
ABSTRACT
Neuroblastoma is a commonly lethal solid tumor of childhood and intensive chemoradiotherapy treatment cures ~50% of children with high-risk disease. The addition of immunotherapy using dinutuximab, a monoclonal antibody directed against the GD2 disialoganglioside expressed on neuroblasts, improves survival when incorporated into front-line therapy and shows robust activity in regressing relapsed disease when combined with chemotherapy. Still, many children succumb to neuroblastoma progression despite receiving dinutuximab-based immunotherapy, and efforts to counteract the immune suppressive signals responsible are warranted. Animal models of human cancers provide useful platforms to study immunotherapies. TH-MYCN transgenic mice are immunocompetent and develop neuroblastomas at autochthonous sites due to enforced MYCN expression in developing neural crest tissues. However, GD2-directed immunotherapy in this model has been underutilized due to the prevailing notion that TH-MYCN neuroblasts express insufficient GD2 to be targeted. We demonstrate that neuroblasts in TH-MYCN-driven tumors express GD2 at levels comparable to human neuroblastomas but rapidly lose GD2 expression when explanted ex vivo to establish tumor cell lines. This occurs in association with a transition from an adrenergic to mesenchymal differentiation state. Importantly, not only is GD2 expression retained on tumors in situ, treatment with a murine anti-GD2 antibody, 14G2a, markedly extends survival in such mice, including durable complete responses. Tumors in 14G2a-treated mice have fewer macrophage and myeloid-derived suppressor cells in their tumor microenvironment. Our findings support the utility of this model to inform immunotherapy approaches for neuroblastoma and potential opportunities to investigate drivers of adrenergic to mesenchymal fate decisions.
Introduction
A variety of immunotherapies are altering the treatment landscape for many cancers, particularly those of hematologic origin. Unfortunately, solid tumor immunotherapies are frequently hindered by the capacity of the tumor microenvironment (TME) to suppress and exclude immune effector cells. Neuroblastoma is a common solid tumor of childhood that carries a 5-year mortality rate of ~50% in its high-risk form despite the use of intensive treatment with surgery, high-dose chemotherapy with stem cell rescue, radiotherapy and biotherapy with retinoids. Like most pediatric cancers, neuroblastomas manifest a low mutation burden and a paucity of neo-antigens for the immune system to target.Citation1–3 Further, its TME is enriched with immune and stromal cells that include tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), T-regulatory cells, and fibroblasts that collectively release immunosuppressive cytokines and attenuate host immune responses.Citation4–6
Despite these barriers, dinutuximab, a chimeric antibody that targets the GD2 disialoganglioside highly expressed on nearly all neuroblastomas, has shown efficacy when combined with immune stimulation with interleukin-2 (IL-2) and granulocyte-macrophage colony-stimulating factor (GM-CSF) in front-line maintenance therapy for patients with high-risk neuroblastoma (HRNB), leading to its FDA approval, and when used in combination with chemotherapy to treat relapsed or refractory neuroblastoma.Citation7–9 Although these clinical successes substantiate the promise of immunotherapy for neuroblastoma, many children still die from tumor progression despite receiving dinutuximab.Citation8–10 A more thorough understanding of the immunosuppressive TME should provide opportunities to improve this therapy. Such mechanistic studies can be achieved in faithful mouse models of cancer, enabling the discovery and preclinical prioritization of novel therapeutic approaches, including combination therapies with GD2-directed antibodies. Despite differences between the murine and human immune systems, immunocompetent transgenic mouse models have been credentialed for the study of immunotherapies and a neuroblastoma model that uses a cancer-relevant oncogenic driver to initiate tumors in immunocompetent mice is paramount to further leverage the activity of dinutuximab.Citation11–13
The Tyrosine-Hydroxylase-MYCN (TH-MYCN) transgenic mouse provides an established model of HRNB. In this genetically engineered model, the MYCN gene, the principal oncogenic driver commonly amplified in human tumors, is highly expressed under the control of a tyrosine hydroxylase promoter to target tumor development in paraspinal or intra-abdominal sympathoadrenal tissues.Citation14 In addition to sharing MYCN overexpression, TH-MYCN tumors resemble human neuroblastomas histologically and genetically.Citation15–18 The model shows strain-dependent differences in phenotype, and, in the most widely utilized 129x1/SvJ background, mice homozygous for the TH-MYCN transgene (TH-MYCN+/+) have 100% lethal tumor penetrance.Citation15 While each tumor arising in these mice shares MYCN overexpression as the initiating event, they have distinct cooperating mutations that promote transformation. TH-MYCN mice have an intact immune system, and thus each tumor undergoes its own unique immunoediting process in situ. Unlike transplantable syngeneic tumor models that replicate tumors from a single tumor-derived cell line and implant them at an orthotopic or remote site, tumors in the TH-MYCN model initiate within autochthonous sympathoadrenal tissues, modeling interactions among neuroblasts, stromal cells and immune cells throughout the course of tumor initiation and progression. The tumor niche is important in influencing cancer progression as evidenced by syngeneic neuroblastoma transplant models in which subcutaneous implantation is associated with reduced tumor growth and fewer infiltrating tumor-suppressive immune cells compared with the same tumor cells injected at orthotopic intra-adrenal sites.Citation19,Citation20
Despite these advantages, an impediment to adoption of the highly penetrant 129x1/SvJ TH-MYCN model for studies into GD2-directed immunotherapies is the prevailing notion that these tumors lack sufficient surface GD2 to serve as an effective tumor antigen, primarily when investigated for syngeneic tumor transplantation studies. In the C57BL/6 background, preferred by many for immunologic studies, tumor penetrance for the TH-MYCN transgene is too low for efficient autochthonous modeling, and tumor-derived cell lines from this strain are found to express insufficient surface GD2 for therapeutic targeting unless they are manipulated pharmacologically or genetically.Citation21 Here, we demonstrate that neuroblastomas arising in 129x1/SvJ TH-MYCN mice express levels of GD2 nearly comparable to that present in human neuroblastomas.Citation16 Importantly, not only is GD2 surface expression present in primary tumors in situ, but this antigen can be targeted using a GD2-directed monoclonal antibody, 14G2a, that is analogous to the human therapeutic antibody, dinutuximab. Therapy with 14G2a extends survival and provides durable complete tumor regression in a subset of treated mice, accompanied by alterations in the TME, including reductions in the frequency of infiltrating monocytic MDSCs. We further show that TH-MYCN neuroblasts explanted from TH-MYCN tumors rapidly lose GD2 expression while propagated in standard tissue culture conditions, and that GD2 loss is associated with a transition of tumor cell lineage from an adrenergic to a mesenchymal state. Indeed, such adrenergic-mesenchymal lineage plasticity has been defined in human neuroblastomas with the mesenchymal state being correlated with therapy resistance and relapse.Citation22,Citation23 Collectively, our work credentials the TH-MYCN model as a relevant platform for immunotherapeutic studies, including GD2-targeting approaches, and may enable further studies into the regulation of mesenchymal state switch that renders tumor cells resistant to cytotoxic therapies and GD2-directed immunotherapies.
Methods
Mice and treatments
129x1/SvJ-Tg TH-MYCN mice were originally obtained by Dr. William Weiss (University of California, San Francisco).Citation14 All mice were bred and housed in the animal facility at the Children’s Hospital of Philadelphia under approved IACUC protocols. Heterozygous TH-MYCN+/- mice were bred and all studies performed on TH-MYCN+/+ offspring in which tumor-onset is fully penetrant. MYCN transgene genotype was determined from DNA isolated from a 1-cm tail snip by qPCR genotyping with the following primers:
Chr18F1 (5’-ACTAATTCTCCTCTCTCTGCCAGTATTTGC-3’),
Chr18R2 (5’-TGCCTTATCCAAAATATAAATGCCCAGCAG-3’), and
OUT1 (5’-TTGGCACACACAAATGTATATACACAATGG-3’), as described previously.Citation24
Homozygous TH-MYCN mice were randomized to receive intraperitoneal injections of 100 μg of anti-GD2 antibody (14G2a; BioLegend), 100 μg of a murine isotype-matched antibody to control for Fc receptor binding (murine IgG2a/κ; Abcam), or phosphate-buffered saline (PBS), twice weekly starting between 14 and 17 days of age and continuing until day 100 of life (n = 15 per group). Group size was powered to detect differences in survival (primary endpoint) and immune cell infiltration assessed on available tumors obtained at the time of sacrifice. All animals assigned to a treatment group were included in survival analyses. Mice were palpated for tumor presence and monitored for signs of morbidity that included poor mobility, hunching, lethargy, weight loss, limb paralysis, neurological changes, dermatitis, or rough hair coat. Mice exhibiting such signs were euthanized and underwent necropsy (termed “terminal” tumors). Group allocation was random, but experiments were not blinded.
To assess the biological impact of 14G2a immunotherapy at the time of response, an additional cohort of TH-MYCN+/+ mice (n = 6) were treated with twice weekly 14G2a antibody beginning between day 14–17 of life and monitored as above. Regressing tumors were detected by abdominal palpation, typically between days 70–75, and harvested for characterization (called “midpoint” tumors).
TH-MYCN+/+ mice that survived to day 200 (alive without tumor symptoms >3 months from the end of therapy; n = 4) were re-challenged with a single flank inoculation of HOM2 cells, a syngeneic TH-MYCN+/+ tumor-derived cell line serially passaged in vivo to enhance tumorigenicity (from Dr. Garrett Brodeur, Children’s Hospital of Philadelphia). Heterozygous TH-MYCN and wild-type (TH-MYCN−/−) 129x1/SvJ mice served as controls (n = 4 each) and received a single flank injection on the same day using the same cell-line preparation. Flanks were shaved and injected with 5 × 106 cells resuspended in Matrigel (Corning Inc.) at a 1:1 ratio by volume. Mice did not receive any treatment after tumor inoculation. Mice were monitored until tumors were established as a firm growing mass >500 mm3 (recorded as days to tumor engraftment) or until 8 weeks if no tumor was established (recorded as non-tumorigenic).
TH-MYCN+/+-derived cell lines
Tumor-bearing mice were euthanized, and tumors removed and dissociated into small tissue fragments using the bottom of a sterile 3-ml syringe pistol in 5 ml of sterile PBS in a 6-cm petri dish. The cellular suspensions were filtered through a 40 µm sterile nylon mesh, washed multiple times, and resuspended in sterile TAC buffer (UltraPure tris hydrochloride; Invitrogen and ammonium chloride; Sigma). Cells were incubated in a 37°C water bath, spun, and resuspended in PBS. These steps were repeated until red blood cells were no longer visible. Pellets were then resuspended in complete IMDM (Iscove’s modified Dulbecco’s media) with 20% fetal bovine serum (FBS), 1% penicillin-streptomycin, 1% L-glutamine, 0.6% recombinant human insulin, human transferrin, and sodium selenite (ITS), and 0.25% gentamicin, transferred into a T75 flask via a 40 µm cell strainer and incubated at 37°C with 5% CO2. Approximately 60% of explanted tumors generate a cell line. Four TH-MYCN+/+ tumors were explanted to derive new cell lines (5144, 5150, 5195, and 5213) that were serially passaged and analyzed by flow cytometry analysis to assess GD2 expression and lineage defining markers over time. In addition, cryopreserved TH-MYCN+/+ tumor-derived cell lines that had been established previously and passaged >3 months (3401, 3261, 3393, and 3392), and 3 isogenic TH-MYCN+/+ tumor cell lines obtained from the Brodeur laboratory (HOM2, G2, and G3B) were evaluated with flow cytometry for GD2 expression and lineage defining markers.
Human neuroblastoma cell lines
Patient-derived neuroblastoma cell lines COGN603, CHLA122, IMR5, NLF, CHLA20, SKNBE1, SKNBE2C, CHLA95, CHLA108, SHEP, and COGN203 were obtained from the cell-line bank at the Children’s Hospital of Philadelphia. Many were previously received from C. Patrick Reynolds (Texas Tech University of Health Sciences; cccells.org). Cells were thawed, plated in complete RPMI (10% FBS + 1% penicillin-streptomycin + 1% L-glutamine), and passaged at ~80% confluency using Versene solution, 0.48 mM EDTA (Gibco). IMR5 (predominantly adrenergic state) and SHEP (predominantly mesenchymal state) cells were used as controls in assessing lineage-defining proteins.
Flow cytometry to assess GD2 expression and characterize tumor-infiltrating leukocytes
Tumor-bearing mice were euthanized at midpoint or terminal timepoints. Tumors (ranging from 1.4 to 5 g in total weight) were cut into fragments of 1–3 mm3 and placed in RPMI Medium 1640 with L-glutamine (Gibco) and 10% heat-inactivated fetal bovine serum (FBS, Life Technologies) before undergoing tissue dissociation with a GentleMACS TM dissociator (Miltenyi Biotech) using the manufacturer’s recommended protocol with media including Collagenase Type IV (STEMCELL Technologies, Cambridge MA) and DNAse I (Sigma-Aldrich). After dissociation, suspensions were passed via a 70 µm filter, washed, and subjected to red blood cell lysis with ACK buffer (Gibco). Single-cell samples were assessed for viability using Trypan Blue 0.4% stain then frozen in fetal bovine serum with 10% dimethyl sulfoxide (Thermo Fisher Scientific) and stored at −80°C before staining for flow cytometry. Multiple flow-cytometry panels were used sequentially as long as sufficient viable cells remained (see consort diagram for tumor sample availability, Fig. S1).
To characterize tumor-infiltrating leukocytes and mesenchymal stem cells (MSCs), primary anti-mouse and anti-human antibodies specific for the following antigens were purchased from BioLegend (alternate names in parentheses; clone names in brackets): CD3 [17A2], CD45 [30-F11], NK1.1 [PK136], CD335 (NKp46) [29A1.4], Ly49A [YE1/48.10.6], CD314 (NKG2D) [CX5], CD159a (NKG2A) [16A11], GD2 [14G2a], MHC I (H-2 K/H-2D) [28-8-6], Rae-1γ [CX1], TGF-1 (LAP) [TW4-2F8], CD11b [M1/70], CD11c [N418], F4/80 [BM8], Ly-6 G/Ly-6C (Gr-1) [RB6-8C5], Ly-6 G [1A8], CD38 [90], CD107a (LAMP-1) [1D4B], TCR γδ [GL3], CD56 [HCD56], CD13 [R3-242], CD73 [TY/11.8]. Additional antibodies include Ly49H [3D10], and MHC Class II (I-A/I-E) [M5/114.15.2] from eBioscience, IL-15 Rα [888220], ULBP-1 [237104], and NCAM1 [809220] from R&D Systems, TGFβR-1 [RM0016-3A11] from Signalway, ULBP-1 from ThermoFisher Scientific, PGP9.5 [31A3] from Novus Biologicals, YAP1 [G6], and Phox2b [B11] from Santa Cruz Bio. PBS57-loaded CD1d tetramers were obtained from the NIH Tetramer Facility (Atlanta, GA). Cells were washed in PBS without serum and incubated with Zombie Aqua viability dye (BioLegend) according to manufacturer’s instructions and subsequently washed with FACS buffer (PBS supplemented with 2.5% FBS; Thermo Fisher Scientific) before Fc blockade with TruStain FcX (BioLegend) and subsequent surface marker staining. Cells were analyzed on a FACSVerseTM flow cytometer (BD Bioscience).
Flow analysis and gating strategy
FCS files were analyzed using FlowJo software (Tree Star: Ashland OR). TH-MYCN neuroblastoma cell lines were gated by selecting live single CD45-NCAM1+ cells, while human neuroblastoma cell lines were gated by selecting live single CD45-CD56+ cells, as previously described.Citation25–28 For further characterization of TH-MYCN neuroblastoma cell lines as well as to distinguish neuroblasts from MSCs, the CD45-NCAM1+ population was evaluated for neuroblast markers including PGP9.5 and Phox2B, in addition to the MSC markers CD13 and CD73. Tumors from mice treated with 14G2a, isotype-control antibody, and PBS were analyzed using the same gating strategy. Frequencies of lymphoid and myeloid cells were determined from live doublet-excluded CD45+ parent cell populations. Among the lymphocyte populations, NK cell frequencies were defined as CD3-NK1.1+ NKp46+ cells, T-cell frequencies were defined as CD3+NK1.1-NKp46- cells, invariant natural killer T cells were defined as CD1d tetramer-binding CD3+, and γδ T cells were defined as γδ TCR+CD3+ cells. NK cell surface expression of activating (Ly49H, IL15Rα, and NKG2D) and inhibitory (Ly49A, NKG2A, and TGFβR-1) receptors were assessed. Among the myeloid populations, dendritic cells (DCs) were defined as F4/80-CD11c+CD11b+, monocytic myeloid-derived suppressor cells (M-MDSCs) were defined as Gr-1+CD11b+Ly6G-, granulocytic myeloid-derived suppressor cells (G-MDSCs) were defined as Gr-1+CD11b+Ly6G+, and macrophages were defined as MHC-II+F4/80+CD11c-.
Detection of lineage-defining proteins
For immunoblots, 30 µg of protein lysate was electrophoresed through a 4–12% Bis-Tris NuPAGE gel and transferred to PVDF membranes using an iBlot gel transfer device. Membranes were blocked for 60 min with a 5% milk in 0.1% Tween-20 in TBS (TTBS) solution followed by TTBS wash, then incubated overnight at 4°C with antibodies for Phox2B (1:1000; Santa Cruz Biotechnology, clone B-11), Yap1 (1:1000; Cell Signaling, polyclonal), or β-tubulin (Sigma Aldrich, clone AA2). Secondary antibody (1:1000–1:2000) was applied for 60–120 min at room temperature, followed by wash steps and detection via horseradish peroxidase chemiluminescent substrate (Millipore). For testing cell lines by immunocytology, 10–20 × 106 live cells were suspended in a 1% agarose solution in isosmotic PBS at 50°C and transferred to a 1.5 mL Eppendorf tube and placed on ice until solid, then removed and fixed with formalin. For testing primary TH-MYCN tumors or tumor cell-line xenografts, tumors were removed in terminal surgeries and formalin-fixed and paraffin embedded. Slides of FFPE materials were deparaffinized, blocked with 5% horse serum, and stained. Yap1 (Cell Signaling Technology, clone D8H1X) and Phox2B (Abcam, clone EPR14423) were both used at 1:50. Slides were counterstained with hematoxylin. Images were captured using a Nikon 80i upright microscope using the 10x and 20x lenses. For assessing lineage markers in the TH-MYCN-derived cell lines by flow-cytometry, live, singlet cells were further characterized with CD13 and CD73 (MSC markers), Phox2B (adrenergic marker), and NCAM-1 and PGP9.5 (markers associated with neuroblastoma), as well as MHC-I.
Statistical analyses
Statistical analyses were performed using Prism software (GraphPad). Survival curves were compared using the method of Kaplan–Meier with a log-rank test for significance. For comparison of groups in flow cytometry analyses, groups were compared using unpaired two-tailed Student’s t-tests. For comparison of viability of cell suspensions, the Mann–Whitney U-test was used. For all, the significance was set as p ≤ .05 and trends as p > .05 and ≤0.10.
Results
GD2 is highly expressed on tumor cells within TH-MYCN neuroblastomas in situ but is lost during propagation as tumor-derived cell lines
End-stage intra-abdominal TH-MYCN+/+ mouse primary tumors were harvested at the time of sacrifice for symptomatic progression (per consort diagram in Fig. S1) and examined for the expression of GD2 on neuroblasts (). GD2 was expressed by the majority of CD45-NCAM1+ cells (median positivity: 86% [range 56–96%]; mean fluorescence intensity (MFI): 7504 [range 2,701–10,013]) from all unmanipulated TH-MYCN tumors (n = 7), using the gating strategy shown in Fig. S2. To further characterize the CD45-NCAM1+GD2+ cells and confirm their identity as neuroblastoma cells, we evaluated the expression of additional neuroblast markers (PGP9.5, Phox2b) and MSC markers (CD13 and CD73). While a moderate percentage (~16%) of these cells expressed both the additional neuroblast marker PGP9.5 and the MSC marker CD73, very few (<1%) expressed both CD73 and CD13, indicating few tumor-infiltrating MSCs and validating this population as overwhelmingly neuroblastoma cells. We next assessed GD2 expression on cell lines previously established and passaged from tumors arising in our TH-MYCN colony (using gating strategy in Fig. S3A). Despite robust GD2 expression on primary tumor cells, tumor-derived cell lines uniformly had <5% GD2 expression though they remained tumorigenic (data not shown). To assess for temporal variability in surface GD2 levels, we passaged these TH-MYCN cell lines weekly, yet GD2 expression remained consistently low. We compared this with GD2 expression on human neuroblastoma cell lines (Fig. S3B). These latter cell lines retained stable and high GD2 expression despite having been passaged extensively in tissue culture, with the exception of one outlier (NLF) with a stable lower frequency of GD2+ cells ( and Fig. S3C).
Figure 1. GD2 and lineage marker expression in TH-MYCN tumors, primary explants and cell lines, and human neuroblastoma cell lines. (a) TH-MYCN+/+ neuroblastomas (NB), previously established and propagated TH-MYCN+/+ and human NB cell lines were examined for surface GD2 expression by flow cytometry in single, live, CD45- cells, and NCAM1+ and CD56+ for mouse and human NBs, respectively. Cell lines were passaged for 3 weeks to assess GD2 stability. (b) Fresh TH-MYCN+/+ tumors were explanted and carried in tissue culture to monitor the GD2+ proportion of tumor cells over time ex vivo. (c) Immunoblot detection of a principal adrenergic marker, Phox2b, principal mesenchymal marker, Yap1, and β-tubulin loading control. IMR5 and SHEP are neuroblastoma cell lines with a predominantly adrenergic or mesenchymal lineage, respectively. TH-MYCN+/+ primary tumors are predominantly adrenergic marker expressing. TH-MYCN+/+ tumor-derived cell lines are largely mesenchymal marker expressing when propagated ex vivo (>3 months, 3000-series cell lines) whereas marker expression is heterogeneous during earlier passage ex vivo (5000-series cell lines). (d) Immunocytochemistry and immunohistochemistry to detect Phox2b and Yap1 from FFPE cell line pellets or primary tumors are shown, concordant with immunoblot results.
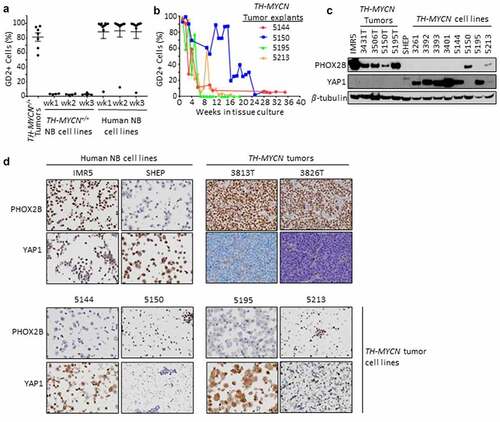
To reconcile these findings, we explanted four separate TH-MYCN primary tumors into tissue culture and serially examined the expression of GD2 over time from explant through outgrowth of a cell line. Nearly all CD45-NCAM1+ live neuroblasts expressed high levels of GD2 at the time of initiation of the culture (with a median of 95% GD2+ NCAM1+ CD45- live cells [range 93–99%]). However, the proportion of such tumor cells expressing GD2 subsequently decreased over time to <10% positive (). The kinetics of GD2 loss varied but all reached a stable low or absent level between 6 and 24 weeks of propagation. Therefore, while human neuroblastoma cells maintain GD2 expression while propagated ex vivo, TH-MYCN-derived cell lines variably lose surface GD2 expression after they are explanted to tissue culture conditions. Given the loss of GD2, we sought to validate that these TH-MYCN cell lines were indeed neuroblastoma cells and not an outgrowth of MSCs or another stromal cell population. These cell lines generally remained tumorigenic and retained MycN expression and were absent CD13/CD73 double-positive cells characteristic of MSCs (data not shown).Citation29
TH-MYCN tumors possess an adrenergic-predominant state, whereas TH-MYCN tumor-derived cell lines possess a mesenchymal-predominant state
Human neuroblasts and cell lines have lineage plasticity and can trans-differentiate between an adrenergic (sympathetic) and mesenchymal (neural-crest-like) state.Citation22,Citation23 Since these epigenetic phenotypes associate with distinct transcriptional outputs, we determined the lineage state of TH-MYCN tumor cells using a consensus adrenergic marker, Phox2b, and a consensus mesenchymal marker, Yap1 by immunoblot, immunocytology, and/or immunohistochemistry.Citation22,Citation23 All TH-MYCN primary tumors expressed abundant Phox2b but were absent Yap1, supporting an adrenergic-predominant state ()) in association with their high GD2 expression. In contrast, all established TH-MYCN tumor-derived cell lines expressed abundant Yap1 and were absent Phox2b, consistent with a mesenchymal-predominant state () and Fig. S4) in association with their low GD2 expression. We next studied TH-MYCN tumor explants at intermediate time-points in tissue culture. The nascent TH-MYCN cell lines 5144 and 5195 both lost surface GD2 expression rapidly () and expressed mesenchymal markers even at early time-points (). However, the two TH-MYCN cell lines with prolonged retention of GD2 expression were found to retain an adrenergic expression pattern (5150) or express markers of each lineage, including retained Phox2b (5213), when assessed at intermediate time-points. Overall, there was striking concordance between GD2 expression and adrenergic lineage in the TH-MYCN models. Further, expression of B4galnt1, the terminal galactosaminyl-transferase in GD2 synthesis, was present in TH-MYCN primary tumors (adrenergic state) but absent in TH-MYCN cell lines (mesenchymal state; Fig. S4B). To assess whether 5213 cells included tumor cells that co-expressed Phox2b and Yap1 or an admixture of both lineage states, we studied cell pellets by immunocytology, which showed a subpopulation of Phox2b expressing cells and a subpopulation of Yap1 expressing cells (). We next determined whether the tissue-culture adopted mesenchymal state could readily revert to the adrenergic state in situ, as human mesenchymal-predominant neuroblastoma cell lines do when re-implanted as tumor xenografts.Citation22 We established Yap1+ TH-MYCN cell lines as allografts in the flank of syngeneic mice and then used immunohistochemistry to detect Phox2b and Yap1, demonstrating that they retained their mesenchymal status (Fig. S4C). In contrast, we processed a primary Phox2b+ TH-MYCN+/+ tumor as a single-cell suspension that was allografted into the flank of a syngeneic TH-MYCN+/- recipient mouse, without any intervening tissue culture propagation. The allografted tumor retained expression of adrenergic lineage markers (Fig. S4D). These data support a lineage switch from adrenergic to mesenchymal state when TH-MYCN murine neuroblasts are cultured ex vivo leading to loss of GD2 expression, likely due to loss of adrenergic transcriptional programs.
Treatment of TH-MYCN tumor-bearing mice with an anti-GD2 antibody induces tumor regression and extends survival
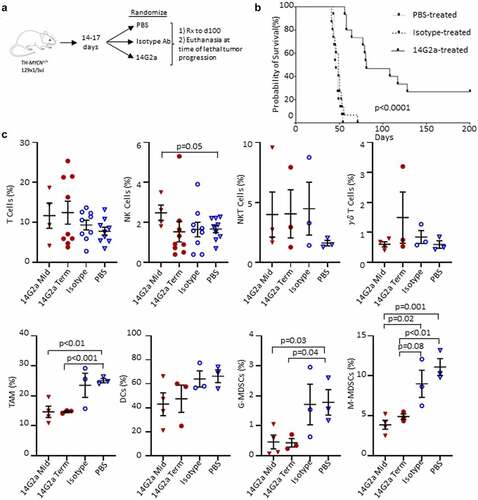
Since TH-MYCN neuroblasts retain high levels of GD2 expression in situ despite losing GD2 expression in tissue culture, we sought to determine the effect of treatment with an anti-GD2 antibody.Citation19 TH-MYCN mice (n = 15 per treatment arm) were randomly assigned to therapy with 14G2a (a murine anti-GD2 antibody), an isotype-matched control antibody, or PBS (). Mice received their assigned treatment twice weekly from day of life 14–17 (~1–2 weeks after tumor initiation, as identified by histologic audits) until they were sacrificed for signs of morbidity from tumor progression (poor mobility, hunching, lethargy, weight loss, limb paralysis, neurological changes, dermatitis, and rough hair coat), or until day 100.Citation30 All mice treated with an isotype-control antibody or PBS had progressive tumor growth and were euthanized while still receiving therapy (median survival time of 46 and 49 days, respectively; ). In contrast, mice treated with 14G2a antibody had significantly extended survival (median survival time of 81 days; p < .001 compared with either control arm) with 7 mice completing assigned treatment through day 100, beyond which they continued to be observed for signs of tumor progression. 14G2a monotherapy led to objective tumor regression in 6 of the 15 mice (40%), and 4 of these mice (27%) had complete regression to palpation that was maintained until day of life 200.
Figure 2. Experimental summary and survival of TH-MYCN+/+ mice and immune cells in the tumor microenvironment. (a) Transgenic TH-MYCN+/+mice were enrolled into 3 treatment groups: PBS, 14G2a, and Isotype. Mice were treated with intraperitoneal (IP) injections twice weekly until death or significant morbidity, and then had tumors isolated in terminal surgeries. (b) Kaplan–Meier survival curve shown by treatment group. 4 mice living at day 200 in the 14G2a-treated group were censored for re-challenge with flank implantation of a TH-MYCN-derived neuroblastoma cell line. Log-rank (Mantel-Cox) test was used to compare survival curves with significance set as p < .05. (c) TH-MYCN+/+ neuroblastoma tumors were isolated from 14G2a midpoint (Mid), 14G2a terminal (Term), Isotype, and PBS treatment groups. Single cell suspensions of tumors were immunostained with fluorescent antibodies specific for markers of T, natural killer (NK), natural killer T (NKT), and gamma delta (γδ) T cells, in addition to markers for tumor associated macrophage (TAM), dendritic cells (DCs), granulocytic myeloid-derived suppressor cells (G-MDSCs) and monocytic myeloid-derived suppressor cells (M-MDSCs). Specified cell frequencies are reported as a percentage of single, live, CD45+ parent populations, by treatment group. Groups were compared with two-tailed Student’s t-test with significance set as p ≤ .05 (trend p ≤ .10).
TH-MYCN mice achieving durable tumor control following anti-GD2 therapy are not protected against re-challenge with syngeneic tumor
Four 14G2a-treated TH-MYCN+/+ mice appeared healthy with no palpable tumor at day 200 (100 days after therapy completion). To assess whether a memory T cell response had developed in these mice in response to neo-epitopes uncovered as a result of the antibody therapy or other immune changes evoked, we re-challenged them with a subcutaneous injection of a tumorigenic TH-MYCN-derived cell line (HOM2 cells). HOM2 cells had been serially passaged in vivo and demonstrated a reduced latency to tumor take and partial retention of GD2 expression (in ~20% of cells). Tumor-free TH-MYCN+/- (n = 4) and TH-MYCN−/− (n = 4; wild-type) mice were concurrently challenged. Tumorigenicity was 100% across all mice, with tumors established at a median of 11.5 and 9 days, respectively, for TH-MYCN+/- and TH-MYCN−/− control mice and 15 days in re-challenged TH-MYCN+/+ mice whose original autochthonous tumors had regressed with anti-GD2 therapy (Fig. S5A). Like other TH-MYCN cell lines, HOM2 cells predominantly express mesenchymal markers as do their established allografted tumors (Fig. S5B).
Characteristics of tumor-infiltrating immune cells in TH-MYCN tumors
TH-MYCN tumors recruit an immunosuppressive microenvironment similar to human neuroblastomas, with a reduction in cytolytic T cells and increase in TAMs, dendritic cells, and MDSCs.Citation31–33 To determine the impact 14G2a antibody therapy had on the TME, we examined the overall frequencies of intratumoral leukocytes in tumors harvested at terminal timepoints among treatment arms. In addition, we sought to identify early immune changes at the time of tumor regression in 14G2a-treated mice (midpoint tumors), as some changes might lessen over time and some terminal timepoint mice completed therapy prior to being sacrificed. Given that mice were enrolled over a 3-month period, we viably froze aliquots of single-cell tumor suspensions before thawing, staining, and running the cells through a flow cytometer for TIL characterization to reduce batch effects. Viability tested prior to freezing and after thawing did not differ (Supplementary Table 1). It is possible that the freeze–thaw event contributed to changes in selective TIL populations or marker expression, yet this would apply across all samples.
Conventional T cell, invariant natural killer T (iNKT) cell, gamma-delta (γδ) T cell, dendritic cell, and NK cell frequencies were not statistically different in 14G2a-treated tumors when compared with isotype-control antibody or PBS-treated tumors (). NK cells were more frequent in midpoint 14G2a-treated tumors when compared with PBS-treated terminal tumors (p = .05) but not isotype-control treated tumors (p = .19). There were reduced monocytic-MDSCs (M-MDSCs) in 14G2a-treated tumors compared with isotype-control antibody at both midpoint (p = .02) and terminal timepoints (trend, p = .08). Additionally, macrophages (TAMs) and granulocytic-MDSCs (G-MDSC) were all present at reduced frequencies at both midpoint and terminal timepoints when compared with PBS treated but not isotype-treated tumors.
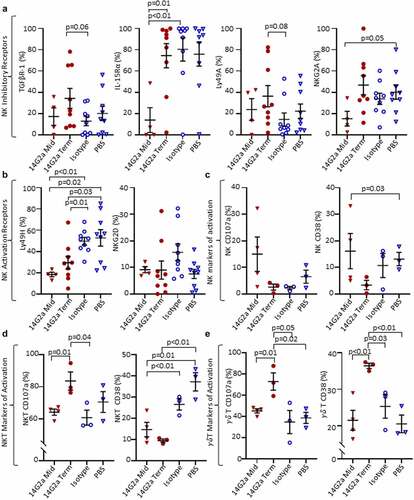
Since NK cells are postulated to be major effectors of the therapeutic response induced by GD2-directed antibody therapy,Citation34,Citation35 we examined their pattern of inhibitory and activating receptors and found a trend toward increased frequencies of intratumoral NK cells expressing the inhibitory TGFβ receptor type 1 (TGFβR1) and Ly49A receptor (p = .06 and 0.08, respectively), and a decrease in the activating Ly49H receptor (p = .01) in the 14G2a-treated group compared with isotype-treated controls. For Ly49H, these changes were also significant when compared with midpoint tumors and compared with PBS-treated tumors (). There were no differences in the proportions of NK cells expressing NKG2D, NKG2A, or interleukin-15 receptor-α (IL-15Rα) among treatment groups at terminal timepoints, although significantly less IL-15Rα expression was observed in midpoint tumor NK cells compared with isotype and 14G2a-treated terminal groups (). Cell surface expression of CD107a, a biomarker of cytotoxic degranulation, was also higher in iNKT and γδ T cells from 14G2a-treated tumors relative to the isotype-control group for both midpoint and terminal tumors (). Similarly, the expression of CD38 remained elevated in γδ T but not NKT cells in the 14G2a terminal groups. It has been suggested that ligation of CD38 may activate T and NK cells and be important for iNKT cell survival.Citation36–38 Overall, while altered TAM and MDSC frequencies suggest that the balance of intratumoral immunosuppressive to effector cells may favor tumor control, NK cell expression of activation and inhibitory receptors are inversely altered, suggesting that tumor-infiltrating NK cells may be more immune tolerant than pro-inflammatory. Conversely, iNKT cells from 14G2a-treated tumors appear to have upregulated CD107a, but iNKT cell frequencies were not altered with treatment. Finally, to evaluate for changes in mice responding to 14G2a-treatment, we evaluated the frequency of intratumoral NK and T cells in mice living ≥ vs. <81 days, the median survival in the 14G2a treatment group. We found that the mean T cell frequency in mice living ≥81 days was greater than that in mice living <81 days (22.73% vs 7.29%, p = .001) and NK-cell frequency was not significantly different in the mice “responding” to 14G2a (0.77% vs 1.9%, p = .32; Fig. S6A). Furthermore, the frequency of intratumoral T cells, but not NK cells, was directly correlated with the length of survival (RCitation2 = 0.64, p = .01; Fig. S6B), while the relationship between intratumoral T-cell frequency and length of survival was not observed in isotype and PBS-treated controls (data not shown).
Figure 3. NK, NKT, and γδ T cell surface markers in the TH-MYCN+/+ TME. Cell surface protein expression from TH-MYCN+/+ neuroblastoma-derived natural killer (NK), invariant natural killer T (NKT), and gamma delta (γδ) T Cells from 14G2a midpoint (Mid), 14G2a terminal (Term), Isotype, and PBS groups. (a) NK Inhibitory receptor expression including transforming growth factor beta receptor-1 (TGFBR-1), interleukin-15 receptor-alpha (IL-15Rα), Ly49A, and NKG2A. (b) NK Activation receptor expression of Ly49H and NKG2D. (c-e) Expression of markers of cellular activation CD38 and CD107a are shown for NK, NKT, and γδ T cells. Two-tailed Student’s t-test performed to compare groups with significance set as p ≤ .05 (trend p ≤ .10).
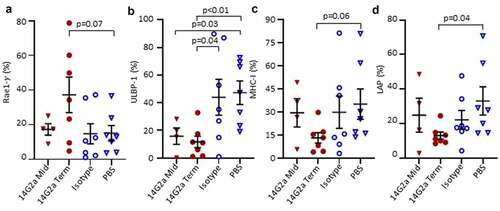
To further probe whether the changes noted in intratumoral NK cells reflected alterations in the susceptibility of neuroblasts to immune control, we examined neuroblast expression of ligands that regulate NK cell reactivity to putative targets, including NK cell activating ligands retinoic acid early-inducible protein 1-gamma (Rae-1γ) and UL16 binding protein 1 (ULBP-1), and NK cell inhibiting ligands major histocompatibility class I (MHC-I) and latency-associated peptide (LAP; ). We found that a lower proportion of 14G2a-treated neuroblasts expressed ULBP-1 relative to isotype control (p = .04), while there was no difference in expression of Rae-1γ. MHC-I expression was lower in 14G2a-treated mice at terminal timepoints relative to PBS control (trend, p = .06) but not isotype. The frequencies of LAP-expressing neuroblasts were lower in 14G2a-treated versus PBS-treated mice (p = .04), but not significantly different when compared with isotype. NK cells are activated via binding of NKG2D to ULBP-1; therefore, the observed decreased expression of ULBP-1 in 14G2a-treated mice tumors may reflect a mechanism for neuroblast immune evasion under increased pressure with 14G2a treatment. CD8+ T cells recognize tumor antigens on MHC-I, and the trend toward decreased MHC-I expression in 14G2a-treated mice may reflect a well-described mechanism of T-cell immune evasion in neuroblastoma, although MHC downregulation would also allow for NK cell activation.Citation39,Citation40 Collectively, these data highlight the complexities of interactions and alterations in the TH-MYCN neuroblastoma TME associated with anti-GD2 treatments.
Figure 4. Surface expression of NK ligands on TH-MYCN+/+ neuroblasts. Tumors from 14G2a midpoint (Mid), 14G2a terminal (Term), Isotype, and PBS-treated TH-MYCN+/+ mice were isolated. Single cell suspensions were immunostained and CD45-, GD2+, live cells were analyzed for cell surface expression of specified NK Ligands. (a-d) Retinoic acid early-inducible protein 1-γ (Rae1-γ), UL16 Binding Protein-1 (ULBP-1), Major Histocompatibility Complex Class I (MHC-I), and Latency Associated Peptide (LAP) expression on live, CD45-, GD2+ neuroblasts is shown. Bars show the mean and standard error of the mean. Groups were compared with Students t-test with significance set as p ≤ .05.
Discussion
The use of models of cancer that faithfully recapitulate human disease has enabled the discovery and preclinical prioritization of novel therapies. Here, we credential the TH-MYCN transgenic mouse for neuroblastoma immunotherapy studies. TH-MYCN mice activate the principal oncogenic driver of neuroblastoma, MYCN, to initiate tumors at autochthonous sympathoadrenal sites whose distinct cooperating mutations influence the immunoediting process. This in situ immunoediting results in the recapitulation of the genomic, microenvironmental, and immunologic heterogeneity that defines cancer. As such, the TH-MYCN model presents distinct advantages over syngeneic transplant models for investigating the activity of immunotherapies, such as dinutuximab. Although the model is well established, it has been underutilized as a platform to test GD2-directed therapies due to the pervasive impression that TH-MYCN tumor cells express insufficient surface GD2 to be targeted. Here, we show that tumors in situ express GD2 levels at high frequencies, and that this expression is therapeutically actionable. We observed a profound survival benefit with 14G2a therapy with near doubling of median survival time, 40% of mice had objective tumor regressions, and 30% had durable complete responses beyond 200 days. In addition, tumors arising in TH-MYCN+/- mice were recently shown to respond to 14G2a therapy combined with cyclophosphamide, similar to findings in children with relapsed neuroblastoma in the COG ANBL1221 chemoimmunotherapy trial.Citation7,Citation19
Notably, 7 of 15 14G2a-treated mice in our trial lived without morbidity beyond cessation of antibody administration at day 100, while none of the mice in the control groups survived beyond day 70. This finding prompted us to consider whether the mice with prolonged survival had gained immunologic memory due to epitope-spreading resulting from anti-GD2 antibody therapy. This was tested in a re-challenge study in the four mice that lived past day 200. Yet, there was no decrease in tumorigenicity or significant change in time to tumor establishment among these groups. This observation suggests a lack of strongly protective anamnestic responses in the re-challenged mice, although we did not perform in-depth analyses to rule out more subtle memory responses to TH-MYCN neuroblast cell lines. It should be noted, however, that this finding is limited by the switch in lineage state in the cell line used for re-challenge, which restricts the interpretation of this finding to mesenchymal GD2-negative cell lines from TH-MYCN mice. Finally, we do not believe that the lack of GD2 expression in this cell line would lead to loss of T cell-mediated protection, as GD2 is a disialoganglioside rather than a protein antigen to which conventional T cells could respond.
We also show that established TH-MYCN tumor-derived cell lines have markedly reduced GD2 expression in the absence of prior exposure to an anti-GD2 immunotherapy, confirming the findings of others.Citation20,Citation21 Importantly, TH-MYCN neuroblasts lose GD2 during the process of explanting the primary tumor and propagating as a cell line, such that serial syngeneic tumor allografts are unlikely to be useful for GD2-directed therapeutic studies, as shown previously.Citation20,Citation21 The majority of GD2 expression was lost in <8 weeks of propagation, suggesting the in situ TME provides a stimulus for GD2 expression, or that tissue culture conditions induce its loss. It has previously been shown that neuroblastomas exist in distinct epigenetic states that can interconvert.Citation23 Indeed, we show that a change in lineage state with conversion from a predominantly adrenergic state to a mesenchymal state accompanies loss of GD2 from the tumor cell surface. This change in state has been genetically associated with NOTCH-family transcription factor signaling, loss of ARID1A expression, and with TNF-α and epidermal growth factor exposure in vitro.Citation41–43 It has previously been suggested that GD2 is expressed more highly at cell–cell junctions and it is possible that disruption of these contacts and removal from the sympatho-adrenal location contribute to GD2 loss in tissue culture.Citation44 Notably, the expression of GD2 synthases, in particular the B4GALNT1 enzyme that catalyzes the final step in synthesis, is confined to sympathoblasts (adrenergic) in single-cell RNAseq studies.Citation45 Plausibly, GD2 is expressed as part of the transcriptional output of the adrenergic state and is lost as a consequence of the mesenchymal lineage change. Our finding that TH-MYCN-derived tumor cells can assume a mesenchymal state both in culture and in reestablished allografts may prove important. Emerging evidence suggests that adrenergic and mesenchymal states have profound differences in inflammatory signaling and immune susceptibility, and an immunocompetent model to study these states is lacking.Citation46 However, these findings also support that allografts of TH-MYCN cell lines that have been engineered to re-express GD2 are unlikely to recapitulate the predominantly adrenergic state of primary tumors and may not be faithful models for GD2-directed immunotherapies.
Dinutuximab is purported to exert its antitumor effect via antibody-dependent cellular cytotoxicity (ADCC) as well as complement-dependent cytotoxicity.Citation9 In our evaluation of the TH-MYCN TME, we did not observe differences in intratumoral NK, T, and iNKT cell frequencies between 14G2a and isotype-control groups at terminal timepoints but did observe an increase in NK cells at the time of response to 14G2a (midpoint tumors) compared with the PBS-treated group, and an association between T cell frequency and survival length in the 14G2a-treated group. NK cells are known to transactivate iNKT cells, and we did observe an increase in CD107a expression in 14G2a-treated intratumoral iNKT cells.Citation47 These findings suggest that the anti-GD2 antibody may result in downstream iNKT cell degranulation. Additionally, it has been shown that activated iNKT cells reshape the immunosuppressive TME through recognition and lysis or reprogramming of TAMs and MDSCs.Citation48 It was therefore interesting to find that macrophage, M-MDSC, and G-MDSC populations were reduced in 14G2a-treated groups. As TAMs and MDSCs are known to provide critical support for neuroblastoma growth and are enriched in metastases, the reduced intratumoral frequency of these cells in the 14G2a-treatment group may represent a beneficial effect of 14G2a contributing to the prolonged survival seen in TH-MYCN mice.Citation33,Citation49–51 Our analyses of the TME was limited by relatively small numbers of mice in each treatment group. However, significant findings including the decreased TAM and MDSC frequency in 14G2a-treated mice compared with controls and an increase in T-cell infiltration in TH-MYCN tumors responding to 14G2a therapy provide useful preliminary data to guide future studies of this immunocompetent mouse model.
Intratumoral NK cells treated with 14G2a had lower expression of the activating receptor Ly49H and greater expression of inhibitory receptors TGFβ-R1 and Ly49A relative to the isotype control antibody cohort. We speculate that this finding indicates that the mice who eventually succumb to disease are ones in whom the 14G2a-induced immune pressure has diminished. This altered NK receptor profile following anti-GD2 antibody treatment provides potential opportunities for enhancing 14G2a-mediated efficacy. For instance, binding of TGFβ to the increased TGFβ-R1 could diminish NK cell gamma interferon- γ (IFN-γ) production and ADCC, thereby contributing to 14G2a resistance.Citation52 Use of TGFβ-R1 blockade with galunisertib has been used in vitro and in immunocompromised mice with improved efficacy of GD2-directed antibody therapy, but not in immunocompetent TH-MYCN mice.Citation53
We also show that a lower frequency of TH-MYCN neuroblasts from 14G2a-treated mice expresses the NK cell activating ligand ULBP-1 than those from isotype-treated control mice. Conversely, a trend toward a larger proportion of neuroblasts expressed Rae-1γ in mice treated with 14G2a relative to the isotype control antibody, although this did not reach statistical significance. Percentages of neuroblasts expressing LAP and MHC-I were decreased in 14G2a-treated terminal tumors relative to PBS-treated terminal tumors. MHC-I downregulation, which is commonly found in human neuroblastoma at diagnosis, has previously been described as a mechanism for T cell immune evasionCitation39. This finding in 14G2a-treated tumors may represent a response to increased immunologic pressure although, as noted previously, decreased MHC-I expression also allows for NK activation.Citation34 The downregulation of MHC-I in this mouse model may therefore reflect the TH-MYCN tumor immune evasion response to an increased frequency of T cells with prolonged survival seen in those treated with 14G2a (Fig. S6B).
The TH-MYCN mouse model offers many advantages for the study of immune:tumor interactions although there are limitations including the low frequency of macrometastases.Citation17 Anti-GD2 therapies against neuroblastoma have been studied in orthotopic models of human cell lines or patient-derived xenografts established in immunodeficient mice.Citation17,Citation54,Citation55 These studies showed promising results regarding the augmented cytotoxic effect of anti-GD2 immunotherapy in combination with soluble IL-15/IL-15Rα and GM-CSF or ex vivo activated human NK cells following tumor resection but are limited by their reduced immune repertoire and non-native TME.Citation54,Citation55 A combination of complementary pre-clinical mouse models may provide an ideal approach to evaluate GD2-directed immunotherapies in neuroblastoma. Collectively, our findings support the utility of the TH-MYCN immunocompetent mouse for pre-clinical assessments of the efficacy of immunotherapies, including combination approaches that may synergize with GD2-targeted therapies. We also demonstrate the usefulness of the model for evaluating TME changes, which may inform future therapeutic approaches, though validating these findings using non-cryopreserved tumor suspensions across larger numbers is warranted given their translational relevance. Such pre-clinical assessments have potential for expediting the discovery of new treatment options for high-risk neuroblastoma.
List of abbreviations
Author contributions
KOM and SK designed, performed experiments, data analysis, created figures, and initial draft of the manuscript; FG designed, performed experiments, and data analysis; AW, CB, PK, RV, and AV provided technical expertise, performed experiments, and data analysis. MDH and HB conceived and supervised the study, were involved in the design and evaluation of experiments, critically reviewed all drafts of the manuscript. All authors contributed to the writing of the manuscript, critically reviewed, and approved the submitted version.
Availability of data and material
All data that support the findings of this study are available from the corresponding authors upon reasonable requests.
Consent for publication
The authors give consent for publication of referenced data. There are no identifiable details or human subject data included in this manuscript.
Ethics approval and consent to participate
All animal studies were carried out under a protocol approved by IACUC at the Children’s Hospital of Philadelphia. Protocol #16-000905.
Supplemental Material
Download Zip (674.3 KB)Acknowledgments
We would like to thank Dr. Garrett M. Brodeur (Division of Oncology, Children’s Hospital of Philadelphia) for kindly sharing the HOM2, G2, and G3B cell lines. We would like to thank the Wistar Institute Histotechnology Facility for assistance with immunohistochemistry on neuroblastoma tumors and cell pellets, and Ocean Malka for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2075204
Additional information
Funding
References
- Hwang WL, Wolfson RL, Niemierko A, Marcus KJ, DuBois SG, Haas-Kogan D. Clinical impact of tumor mutational burden in neuroblastoma. J Natl Cancer Inst. 2019;111:695–13. doi:10.1093/jnci/djy157.
- Noskova H, Kyr M, Pal K, Merta T, Mudry P, Polaskova K, Ivkovic TC, Adamcova S, Hornakova T, Jezova M, et al. Assessment of tumor mutational burden in pediatric tumors by real-life whole-exome sequencing and in silico simulation of targeted gene panels: how the choice of method could affect the clinical decision? Cancers. 2020;12:230. doi:10.3390/cancers12010230.
- Coughlan D, Gianferante M, Lynch CF, Stevens JL, Harlan LC. Treatment and survival of childhood neuroblastoma: evidence from a population-based study in the United States. Pediatr Hematol Oncol. 2017;34:320–330. doi:10.1080/08880018.2017.1373315.
- Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, Corrias MV, Moretta A, Bottino C. Neuroblastoma-Derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol. 2013;190:5321–5328. doi:10.4049/jimmunol.1202693.
- Brandetti E, Veneziani I, Melaiu O, Pezzolo A, Castellano A, Boldrini R, Ferretti E, Fruci D, Moretta L, Pistoia V, et al. MYCN is an immunosuppressive oncogene dampening the expression of ligands for NK-cell-activating receptors in human high-risk neuroblastoma. OncoImmunology. 2017;6:e1316439. doi:10.1080/2162402X.2017.1316439.
- Hashimoto O, Yoshida M, Koma Y, Yanai T, Hasegawa D, Kosaka Y, Nishimura N, Yokozaki H. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development: the role of non-tumour stromal cells in neuroblastoma progression. J Pathol. 2016;240:211–223. doi:10.1002/path.4769.
- Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, Parisi MT, Servaes SEN, Diccianni MB, Sondel PM, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol. 2017;18:946–957. doi:10.1016/S1470-2045(17)30355-8.
- Mody R, Yu AL, Naranjo A, Zhang FF, London WB, Shulkin BL, Parisi MT, Servaes SEN, Diccianni MB, Hank JA, et al. Irinotecan, temozolomide, and dinutuximab with GM-CSF in children with refractory or relapsed neuroblastoma: a report from the children’s oncology group. J Clin Oncol Off J Am Soc Clin Oncol. 2020;38:2160–2169. doi:10.1200/JCO.20.00203.
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:7554–7564. doi:10.1056/NEJMoa0911123.
- Yu AL, Gilman AL, Ozkaynak MF, Naranjo A, Diccianni MB, Gan J, Hank JA, Batova A, London WB, Tenney SC, et al. Long-term follow-up of a phase iii study of ch14.18 (dinutuximab) + cytokine immunotherapy in children with high-risk neuroblastoma: COG Study ANBL0032. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27:2179–2189. doi:10.1158/1078-0432.CCR-20-3909.
- Hanahan D. Transgenic mice as probes into complex systems. Science. 1989;246:1265–1275. doi:10.1126/science.2686032.
- Roussel MF, Stripay JL. Modeling pediatric medulloblastoma. Brain Pathol. 2019:12803. doi:10.1111/bpa.12803.
- Feuring-Buske M, Gerhard B, Cashman J, Humphries RK, Eaves CJ, Hogge DE. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia. 2003;17:760–763. doi:10.1038/sj.leu.2402882.
- Weiss WA. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–2995. doi:10.1093/emboj/16.11.2985.
- Rasmuson A, Segerström L, Nethander M, Finnman J, Elfman LHM, Javanmardi N, Nilsson S, Johnsen JI, Martinsson T, Kogner P, et al. Tumor development, growth characteristics and spectrum of genetic aberrations in the TH-MYCN mouse model of neuroblastoma. PLoS ONE. 2012;7:e51297. doi:10.1371/journal.pone.0051297.
- Hackett CS, Hodgson JG, Law ME, Fridlyand J, Osoegawa K, de Jong PJ, Nowak NJ, Pinkel D, Albertson DG, Jain A, et al. Genome-wide array CGH analysis of murine neuroblastoma reveals distinct genomic aberrations which parallel those in human tumors. Cancer Res. 2003;63(17):5266–5273.
- Teitz T, Stanke JJ, Federico S, Bradley CL, Brennan R, Zhang J, Johnson MD, Sedlacik J, Inoue M, Zhang ZM, et al. Preclinical models for neuroblastoma: establishing a baseline for treatment. PloS One. 2011;6:e19133. doi:10.1371/journal.pone.0019133.
- Moore HC, Wood KM, Jackson MS, Lastowska MA, Hall D, Imrie H, Redfern CPF, Lovat PE, Ponthan F, O’Toole K, et al. Histological profile of tumours from MYCN transgenic mice. J Clin Pathol. 2008;61:1098–1103. doi:10.1136/jcp.2007.054627.
- Webb ER, Lanati S, Wareham C, Easton A, Dunn SN, Inzhelevskaya T, Sadler FM, James S, Ashton-Key M, Cragg MS, et al. Immune characterization of pre-clinical murine models of neuroblastoma. Sci Rep. 2020;10:16695. doi:10.1038/s41598-020-73695-9.
- Kroesen M, Brok IC, Reijnen D, van Hout-kuijer MA, Zeelenberg IS, Den Brok MH, Hoogerbrugge PM, Adema GJ. Intra-adrenal murine TH-MYCN neuroblastoma tumors grow more aggressive and exhibit a distinct tumor microenvironment relative to their subcutaneous equivalents. Cancer Immunol Immunother CII. 2015;64:563–572. doi:10.1007/s00262-015-1663-y.
- Voeller J, Erbe AK, Slowinski J, Rasmussen K, Carlson PM, Hoefges A, VandenHeuvel S, Stuckwisch A, Wang X, Gillies SD, et al. Combined innate and adaptive immunotherapy overcomes resistance of immunologically cold syngeneic murine neuroblastoma to checkpoint inhibition. J Immunother Cancer. 2019;7:344. doi:10.1186/s40425-019-0823-6.
- Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugene C, Raynal V, Etchevers HC, Thomas S, Lermine A, Daudigeos-Dubus E, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet. 2017;49:2179–2189. doi:10.1038/ng.3921.
- van Groningen T, Koster J, Valentijn LJ, van Groningen T, Zwijnenburg DA, Akogul N, Hasselt NE, Broekmans M, Haneveld F, Nowakowska NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. 2017;49(8):1261–1266. doi:10.1038/ng.3899.
- Haraguchi S, Nakagawara A. A simple PCR method for rapid genotype analysis of the TH-MYCN transgenic mouse. PloS One. 2009;4:e6902. doi:10.1371/journal.pone.0006902.
- Winter C, Pawel B, Seiser E, Zhao H, Raabe E, Wang Q, Judkins AR, Attiyeh E, Maris JM. Neural cell adhesion molecule (NCAM) isoform expression is associated with neuroblastoma differentiation status. Pediatr Blood Cancer. 2008;51:10–16. doi:10.1002/pbc.21475.
- Phimister E, Kiely F, Kemshead JT, Patel K. Expression of neural cell adhesion molecule (NCAM) isoforms in neuroblastoma. J Clin Pathol. 1991;44:580–585. doi:10.1136/jcp.44.7.580.
- Bryson GJ, Lear D, Williamson R, Wong RCW. Detection of the CD56+/CD45- immunophenotype by flow cytometry in neuroendocrine malignancies. J Clin Pathol. 2002;55:535–537. doi:10.1136/jcp.55.7.535.
- Bozzi F, Collini P, Aiello A, Barzanò E, Gambirasio F, Podda M, Meazza C, Ferrari A, Luksch R. Flow cytometric phenotype of rhabdomyosarcoma bone marrow metastatic cells and its implication in differential diagnosis with neuroblastoma. Anticancer Res. 2008;28:1565–1569.
- Theodorakos I, Paterakis G, Papadakis V, Vicha A, Topakas G, Jencova P, Karchilaki E, Taparkou A, Tsagarakis N, Polychronopoulou S. Interference of bone marrow CD56+ mesenchymal stromal cells in minimal residual disease investigation of neuroblastoma and other CD45- /CD56+ pediatric malignancies using flow cytometry. Pediatr Blood Cancer. 2019;66:e27799. doi:10.1002/pbc.27799.
- Hansford LM, Thomas WD, Keating JM, Burkhart CA, Peaston AE, Norris MD, Haber M, Armati PJ, Weiss WA, Marshall GM, et al. Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Proc Natl Acad Sci. 2004;101:12664–12669. doi:10.1073/pnas.0401083101.
- Carlson L-M, Rasmuson A, Idborg H, Segerström L, Jakobsson P-J, Sveinbjörnsson B, Kogner P. Low-dose aspirin delays an inflammatory tumor progression in vivo in a transgenic mouse model of neuroblastoma. Carcinogenesis. 2013;34:1081–1088. doi:10.1093/carcin/bgt009.
- Layer JP, Kronmüller MT, Quast T, van den Boorn-Konijnenberg D, Effern M, Hinze D, Althoff K, Schramm A, Westermann F, Peifer M, et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology. 2017;6:e1320626. doi:10.1080/2162402X.2017.1320626.
- Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, Hadjidaniel M, Liu CWY, Metelitsa LS, Pique-Regi R, et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30:3525–3532. doi:10.1200/JCO.2011.40.9169.
- Tarek N, Le Luduec J-B, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, Modak S, Heller G, Dupont B, Cheung NK, et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest. 2012;122(9):3260–3270. doi:10.1186/s40425-019-0823-6.
- Zobel MJ, Zamora AK, Wu H, Sun J, Lascano D, Malvar J, Wang L, Sheard MA, Seeger RC, Kim ES, et al. Initiation of immunotherapy with activated natural killer cells and anti-GD2 antibody dinutuximab prior to resection of primary neuroblastoma prolongs survival in mice. J Immunother Cancer. 2020;8:e001560. doi:10.1136/jitc-2020-001560.
- Funaro A, Spagnoli GC, Ausiello CM, Alessio M, Roggero S, Delia D, Zaccolo M, Malavasi F. Involvement of the multilineage CD38 molecule in a unique pathway of cell activation and proliferation. J Immunol Baltim Md 1950. 1990;145:2390–2396.
- Chen Y-G, Chen J, Osborne MA, Chapman HD, Besra GS, Porcelli SA, Leiter EH, Wilson SB, Serreze DV. CD38 Is required for the peripheral survival of immunotolerogenic CD4 + invariant NK T cells in nonobese diabetic mice. J Immunol. 2006;177:10–16. doi:10.4049/jimmunol.177.5.2939.
- Sconocchia G, Titus JA, Mazzoni A, Visintin A, Pericle F, Hicks SW, Malavasi F, Segal DM. CD38 triggers cytotoxic responses in activated human natural killer cells. Blood. 1999;94:3864–3871. doi:10.1182/blood.V94.11.3864.
- Cornel AM, Mimpen IL, Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers. 2020;12:1760. doi:10.3390/cancers12071760.
- Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, Cerboni C, Santoni A. NKG2D and Its Ligands: ‘One for All, All for One’. Front Immunol. 2018;9:476. doi:10.3389/fimmu.2018.00476.
- Huang Y, Tsubota S, Nishio N, Takahashi Y, Kadomatsu K. Combination of tumor necrosis factor-α and epidermal growth factor induces the adrenergic-to-mesenchymal transdifferentiation in SH-SY5Y neuroblastoma cells. Cancer Sci. 2021;112(2):715–724. doi:10.1111/cas.14760.
- Shi H, Tao T, Abraham BJ, Durbin AD, Zimmerman MW, Kadoch C, Look AT. ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci Adv. 2020;6:eaaz3440. doi:10.1126/sciadv.aaz3440.
- van Groningen T, Akogul N, Westerhout EM, van Groningen T, Chan A, Hasselt NE, Zwijnenburg DA, Broekmans M, Stroeken P, Haneveld F, et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat Commun. 2019;10:1530. doi:10.1038/s41467-019-09470-w.
- Heiss P, Bermatz S, Wehnes H, Heinzmann U, Senekowitsch-Schmidtke R. Distribution of disialoganglioside GD2-antigen and binding of anti-GD2 antibodies on spheroids of neuroblastoma cell line. Anticancer Res. 1997;17:3145–3147.
- Kildisiute G, Kholosy WM, Young MD, Roberts K, Elmentaite R, van Hooff SR, Pacyna CN, Khabirova E, Piapi A, Thevanesan C, et al. Tumor to normal single-cell mRNA comparisons reveal a pan-neuroblastoma cancer cell. Sci Adv. 2021;7. doi:10.1126/sciadv.abd3311.
- Wolpaw AJ, Grossmann LD, Dong MM, Dessau JL, Dong MM, Aaron BJ, Brafford PA, Volgina D, Pascual-Pasto G, Rodriguez-Garcia A, et al. Epigenetic state determines inflammatory sensing in neuroblastoma. Proc Natl Acad Sci USA.2021;19(6). doi: 10.1073/pnas.2102358119.
- Wolf BJ, Choi JE, Exley MA. Novel approaches to exploiting invariant NKT cells in cancer immunotherapy. Front Immunol. 2018;9:384. doi:10.3389/fimmu.2018.00384.
- Song L, Asgharzadeh S, Salo J, Engell K, Wu H-W, Sposto R, Ara T, Silverman AM, DeClerck YA, Seeger RC, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. 2009;119:1524–1536. doi:10.1172/JCI37869.
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi:10.1038/nature07205.
- Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–355. doi:10.1016/j.semcancer.2008.03.004.
- Dysthe M, Parihar R. Myeloid-Derived suppressor cells in the tumor microenvironment. In Birbrair A, editor. Tumor Microenvironment. Cham, Switzerland: Springer International Publishing; 2020. p. 117–140. doi:10.1007/978-3-030-35723-8_8.
- Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, Allard J, Wei M, Mao H, Byrd JC, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. 2008;181:3784–3792. doi:10.4049/jimmunol.181.6.3784.
- Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, Xu Y, Wang L, Sposto R, Kim ES, et al. TGFβR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-gd2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23:804–813. doi:10.1158/1078-0432.CCR-16-1743.
- Nguyen R, Moustaki A, Norrie JL, Brown S, Akers WJ, Shirinifard A, Dyer MA. Interleukin-15 enhances anti-GD2 antibody-mediated cytotoxicity in an orthotopic PDX model of neuroblastoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25:7554–7564. doi:10.1158/1078-0432.CCR-19-1045.
- Barry WE, Jackson JR, Asuelime GE, Wu H-W, Sun J, Wan Z, Malvar J, Sheard MA, Wang L, Seeger RC, et al. Activated natural killer cells in combination with Anti-GD2 antibody dinutuximab improve survival of mice after surgical resection of primary neuroblastoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25:325–333. doi:10.1158/1078-0432.CCR-18-1317.