chemotherapy combined with immune checkpoint inhibitor (ICI) therapy and ICI two-drug combination therapy are the current treatment trend.Citation42 LILRB4 expression has been shown to highly associated with CTLA-4 expression in chronic myelomonocytic leukemia (CMML) samples.Citation43 In this study, we observed that with the positive correlation between LILRB4 and PD-1, the combination therapy of anti-LILRB4/gp49B and anti-PD-1 mAbs showed higher anti-tumor metastasis ability compared to the monotherapy. These results suggested ICI targeting LILRB4 may provide a novel combination therapeutic approach in several cancer diseases.
A previous study reported that with intradermal injection of B16F10 cells, gp49B was expressed on most types of tumor-infiltrating immune cells, including CD3 T cell, macrophages, neutrophils DCs, NK cells. In addition, LILRB4 is highly expressed on Treg cells, exhausted CD8 T cells, and CD11b+ TAMs in the B16F10 model. Sharma et al. also reported that frequencies of CD8 T cells and CD4 Teff cells were increased, while the Treg frequency was decreased in tumors.Citation41 In this study, we found that gp49B is constitutively expressed on splenic MDSCs and DCs in WT control mice (Supplemental Figure 7A). After tumor challenge with LLC cells by iv injection, the expression of gp49B was increased on splenic and lung-infiltrated MDSCs, a subset of CD4/8 T cells and DCs (Supplemental Figure 7, B and C). The expression level of gp49B was not highly expressed on CD4 and CD8 cells, possibly due to the use of a different tumor challenge mode and a different tumor type. Although with weak expression of gp49B, the frequency of Treg population decreased, while the frequency of CD4 T cells increased in splenocytes and lung-infiltrated cells from gp49B−/− tumor-bearing mice. The frequency of the DC population was comparable the in lung of WT and gp49B−/− tumor-bearing mice (Supplemental Figure 8 and 9).
In the TME, M-MDSCs rapidly differentiate to TAMs.Citation9 An LILRB4 analysis of tumor-infiltrating myeloid cells in a murine colon adenocarcinoma MC38 tumor model by CyTOF showed that myeloid-expressed LILRB4 is major on TAMs with immunosuppressive markers, such as Arg-1, CD206, and CD163.Citation41 Our data showed that tumor- and lung-infiltration of M-MDSCs was decreased in gp49B−/− tumor-bearing mice ( and Supplemental Figure 8), while population frequencies of splenic and BM M-MDSCs were not significantly different between WT and gp49B−/− mice inoculated with or without LLC cells (Supplemental Figure 9–11). These results suggested that gp49B may play an important role in the recruitment of M-MDSCs in TME. The mechanism of LILRB4-mediated MDSC recruitment in TME needs to be further elucidated.
Polarization and reprogramming of MDSCs, which are able to be polarized from an M1 phenotype to an M2 phenotype or vice versa, affect tumor progression.Citation7 Predominantly, tumor-associated MDSCs exhibit M2-like phenotype with pro-tumor activities. M2 phenotype MDSCs activate Treg to suppress Teff, whereas M1 counterparts have the opposing actions. Herein, we showed that the MDSCs from gp49B−/− tumor-bearing mice inhibit Treg activation and activate CD4 Teff proliferation, suggesting that LILRB4 could regulate the polarization of tumor-associated MDSCs. Several studies have shown that STAT3 activation results in the polarization of M2-TAM, and also results in the production of factors that promote tumor invasiveness, such as VEGFA and MMPs.Citation44,Citation45 Therefore, we propose that LILRB4 signaling may activate STAT3 to promote M2-polarization and induce the expression of VEGFA and MMP-9. In addition to expression on tumor-infiltrating immune cells, LILRB4 expression is also increased on human non-small cell lung cancer (NSCLC) cells, and LILRB4 overexpression enhances the migration and invasion of NSCLC cells by activating the ERK1/2-EMT/VEGFA axis.Citation46 These results suggest that LILRB4 is involved in tumorigenesis by regulating gene expression associated with migration, invasion, and angiogenesis.
Exosomes have been shown to affect the tumor environment by influencing the extracellular matrix and immune system.Citation47 miRNA-carrying exosomes released from cancer cells, immune cells, and mesenchymal cells in the tumor environment influence cancer progression and metastasis.Citation36,Citation48,Citation49 Several studies have reported that dysregulation of miR-1, miR-133, and miR-206 has been observed in a number of cancer disease.Citation50–53 miR-1 and miR-206 inhibit cancer cell migration and tumor angiogenesis by targeting MET proto-oncogene, receptor tyrosine kinase (MET), and chemokine ligand-2 (CCL2)/VEGFA, respectively.Citation54–56 Ectopic miR-1 and miR-133a have been shown to inhibit cell migration and invasion abilities by targeting transgelin 2 (TAGLN2), which may have an oncogenic function in bladder cancer.Citation57 Our data showed that gp49B−/− MDSCs and LILRB4−/− M2-macrophage-derived exosomes, containing high expression levels of miR-1a-3p, miR-206-3p, inhibit PC9 cell migration (). In addition, the expression levels of miR-1a-3p, miR-206-3p, and miR-133a-3p were comparable in LLC cells co-cultured with WT or gp49B−/− BM-MDSCs (Supplemental Figure 12). Here, we suggest that LILRB4 contributes to inhibition of miR-1 and miR-206 expression in MDSC and M2-macrophage, and that LILRB4 may regulate miR-133a expression in other immune cells.
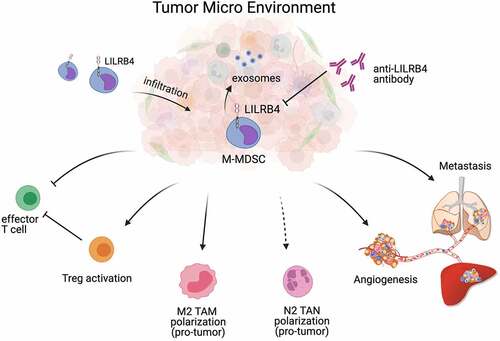
In conclusion, we have identified that LILRB4 serves as a key player in regulating the immunosuppressive function of MDSC and repressing anti-tumor miR-1 family miRNAs expression in the TME (), and further studies need to be conducted to investigate the content of exosomes regulated by LILRB4 after antibody treatment, and the detail mechanism of LILRB4-mediated miRNA expression.
Figure 7. The tumor-promoting function of LILRB4/gp49B in MDSC-mediated tumor metastasis. LILRB4/gp49B facilitates M-MDSCs infiltration in tumor environment, and promotes MDSC-mediated tumor metastasis by promoting the activation of Treg cells, inhibiting Teff cells, enhancing cancer cell migration, and accelerating angiogenesis. In addition, LILRB4/gp49B suppresses anti-tumor miR-1 family microRNAs (miRNAs) expressed in MDSC-derived exosomes.
Ethics approval and consent to participate
The animal experimental protocol was formulated in accordance with the guidelines of Law on the Concerning the Conservation and Sustainable Use of Biological Diversity through Regulations on the Use of Living Modified Organisms, and was approved by Animal Experiment Ethics Review Committee of Tohoku University.
Supplemental Material
Download Zip (2.9 MB)Acknowledgments
We thank W. M. Yokoyama (Washington University School of Medicine, St. Louis) for providing the hybridoma. This research was partially supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP21am0101078. The illustrative diagram in was created with Biorender.com.
Disclosure statement
A patent has been applied for by Tohoku University with MTS, SE, and TT as named inventors to Japan Patent Office at the application number PCT/JP2020/030175 (WO/2021/029318). Other authors declare no conflict of interest.
Supplementary materiyal
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2060907
Additional information
Funding
References
- Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32(19–20):1267–15. doi:10.1101/gad.314617.118.
- El-Kenawi A, Hanggi K, Ruffell B. The immune microenvironment and cancer metastasis. Cold Spring Harb Perspect Med. 2020;10:a037424. doi:10.1101/cshperspect.a037424.
- Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73–86. doi:10.1038/nri3789.
- Lim HX, Kim TS, Poh CL. Understanding the differentiation, expansion, recruitment and suppressive activities of myeloid-derived suppressor cells in cancers. Int J Mol Sci. 2020;21(10):3599. doi:10.3390/ijms21103599.
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13(10):739–752. doi:10.1038/nrc3581.
- Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200(2):422–431. doi:10.4049/jimmunol.1701019.
- Yang WC, Ma G, Chen SH, Pan PY. Polarization and reprogramming of myeloid-derived suppressor cells. J Mol Cell Biol. 2013;5(3):207–209. doi:10.1093/jmcb/mjt009.
- Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. 2008;83(5):1136–1144. doi:10.1189/jlb.0907611.
- Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–220. doi:10.1016/j.it.2016.01.004.
- Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, Utikal J, Umansky V. myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. 2018;9:1310. doi:10.3389/fimmu.2018.01310.
- Law AMK, Valdes-Mora F, Gallego-Ortega D. myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. 2020;9(3):561. doi:10.3390/cells9030561.
- de Goeje PL, Bezemer K, Heuvers ME, Dingemans AC, Groen HJ, Smit EF, Hoogsteden HC, Hendriks RW, Aerts JG, Hegmans JP, et al. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology. 2015;4(7):e1014242. doi:10.1080/2162402X.2015.1014242.
- Colonna M, Navarro F, Lopez-Botet M. A novel family of inhibitory receptors for HLA class I molecules that modulate function of lymphoid and myeloid cells. Curr Top Microbiol Immunol. 1999;244:115–122. doi:10.1007/978-3-642-58537-1_10.
- Vlad G, Chang CC, Colovai AI, Berloco P, Cortesini R, Suciu-Foca N. Immunoglobulin-like transcript 3: a crucial regulator of dendritic cell function. Hum Immunol. 2009;70(5):340–344. doi:10.1016/j.humimm.2009.03.004.
- Katz HR. Inhibition of pathologic inflammation by leukocyte Ig-like receptor B4 and related inhibitory receptors. Immunol Rev. 2007;217(1):222–230. doi:10.1111/j.1600-065X.2007.00522.x.
- Kang X, Kim J, Deng M, John S, Chen H, Wu G, Phan H, Zhang CC. Inhibitory leukocyte immunoglobulin-like receptors: immune checkpoint proteins and tumor sustaining factors. Cell Cycle. 2016;15(1):25–40. doi:10.1080/15384101.2015.1121324.
- Kuroiwa A, Yamashita Y, Inui M, Yuasa T, Ono M, Nagabukuro A, Matsuda Y, Takai T. Association of tyrosine phosphatases SHP-1 and SHP-2, inositol 5-phosphatase SHIP with gp49B1, and chromosomal assignment of the gene. J Biol Chem. 1998;273(2):1070–1074. doi:10.1074/jbc.273.2.1070.
- van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother. 2017;66(8):1079–1087. doi:10.1007/s00262-017-2023-x.
- Park M, Raftery MJ, Thomas PS, Geczy CL, Bryant K, Tedla N. Leukocyte immunoglobulin-like receptor B4 regulates key signalling molecules involved in FcgammaRI-mediated clathrin-dependent endocytosis and phagocytosis. Sci Rep. 2016;6(1):35085. doi:10.1038/srep35085.
- Lu HK, Rentero C, Raftery MJ, Borges L, Bryant K, Tedla N. Leukocyte Ig-like receptor B4 (LILRB4) is a potent inhibitor of FcgammaRI-mediated monocyte activation via dephosphorylation of multiple kinases. J Biol Chem. 2009;284(50):34839–34848. doi:10.1074/jbc.M109.035683.
- Mori Y, Tsuji S, Inui M, Sakamoto Y, Endo S, Ito Y, Fujimura S, Koga T, Nakamura A, Takayanagi H, et al. Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J Immunol. 2008;181(7):4742–4751. doi:10.4049/jimmunol.181.7.4742.
- Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, He L, Chen Y, Chen H, Luo W. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. 2018;562(7728):605–609. doi:10.1038/s41586-018-0615-z.
- Xu Z, Chang CC, Li M, Zhang QY, Vasilescu EM, D’Agati V, Floratos A, Vlad G, Suciu-Foca N. ILT3.Fc-CD166 interaction induces inactivation of p70 S6 kinase and inhibits tumor cell growth. J Immunol. 2018;200(3):1207–1219. doi:10.4049/jimmunol.1700553.
- Kasai S, Inui M, Nakamura K, Kakizaki Y, Endo S, Nakamura A, Ito S, Takai T. A novel regulatory role of gp49B on dendritic cells in T-cell priming. Eur J Immunol. 2008;38(9):2426–2437. doi:10.1002/eji.200737550.
- Su MT, Inui M, Wong YL, Takahashi M, Sugahara-Tobinai A, Ono K, Miyamoto S, Murakami K, Itoh-Nakadai A, Kezuka D, et al. Blockade of checkpoint ILT3/LILRB4/gp49B binding to fibronectin ameliorates autoimmune disease in BXSB/ Yaa mice. Int Immunol. 2021;33(8):447–458. doi:10.1093/intimm/dxab028.
- Wong YL, Su MT, Sugahara-Tobinai A, Itoi S, Kezuka D, Endo S, Inui M, Takai T. Gp49B is a pathogenic marker for auto-antibody-producing plasma cells in lupus-prone BXSB/Yaa mice. Int Immunol. 2019;31(6):397–406. doi:10.1093/intimm/dxz017.
- Kusmartsev SA, Li Y, Chen SH. Gr-1 + myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000;165(2):779–785. doi:10.4049/jimmunol.165.2.779.
- Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–678. doi:10.1038/s41587-020-0546-8.
- Umansky V, Blattner C, Gebhardt C, Utikal J. the role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines (Basel). 2016;4(4). doi:10.3390/vaccines4040036.
- Pang X, Fan HY, Tang YL, Wang SS, Cao MX, Wang HF, Dai -L-L, Wang K, Yu X-H, Wu J-B, et al. Myeloid derived suppressor cells contribute to the malignant progression of oral squamous cell carcinoma. PLoS One. 2020;15(2):e0229089. doi:10.1371/journal.pone.0229089.
- Rashid MH, Borin TF, Ara R, Piranlioglu R, Achyut BR, Korkaya H, Liu Y, Arbab A. Critical immunosuppressive effect of MDSCderived exosomes in the tumor microenvironment. Oncol Rep. 2021;45(3):1171–1181. doi:10.3892/or.2021.7936.
- Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125(9):3365–3376. doi:10.1172/JCI80006.
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. doi:10.1038/nrc2444.
- Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421. doi:10.1016/j.ccr.2004.08.031.
- Lobb RJ, Lima LG, Moller A. Exosomes: key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev Biol. 2017;67:3–10. doi:10.1016/j.semcdb.2017.01.004.
- Sun Z, Shi K, Yang S, Liu J, Zhou Q, Wang G, Song J, Li Z, Zhang Z, Yuan W, et al. Effect of exosomal miRNA on cancer biology and clinical applications. Mol Cancer. 2018;17(1):147. doi:10.1186/s12943-018-0897-7.
- Geis-Asteggiante L, Belew AT, Clements VK, Edwards NJ, Ostrand-Rosenberg S, El-Sayed NM, Fenselau C. Differential content of proteins, mRNAs, and miRNAs suggests that MDSC and their exosomes may mediate distinct immune suppressive functions. J Proteome Res. 2018;17(1):486–498. doi:10.1021/acs.jproteome.7b00646.
- Deng Z, Rong Y, Teng Y, Zhuang X, Samykutty A, Mu J, Zhang L, Cao P, Yan J, Miller D, et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene. 2017;36(5):639–651. doi:10.1038/onc.2016.229.
- Nair R, Salinas-Illarena A, Baldauf HM. New strategies to treat AML: novel insights into AML survival pathways and combination therapies. Leukemia. 2021;35(2):299–311. doi:10.1038/s41375-020-01069-1.
- Gui X, Deng M, Song H, Chen Y, Xie J, Li Z, He L, Huang F, Xu Y, Anami Y, et al. Disrupting LILRB4/APOE interaction by an efficacious humanized antibody reverses T-cell suppression and blocks AML development. Cancer Immunol Res. 2019;7(8):1244–1257. doi:10.1158/2326-6066.CIR-19-0036.
- Sharma N, Atolagbe OT, Ge Z, Allison JP. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J Exp Med. 2021;218(7):e20201811. doi:10.1084/jem.20201811.
- Ma X, Zhang Y, Wang S, Wei H, Yu J. Immune checkpoint inhibitor (ICI) combination therapy compared to monotherapy in advanced solid cancer: a systematic review. J Cancer. 2021;12(5):1318–1333. doi:10.7150/jca.49174.
- Chien KS, Class CA, Montalban-Bravo G, Wei Y, Sasaki K, Naqvi K, Ganan-Gomez I, Yang H, Soltysiak KA, Kanagal-Shamanna R, et al. LILRB4 expression in chronic myelomonocytic leukemia and myelodysplastic syndrome based on response to hypomethylating agents. Leuk Lymphoma. 2020;61(6):1493–1499. doi:10.1080/10428194.2020.1723014.
- Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. doi:10.1038/ni.1937.
- Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, Yang J, Pan J, Hu S, Zhang C, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018;17(4):428–438. doi:10.1080/15384101.2018.1444305.
- Li J, Gao A, Zhang F, Wang S, Wang J, Wang J, Han S, Yang Z, Chen X, Fang Y, et al. ILT3 promotes tumor cell motility and angiogenesis in non-small cell lung cancer. Cancer Lett. 2021;501:263–276. doi:10.1016/j.canlet.2020.10.048.
- Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, Tao Y, He Z, Chen C, Jiang Y, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther. 2020;5(1):145. doi:10.1038/s41392-020-00261-0.
- Dilsiz N. Role of exosomes and exosomal microRNAs in cancer. Future Sci OA. 2020;6:FSO465. doi:10.2144/fsoa-2019-0116.
- Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl). 2013;91(4):431–437. doi:10.1007/s00109-013-1020-6.
- Xie M, Dart DA, Owen S, Wen X, Ji J, Jiang W. Insights into roles of the miR-1, −133 and −206 family in gastric cancer (Review). Oncol Rep. 2016;36(3):1191–1198. doi:10.3892/or.2016.4908.
- Han C, Yu Z, Duan Z, Kan Q. Role of microRNA-1 in human cancer and its therapeutic potentials. Biomed Res Int. 2014;2014:428371. doi:10.1155/2014/428371.
- Pan JY, Sun CC, Bi ZY, Chen ZL, Li SJ, Li QQ, Wang Y-X, Bi -Y-Y, Li D-J. miR-206/133b cluster: a weapon against lung cancer? Mol Ther Nucleic Acids. 2017;8:442–449. doi:10.1016/j.omtn.2017.06.002.
- Nohata N, Hanazawa T, Enokida H, Seki N. microRNA-1/133a and microRNA-206/133b clusters: dysregulation and functional roles in human cancers. Oncotarget. 2012;3(1):9–21. doi:10.18632/oncotarget.424.
- Chen X, Tong ZK, Zhou JY, Yao YK, Zhang SM, Zhou JY. MicroRNA-206 inhibits the viability and migration of human lung adenocarcinoma cells partly by targeting MET. Oncol Lett. 2016;12(2):1171–1177. doi:10.3892/ol.2016.4735.
- Han C, Zhou Y, An Q, Li F, Li D, Zhang X, Yu Z, Zheng L, Duan Z, Kan Q, et al. MicroRNA-1 (miR-1) inhibits gastric cancer cell proliferation and migration by targeting MET. Tumour Biol. 2015;36(9):6715–6723. doi:10.1007/s13277-015-3358-6.
- Shen H, Yu X, Yang F, Zhang Z, Shen J, Sun J, Choksi S, Jitkaew S, Shu Y. Reprogramming of normal fibroblasts into cancer-associated fibroblasts by miRNAs-Mediated CCL2/VEGFA signaling. PLoS Genet. 2016;12(8):e1006244. doi:10.1371/journal.pgen.1006244.
- Yoshino H, Chiyomaru T, Enokida H, Kawakami K, Tatarano S, Nishiyama K, Nohata N, Seki N, Nakagawa M. The tumour-suppressive function of miR-1 and miR-133a targeting TAGLN2 in bladder cancer. Br J Cancer. 2011;104(5):808–818. doi:10.1038/bjc.2011.23.