
ABSTRACT
Interleukin-1α (IL-1α) plays an important role in inflammation and hematopoiesis. Many tumors have increased IL-1α expression. However, the immune regulatory role of secreted IL-1α in tumor development and whether it can be targeted for cancer therapy are still unclear. Here, we found that tumoral-secreted IL-1α significantly promoted hepatocellular carcinoma (HCC) development in vivo. Tumoral-released IL-1α were found to inhibit T and NK cell activation, and the killing capacity of CD8+ T cells. Moreover, MDSCs were dramatically increased by tumoral-released IL-1α in both spleens and tumors. Indeed, higher tumoral IL-1α expression is associated with increased tumoral infiltration of MDSCs in HCC patients. Further studies showed that tumoral-released IL-1α promoted MDSC recruitment to the tumor microenvironment through a CXCR2-dependent mechanism. Depletion of MDSCs could diminish the tumor-promoting effect of tumoral-released IL-1α. On the contrary, systemic administration of recombinant IL-1α protein significantly inhibited tumor development by activating T cells. In fact, IL-1α protein could promote T cell activation and enhance the cytotoxicity of CD8+ T cells in vitro. Thus, our study demonstrated that tumoral-released IL-1α promoted tumor development through recruiting MDSCs to inhibit T cell activation, while systemic IL-1α directly promoted anti-tumor T cell responses. We further identified calpain 1 as the major intracellular protease mediating tumoral IL-1α secretion. Calpain 1 KO tumors had diminished IL-1α release and reduced tumor development. Thus, our findings provide new insights into the functions of secreted IL-1α in tumor immunity and its implications for immunotherapy.
Introduction
IL-1α is an important cytokine in IL-1 family, and it is constitutively expressed in a variety of cells. Upon environmental stimulation, both hematopoietic and non-hematopoietic cells can secret IL-1α to promote inflammation.Citation1 IL-1α is translated into 31 kDa precursor form (pro-IL-1α). Pro-IL-1α has a functional nuclear localization sequence (NLS) in the N-terminal domain for nuclear translocation, which is retained in the propiece (N-terminal cleavage product) of IL-1α after the cleavage by calpains or other proteases.Citation2 Pro-IL-1α and IL-1α-propiece can both translocate to the nucleus and bind to the chromatin to exert many functions, such as DNA damage repair, cell growth, differentiation, and adhesion.Citation3,Citation4 Pro-IL-1α is myristoylated and then anchors to the cell membrane via a lectin-like interaction.Citation5 Membrane IL-1α is bioactive through IL-1 receptor signaling pathway and is found to promote anti-tumor immune response.Citation6 The pro-IL-1α can also be cleaved to generate the secreted mature form (17 kDa).Citation7 IL-1α secretion is rarely detected in healthy human body fluids.Citation8 However, pro-IL-1α and mature IL-1α are thought to be released from necrotic cells to initiate inflammation.Citation9 Therefore, IL-1α is a dual function cytokine that exerts its function intracellularly and also binds to its cell membrane receptor to perform extracellular functions.
IL-1α plays a vital role in inflammatory diseases and cancer.Citation10 IL-1α released from necrotic cells activates IL-1 receptor to trigger the proinflammatory cytokine expression, chemokine secretion and neutrophil recruitment.Citation11 Moreover, extracellular IL-1α can trigger its own release by activating surrounding cells, resulting in the aggravation of local inflammation.Citation12 The proinflammatory role of IL-1α was confirmed in dextran sodium sulfate-induced acute colon inflammation in mice.Citation13 Moreover, previous studies suggest that IL-1α promotes the development of chronic inflammatory diseases, including rheumatoid arthritis, psoriasis, atherosclerosis, chronic hepatitis, and cardiovascular diseases.Citation14
Many forms of tumorigenesis are associated with chronic inflammation, and inflammatory cells and cytokines play major roles in the progression of cancer.Citation15 IL-1α binding to IL-1 receptor activates MyD88 signaling pathway to initiate inflammation which induces carcinogenesis and promotes tumor development.Citation16 In head and neck squamous cell carcinoma (HNDCC) and gastric carcinoma, the expression of IL-1α was elevated in tumors of patients with distant metastases or poor prognosis.Citation17 IL-1α single nucleotide polymorphism (SNP) rs17561 that results in A114S substitution produces the pro-IL-1α mutant form that can be cleaved more easily to increase the secretion of IL-1α, and this ratchets up the risk of tumorigenesis and metastasis.Citation18 Recently, tumor-derived IL-1α was found to act on infiltrating myeloid cells to induce the expression of a critical tumor survival factor, the cytokine TSLP, therefore promoted the development of breast cancer.Citation19 On the other hand, studies also reported that overexpression of IL-1α in cytosol or on membrane of fibrosarcoma or lymphoma cells reduced tumorigenicity and inhibited tumor growth. IL-1α exerts anti-tumor immune response through CD8+ T cells, NK cells and macrophages.Citation20–22 IL-1 signaling can also contribute to recruitment and activation of antigen presenting cells to the lymph nodes where they can induce the activation of T cells.Citation23 Moreover, IL-1α can increase the therapeutic efficacy of cetuximab, an EGFR inhibitor, via a T cell-dependent mechanism in HNSCC.Citation24 Therefore, IL-1α release could both promote and inhibit tumor development and the underlying mechanism is not clear.
Although both pro-IL-1α and mature IL-1α can be released to the extracellular space, the mature IL-1α has been suggested to be more biologically active. Calpains, the major intracellular proteases that can cleave pro-IL-1α to generate mature IL-1α, are a group of 15 Ca2+-dependent cysteine proteases.Citation25 It is not known which calpain is most responsible for cleaving IL-1α and whether this calpain can be targeted to regulate IL-1α release and tumor development. Many pathways of IL-1 active release have been proposed in the recent studies.Citation26 IL-1 antagonist and blocking antibodies are being tested in clinical trials for tumor therapy. Monotherapy with an anti-IL-1α blocking antibody showed promising results in patients with advanced non-small cell lung cancer, ovarian cancer, and other refractory cancers, as well as unresectable or metastatic colon cancer.Citation27 It is essential to understand the function of the IL-1α release in tumor development in order to design more effective therapeutic strategies.
In the current study, we aim to investigate the function of tumoral-released and systemically administered IL-1α during tumor development. We found that tumoral-released IL-1α promoted tumor growth via myeloid-derived suppressor cells (MDSCs) recruitment and inhibition of anti-tumor immune response, while systemic administration of IL-1α protein suppressed tumor development and directly activated anti-tumor immunity. In order to specifically inhibit tumoral IL-1α release, we generated calpain 1-deficient tumors and discovered that calpain 1-deficient tumors had significantly reduced IL-1α release and inhibited tumor development. Our findings demonstrate for the first time the opposite roles of tumoral and systemic IL-1α release in tumor development. Moreover, as the major intracellular protease to generate secreted mature IL-1α, calpain 1 could be targeted for inhibiting tumoral IL-1α release and tumor growth.
Materials and methods
Mice
Male C57BL/6 mice (6–8 weeks old) were obtained from Shanghai Laboratory Animal Center (Shanghai, China) or Invivos Pte Ltd. (Singapore). All mice were kept in a specific pathogen-free facility. The study was in accordance with the National Animal Care and Use Committee and approved by the Animal Welfare and Ethics Committee of Soochow University and National University of Singapore Institutional Animal Care and Use Committee.
Cells and lentiviral constructs
Hepa1-6 and 293 T cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and were cultured with 10% FBS DMEM medium (Gibco, Carlsbad, CA). Mouse pro-IL-1α (1–270aa, 31 kDa) and secreted IL-1α (110–270aa, 18 kDa) were amplified by PCR and cloned into lentiviral plasmid (kindly provided by Dr. Yun Zhao, Soochow university, Suzhou, China). Lentivirus was packaged in 293 T cells and then used to infect hepa1-6 cells. Empty vector was used as vector control. Hepa1-6 cells stably expressing pro-IL-1α, secreted IL-1α or vector control were established via sorting YFP-positive cells by FACS Aria III flow cytometer (BD Biosciences, San Jose, CA).
The cancer genome atlas profiling data sets
HCC data sets from 434 patients in The GEPIA database (http://cancergenome.nih.gov) were used to explore the relationship between IL1A, CAPN1 expression and survival time in HCC patients. Kaplan–Meier survival plots were obtained using the GEPIA online tool (http://gepia.cancer-pku.cn/detail.php?gene=&clicktag=survival#iframe). The prediction of the immune cell fractions by CIBERSORT was performed with the public server (https://cibersort.stanford.edu).
Confocal microscopy
Hepa1-6 cells stably expressing secreted IL-1α or vector were plated and observed in borosilicate cover-glass (Thermo Scientific, Waltham, MA). Then the cells were fixed, permeabilized, and stained with rabbit anti-mouse IL-1α antibody (Abcam, Cambridge, MA) and secondary PE-conjugated goat anti-rabbit IgG antibody (MultiSciences Biotech, Hangzhou, China) as described previously.Citation6 After counterstaining with DAPI (Sigma, St. Louis, MO), cells were observed with a Nikon A1 confocal microscope spectral detector (Nikon, Japan).
Immunohistochemistry (IHC)
HCC tissue microarray slides containing tumor and adjacent paracancer tissues (70 cases) were purchased from Outdo Biotech (Shanghai, China). The slides were stained with rabbit anti-IL-1α antibody (Abcam, Cambridge, UK) and HRP-conjugated anti-rabbit IgG secondary antibody (Santa Cruz, Dallas, TX). The DAB peroxidase substrate kit (Beyotime, Haimen, China) was used for enzymatic reaction. And the slides were scored by evaluating the percentage of positive cells and the staining intensity. The percentage of positive cells was quantified as follows: Grade 0, <5% cells; 1, 6–25% cells; 2, 26–50% cells; 3, 51–75% cells; and 4, 76–100% cells. The staining intensity was defined as follows: 0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining. The final IHC score was calculated by multiplying the grade of percentage of positive cells and the staining intensity: 0, total score = 0; 1+, total score = 1–4; 2+, total score = 5–8; 3+, total score = 9–12.
Cell PROLIFERATION
Cells stably expressing secreted IL-1α or vector control were seeded into 96-well plate (3000 per well/100 μL) and incubated for 24, 48 and 72 hours. CCK-8 reagent (10 μL per well) (Dojindo, Gaithersburg, MD) was added and incubated for 4 hours at 37°C. The OD value was detected at 450 nm by a microplate reader (BioTek, Winooski, VT).
Establishment of HCC murine models
Hepa1-6 cells (1 × 106/100 μL) were injected subcutaneously into C57BL/6 mice to establish subcutaneous HCC model. Mice were randomly divided into two groups and were injected with secreted IL-1α- and vector control-hepa1-6 cells respectively. The growth of subcutaneous tumor was measured with digital calipers every 3 days and the mice were sacrificed 3 weeks after tumor injection. Tumor volume was calculated by the following formula: tumor volume = 0.5× width2× length. Hepa1-6 cells (1 × 106/2 mL) were either hydrodynamically injected into mice or orthotopically implanted (1 × 106/25 μL) in the liver to establish orthotopic HCC model.Citation28 Mice were randomly divided into two groups and were injected with secreted IL-1α- and vector control-hepa1-6 cells respectively. In some experiments, mice were first hydrodynamically injected with hepa1-6 cells (1 × 106/2 mL). One day after tumor injection, mice were randomly divided into two groups and injected intraperitoneally with 100 ng recombinant IL-1α protein (R&D systems, Minneapolis, MN) every week. Control group mice were treated with equal volume of PBS. After 3 weeks of tumor inoculation, mice were sacrificed and the tumor nodules in the liver were counted or measured.
Flow cytometry analysis
Single-cell suspensions were generated from spleen and liver of tumor-bearing mice. The following anti-mouse antibodies were used: anti-CD45-BUV395, anti-CD3E-PE/CF594, anti-CD44-PE, anti-CD4-APC/H7, anti-CD8-APC, anti-CD69-FITC, anti-CD86-FITC, anti-CD11c-PE/Cy7, anti-Gr1-PE, anti-CD11b-APC, anti-CD19-PE/Cy7, anti-NK1.1-PerCP/Cy5.5 (BD Bioscience, San Diego, CA), anti-Ly6C-BV421, anti-Ly6G-AF700, anti-CD4-FITC, anti-CD62L-PerCP/Cy5.5, anti-NKG2D-FITC, anti-F4/80-PerCP/Cy5.5, anti-IFN-γ-PE and anti-TNF-α-PE/Cy7 (Biolegend, San Diego, CA). Anti-mouse CD16/32 FcR block antibody was purchased from Biolegend (San Diego, CA). For intracellular cytokine staining, cells were stimulated with Cell Activation Cocktail (Biolegend, San Diego, CA) for 4 hours and stained with surface antibodies. Then cells were fixed, permeabilized and stained with intracellular cytokine antibodies. Cells were analyzed using a FACS Canto II flow cytometer (BD Biosciences, San Jose, CA).Citation29 Data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Cytokine analysis
The supernatant of hepa1-6 cells stably expressing secreted IL-1α or vector control was examined for IL-1α levels by the mouse IL-1α ELISA kit (Biolegend, San Diego, CA) according to the manufacturer’s instructions.
Cytotoxicity assay
The CytoTox 96 non-radioactive cytotoxicity assay kit (Promega, Madison, WI) was used to measure the cytotoxic activity of CD8+ T cells against tumor cells. After 1 week of tumor inoculation, splenocytes were harvested from tumor-bearing mice and stimulated with hepa1-6 cell lysates plus rhIL-2 (100 U/mL) for 4 days. For assays of examining the in vitro function of IL-1α, splenocytes were harvested from mice and stimulated with hepa1-6 cell lysates plus rhIL-2 (100 U/mL) with or without 25 ng/ml recombinant IL-1α proteins (R&D Systems, Minneapolis, MN) for 4 days. CD8+ T cells were sorted from splenocytes by negative selection using immunomagnetic beads (StemCell Technologies, Vancouver, BC) according to the manufacturer’s protocol. Then the cytotoxicity assay was performed using hepa1-6 cells as the target cells.
Transwell assay
MDSCs were cultured as previously described.Citation30 Briefly, BM cells were cultured in the presence of 10 ng/mL GM-CSF (Peprotech, Rocky Hill, NJ) and 10 ng/mL IL-4 (Peprotech, Rocky Hill, NJ) for 4 days to induce the differentiation to MDSCs. Supernatants from control or IL-1α-overexpressing hepa1-6 cells were added into 24-well plates with Transwell polycarbonate-permeable supports (pore size = 8.0 μm; Merck Millipore, Burlington, MA). 2 × 105 MDSCs were preincubated with or without CXCR2 antagonist, SB265610 (Sigma, St. Louis, MO), for 30 min and seeded on the upper chambers of the inserts. After incubation for 18 hours, the MDSCs in the bottom compartment were counted.
Depletion of MDSCs
Gemcitabine as a chemotherapy drug was reported to deplete MDSCs in vivo.Citation31 Mice were injected intrahepatically with hepa1-6 cells stably expressing secreted IL-1α. One day after tumor injection, mice were randomly divided into two groups and were treated with gemcitabine (GEM, LC Laboratories, Woburn, MA) at a dose of 100 mg/kg mouse body weight every 3 days. Control group mice were treated with equal volume of double-distilled water. The depletion of MDSCs was confirmed by flow cytometry on day 13 after the initial injection.
To deplete the MDSCs with anti-Gr-1 antibody, mice were injected intrahepatically with hepa1-6 cells stably expressing secreted IL-1α. One day after tumor injection, mice were randomly divided into two groups and were treated with 200 ug rat anti-mouse Gr-1 antibody (Bio X Cell, Lebanon, NH) every 3 days. Control group mice were treated with equal amount of rat IgG2b isotype control (Bio X Cell).
SB265610 treatment
Mice were injected intrahepatically with hepa1-6 cells stably expressing secreted IL-1α. One day after tumor injection, mice were randomly divided into two groups and were treated with CXCR2 inhibitor SB265610 (Sigma, St. Louis, MO) at a dose of 2 mg/kg mouse body weight every 3 days. Control group mice were treated with equal volume of DMSO (Sigma, St. Louis, MO).
CRISPR/Cas9 transfection
For the generation of Calpain 1 knockout cells using CRISPR-Cas9 gene editing, sgRNA targeting Calpain 1 gene (Santa Cruz, Dallas, TX) was transfected into pro-IL-1α-expressing hepa1-6 cells using Lipofectamine CRISPRMAX Cas9 Transfection Reagent (Thermo Scientific, Waltham, MA). HDR plasmid encoding RFP were co-transfected to be used for further cell sorting. Three days after transfection, cells expressing RFP were sorted into 96 well plates using MoFlo XDP (Beckman Coulter, Indianapolis, IN) to generate stable single cell clones. The sgRNA sequences for calpain 1 knockout were sequence 1: CCTTCGAGGACTTTACCGGT; sequence 2: CGAGACTATTCTGCACCGAG; Sequence 3: GTCAAACCCCCAGTTCATCG.
Reverse transcription and quantitative polymerase chain reaction (RT-qPCR)
Total RNA extraction was performed using the RNeasy Mini Kit (Qiagen, Hilden, Germany). 1 µg of RNA for each sample was reverse transcribed to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) on a PCR machine (Bio-Rad, Hercules, CA). qPCR was performed using the GoTaq qPCR master mix (Promega, Madison, WI) on the ABI 7500 Real Time PCR system (Applied Biosystems, Waltham, MA). The relative expression to GAPDH was calculated according to control cells by the comparative threshold cycle method. Primers sequences are listed in supplementary Table S1.
SDS-PAGE and western blot analysis
Cells were lysed, and total protein was extracted using the NP40 lysis buffer mixed with protease and phosphatase inhibitors cocktail tablets (Roche, Switzerland). BCA assay was performed using Pierce BCA Protein Assay Kit (Thermo Fisher, Waltham, MA) to measure protein concentration in the samples. Standardized amounts from 25 µg to 100 µg of protein for each sample were loaded into 10-well gels to run SDS-PAGE. The gels were transferred to 0.2 µm polyvinylidene difluoride (PVDF) transfer membranes (Thermo Fisher, Waltham, MA). Membranes were blocked with 5% skimmed milk (Sigma, St. Louis, MO) for an hour and incubated with respective primary antibodies overnight. Anti-mouse IL-1α antibody (Abcam, Cambridge, UK), anti-mouse Calpain 1 antibody (Thermo Fisher, Waltham, MA) and anti-mouse β-actin antibodies (Santa Cruz, Dallas, TX) were used as primary antibodies followed by HRP-conjugated goat anti-rabbit IgG (Santa Cruz, Dallas, TX). Finally, western blot substrates, SignalFire Enhanced Chemiluminescent and Elite ECL Reagents (Cell Signaling Technology, Danvers, MA), were subsequently used for protein detection.
Calpain activity assay
Cell lysates were prepared using the same method described for SDS-PAGE. 50 µg of protein sample was added into the 96-well plate to test for calpain activity. 50 µL of Calpain-Glo Reagent (Promega, Madison, WI) was added into each well. The plate was wrapped with aluminum foil and incubated on a plate shaker at 500 rpm for 15 minutes. Luminescence was recorded using the Gen5TM All-In-One Microplate Reader (BioTek, Winooski, VT).
T cell activation assay
The plates were pre-coated with anti-CD3 (2 μg/mL) and anti-CD28 (0.4 μg/mL) antibodies (Biolegend, San Diego, CA) overnight. Naïve T cells were sorted from splenocytes using FACS Aria III flow cytometer and were co-cultured with transduced hepa1-6 cells or IL-1α protein for 48 hours in plates. In the co-culture system, naïve T cells (2 × 105/well) and transduced hepa1-6 cells (1 × 105/well, irradiated with 50 Gy) were seeded in 96-well plate. In the transwell system, transduced hepa1-6 cells (1 × 105) were placed in the upper chamber and naïve T cells (4 × 105) were seeded in the lower chamber of 24-well plate. For assays examining the in vitro function of IL-1α, naïve T cells (2 × 105/well) were stimulated with 25 ng/ml recombinant IL-1α proteins (R&D Systems, Minneapolis, MN) in 96-well plate. After 48 hours of incubation, the activation status of T cells was analyzed using flow cytometry.
Statistical analysis
All statistical analyses were performed using Student’s t test (unpaired, two-tailed) between two groups and One-way ANOVA for more than two groups. Data were analyzed with GraphPad prism 7 software (Graphpad, San Diego, CA). P value < .05 was considered as statistically significant (*), P < .01 and P < .001 were shown as ** and ***, respectively.
Results
Tumoral-secreted IL-1α promotes tumor growth in vivo
In the HCC patient samples, we found that IL-1α was highly expressed in the adjacent tissues compared to the tumor tissues (Fig. S1A & B). IL-1α expression levels are also negatively associated with patient overall survival in HCC patients (Fig. S1C). These results indicated that IL-1α may play an important role in the HCC development. To specifically investigate the effect of secreted IL-1α on tumor growth, we constructed hepa1-6 cells stably expressing secreted IL-1α or vector control (). IL-1α secretion from tumor cells was examined and confirmed by confocal microscopy () and ELISA with the culture supernatants (). To assess the direct effect of secreted IL-1α on tumor cell growth, we examined the proliferation of the cells and showed that tumoral-secreted IL-1α had no direct effect on the cell proliferation in vitro ().
Figure 1. Tumoral-secreted IL-1α promotes tumor growth in murine HCC models.
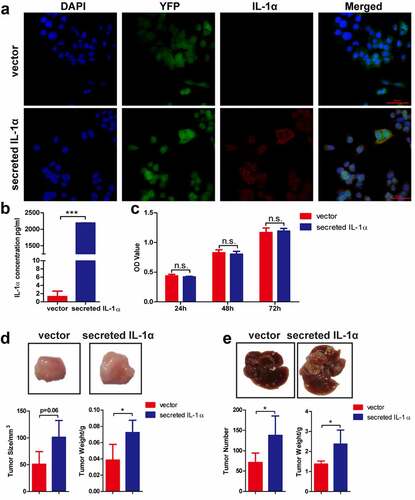
To dissect the role of tumoral secreted IL-1α in vivo, we first established subcutaneous HCC model (). Tumoral-secreted IL-1α significantly promoted tumor growth compared to vector control group in subcutaneous HCC model. Next, we established orthotopic HCC model via hydrodynamical injection of secreted IL-1α- or vector control-hepa1-6 cells into C57BL/6 mice. Both the number of tumor nodules and the liver weights were significantly increased in tumoral-secreted IL-1α group compared to vector control group (). These results demonstrated that tumoral secreted-IL-1α promoted HCC development.
Tumoral-secreted IL-1α inhibits T and NK cell activation in vivo
To investigate the mechanism of tumor-promoting activity of tumoral-secreted IL-1α, we collected splenocytes and intrahepatic leukocytes from mice one week after orthotopical tumor cell injection to analyze the immune cell subsets and their activation status via flow cytometry analysis (, Fig. S2). The percentages and absolute numbers of T cells, B cells and macrophages were not affected by tumoral-secreted IL-1α (Fig. S2). However, tumoral-secreted IL-1α decreased the percentages of activated (CD69+) and effector CD8+ T cells in the liver, while there was only a slight decrease in percentages of activated liver CD4+ T cells (). Moreover, the percentages of naïve CD4+ T and CD8+ T cells were increased and percentages of effector CD4+ T and CD8+ T cells were decreased in both spleen and liver by the expression of tumoral-secreted IL-1α. Additionally, the percentage of memory CD8+ T cells was also decreased in the spleen by tumoral-secreted IL-1α. We further investigated the T cells activation status 3 weeks after tumor injection. The results showed that the tumoral-secreted IL-1α had no significant effect on CD69 expression on T cells, as well as the effector, memory and naive T cell percentages in both spleen and liver (Fig. S3A-B). Notably, we found that tumoral-secreted IL-1α inhibited IFN-γ production by CD8+ T cells in spleen (Fig. S3C). The liver infiltrating TNF-α-producing CD4+ and CD8+ T cells, as well as the IFN-γ-producing CD8+ T cells were also decreased by tumoral-secreted IL-1α (Fig. S3D). Taken together, these results indicated that tumoral-secreted IL-1α inhibited the activation of CD4+ and CD8+ T cells.
Figure 2. Tumoral-secreted IL-1α inhibits T and NK cell activation in vivo.
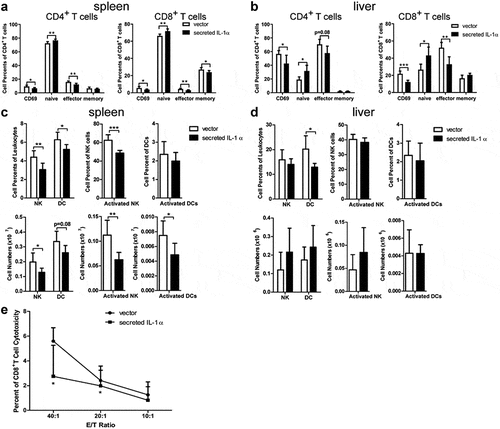
Furthermore, the percentages and total numbers of NK and activated NK cells were decreased in spleens of the secreted IL-1α group ( &d). Tumoral-secreted IL-1α also decreased the percentages and numbers of DCs in spleen and liver. The number of activated DCs was also decreased in spleens of the secreted IL-1α group compared to the control group. To further confirm that the anti-tumor T cell function was inhibited by tumoral-secreted IL-1α, we examined the cytotoxicity of CD8+ T cells against tumor cells (). CD8+ T cells isolated from the mice of secreted IL-1α group exhibited significantly lower killing capacity against tumor cells compared to those from the control mice. Taken together, these results demonstrated that tumoral-secreted IL-1α exerted its tumor-promoting effect through inhibiting the activation of T and NK cells.
Tumoral-secreted IL-1α increases MDSCs in vivo
MDSCs suppress immune responses and promote tumor development in many tumor models.Citation32 Our previous study showed that secreted IL-1α could promote the infiltration and proliferation of MDSCs in the liver through its function on hepatocytes during inflammatory response.Citation33 To investigate whether tumoral-secreted IL-1α exerts the tumor-promoting effect through MDSCs, we analyzed CD11b+Gr1+ MDSCs in orthotopic tumor-bearing mice by flow cytometry analysis (). The percentages and numbers of MDSCs in spleen and liver were not increased by tumoral-secreted IL-1α at 1 week after tumor injection (). However, they were significantly increased in both spleen and liver 3 weeks after tumor injection in the secreted IL-1α group (). We further assessed the presence of the two main MDSC subsets, PMN-MDSCs (CD11b+Ly6G+Ly6CLow) and M-MDSCs (CD11b+Ly6G−Ly6CHigh) in spleen and liver (). The results showed that both the percentages and total numbers of the two MDSCs subsets were increased by tumoral-secreted IL-1α expression.
Figure 3. Tumoral-secreted IL-1α increases MDSCs in vivo.
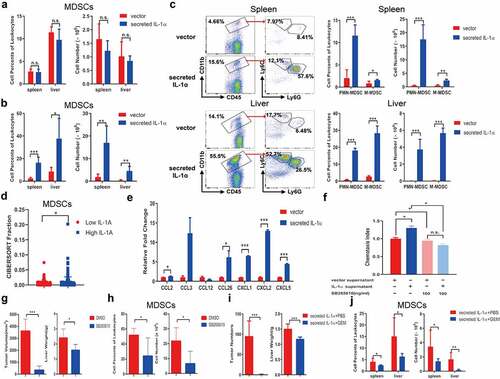
To provide the clinical relevance of the results found in our murine HCC models, we adopted CIBERSORTx to evaluate the tumor-infiltrating fractions of the cells in HCC patients.Citation34 The results showed that in HCC patients, higher tumoral IL-1α expression is associated with increased tumoral infiltration of MDSCs (). These results suggested that tumoral-secreted IL-1α could recruit MDSCs to inhibit T cell functions and promote HCC development.
IL-1 receptor signaling has been reported to be involved in MDSCs expansion, migration and function.Citation35 To elucidate the mechanism of how tumoral-secreted IL-1α promotes MDSC accumulation in the tumor microenvironment, various chemokines recruiting MDSCs were assessed in the control and IL-1α-overexpressing hepa1-6 cells. The results showed that IL-1α promoted CCL2, CCL26, CXCL1, CXCL2, CXCL5 expression while had no effect on CCL3 and CCL12 (), suggesting IL-1α might induce the migration of MDSCs through upregulating chemokine expressions in tumor cells. Transwell experiment confirmed that supernatants of IL-1α overexpressing hepa1-6 cells significantly promoted MDSCs migration compared with those from control cells (). Since CXCL1, CXCL2 and CXCL5 shared the same chemokine receptor CXCR2, we preincubated MDSCs with CXCR2 inhibitor SB265610 and found that CXCR2 blockade diminished the effect of IL-1α on MDSC migration. In vivo study also confirmed that SB265610 treatment significantly reduced tumor growth (). The tumoral MDSC infiltration was also decreased after CXCR2 blockade (). Moreover, both tumoral infiltrating CD4+ and CD8+ T cells exhibited increased TNF-α production after SB265610 treatment (Fig. S4A). These results demonstrated that tumoral-released IL-1α promoted MDSC recruitment to the tumor microenvironment through a CXCR2-dependent mechanism. Additionally, we depleted MDSCs by administration of gemcitabine in mice (). Depletion of MDSCs dramatically inhibited tumor growth in tumors expressing secreted IL-1α, suggesting the tumor-promoting effect of tumoral-secreted IL-1α was dependent on the presence of MDSCs. MDSC depletion was confirmed by flow cytometry analysis in both spleen and liver (). Additionally, we applied an alternative method to deplete MDSCs in vivo by injecting the anti-mouse Gr-1 antibody. The results showed that the majority of MDSCs were depleted after anti-Gr-1 antibody treatment (Fig. S4B). Moreover, MDSC depletion in HCC tumor-bearing mice expressing secreted IL-1α significantly reduced tumor growth (Fig. S4C). Flow cytometry analysis showed that the percentage TNF-α-producing CD4+ T cells was increased in the spleen after anti-Gr-1 treatment. The liver infiltrating TNF-α-producing CD4+ and CD8+ T cells, as well as the IFN-γ-producing CD8+ T cells were also increased by anti-Gr-1 treatment (Fig. S4D). In summary, these findings demonstrated that tumoral-secreted IL-1α suppressed immune response and facilitated tumor growth through its promoting effect on MDSCs.
Systemic IL-1α administration exerts an anti-tumor effect and activates T cells
To determine whether the systemic IL-1α expression could have the same effect as the tumoral-secreted IL-1α, we administered recombinant IL-1α protein (secreted form) intraperitoneally to the tumor-bearing mice with PBS as control (). We found that systemic administration of secreted IL-1α protein suppressed tumor growth (). Both the number of tumor nodules and the liver weight were decreased by systemic IL-1α administration. Moreover, this anti-tumor activity of the systemically administered IL-1α was dose-dependent (Fig. S5A). To determine whether the recombinant IL-1α protein could directly inhibit tumor growth, we examined tumor cell proliferation upon treatment with various concentrations of recombinant IL-1α protein at different time points (Fig. S5B). The results showed that recombinant IL-1α protein does not have direct effect on tumor cells, indicating that systemically administered IL-1α might modulate immune responses to exert its anti-tumor activity in vivo.
Figure 4. Systemic administration of recombinant IL-1α inhibits tumor development.
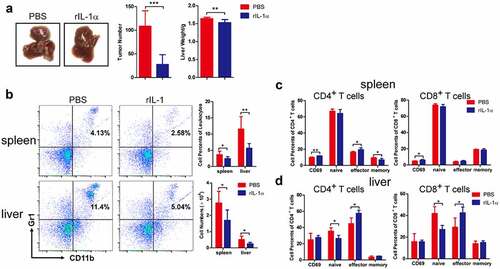
To address this hypothesis, we analyzed the immune cell phenotypes of tumor-bearing mice via flow cytometry analysis. Compared to PBS control group, the percentages and cell numbers of MDSCs were significantly lower in both spleen and liver in rIL-1α group 3 weeks after tumor cells injection (), while there was no difference in MDSC percentages and numbers between rIL-1α and control mice 1 week after tumor cell injection (Fig. S5C). Administration of rIL-1α protein did not affect the percentages and total numbers of CD4+ T and CD8+ T cells (Fig. S5D). However, it up-regulated the percentages of activated (CD69+) CD4+ T and CD8+ T in spleen and the percentages and numbers of CD4+ T effector and CD8+ T effector cells in liver ( &d). These results suggested that increased systemic IL-1α levels inhibited tumor growth and activated T cells.
To investigate whether there are differential effects of tumoral-secreted IL-1α and recombinant IL-1α protein directly on T cells activation, we co-cultured naïve T cells stimulated with anti-CD3/anti-CD28 with hepa1-6 cells stably expressing secreted IL-1α or recombinant IL-1α protein (). Flow cytometry analysis revealed tumoral-secreted IL-1α significantly increased the percentages of both CD4+ and CD8+ T effector cells either in a mixed co-culture system with the tumor cells secreting IL-1α () or in a Transwell co-culture system with the same cells (). We also noticed that co-culturing hepa1-6 expressing secreted IL-1α with T cells in a cell-cell contact manner significantly enhanced the activation of CD4+ and CD8+ T cells, while this effect of secreted IL-1α was less prominent in the Transwell co-culture system. Interestingly, T cells, especially CD4+ T cells, of control group showed less activation in the cell-cell contact system than in the Transwell system, suggesting there was a T cell suppression derived from direct contact with tumor cells. These results implicated that IL-1α signaling might overcome the suppressive effects of tumor cells on T cells.
Figure 5. Secreted IL-1α promotes T cell activation in vitro.
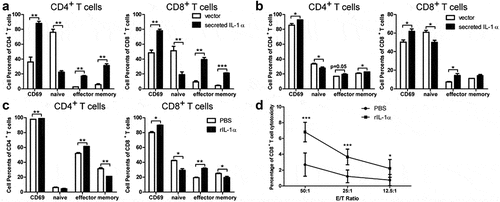
The percentages of activated and effector CD4+ T and CD8+ T cells were also increased by treatment with recombinant IL-1α protein (). To further demonstrate the cytotoxicity of CD8+ T cells could also be enhanced by secreted IL-1α, we performed the cytotoxicity assay using CD8+ T cells against hepa1-6 cells (). The result showed that secreted IL-1α enhanced the cytotoxicity of CD8+ T cells. Therefore, either tumoral-secreted IL-1α or recombinant IL-1α protein could promote T cell activation in vitro. Their differential effects on tumor growth in vivo could be due to the indirect functions on T cells via MDSCs.
Calpain 1 activity is required for tumoral-secreted IL-1α-mediated tumor progression
To further confirm the role of tumoral-secreted IL-1α and explore the strategy to specifically inhibit tumoral IL-1α secretion, we knocked out the calpain 1 gene in hepa1-6 cells stably expressing pro-IL-1α (). Calpains are the main intracellular proteases that cleavage pro-IL-1α into its secreted form. We analyzed the associations between different calpains with the HCC patient survival and found that high level of calpain 1 expression was associated with poor patient prognosis (Fig. S6). Calpain 1 knockout (KO) by Crispr/Cas9 technology was confirmed by RT-qPCR () and western blot (). We also confirmed that Calpain 1 KO significantly reduced the calpain activity () and IL-1α secretion () in pro-IL-1α-expressing hepa1-6 cell line. Western blot results also showed that no mature form of IL-1α can be detected in the cells after Calpain 1 KO (Fig. S7). Moreover, Calpain 1 KO had no effect on tumor cell proliferation, whereas both the early and late apoptosis were significantly inhibited in calpain 1 KO cells (& f).
Figure 6. Calpain 1 is essential for tumoral-secreted IL-1α-mediated HCC progression.
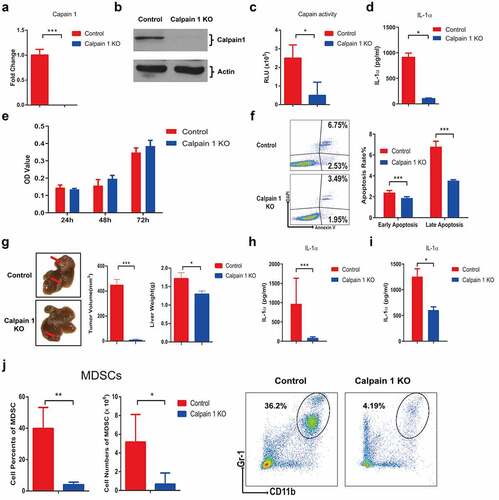
Next, we established murine orthotopic HCC model using the Calpain 1 KO cells (). The results showed that Calpain 1 KO group had significantly reduced tumor growth compared to the control group (). IL-1α levels were also significantly decreased in both tumor () and serum () of the Calpain 1 KO group compared with the control group. The percentage and absolute number of MDSCs in the liver were also reduced in the Calpain 1 KO group (). These results suggested that cleavage of pro-IL-1α by Calpain 1 was essential for secreted IL-1α-mediated HCC progression.
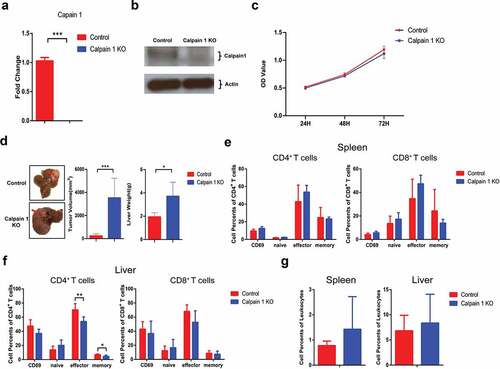
To confirm that Calpain 1 promoted HCC development through regulating IL-1α maturation, we performed Calpain 1 KO in parental hepa1-6 cells. The successful KO of Calpain 1 was confirmed by RT-qPCR () and western blot (). In vitro study showed that Calpain 1 KO had no effect on hepa1-6 cell proliferation (). Intriguingly, Calpain 1 KO in parental hepa1-6 cells significantly promoted HCC growth in vivo (). Flow analysis showed that Calpain 1 KO had no effects on T cells activation in the spleen (). In the liver, Calpain 1 KO significantly decreased the percentages of effector and memory CD4+ T cells, whereas CD8+ T cells were not affected (). Additionally, Calpain 1 KO in parental hepa1-6 cells had no impact on MDSC tumoral infiltration, suggesting Calpain 1 regulated tumor growth independent of MDSCs in the absence of IL-1α expression (). Taken together, these results further demonstrated that the tumor-promoting function of Calpain 1 is via regulating IL-1α maturation.
Figure 7. Calpain 1 KO promoted HCC development in parental hepa1-6 cells.
Discussion
Chronic inflammation is closely associated with tumor development, progression and metastasis, especially in solid tumors such as HCC.Citation36,Citation37 IL-1α is a pleiotropic cytokine and plays an important role in inflammatory immune responses and hemopoiesis.Citation38 The expression of IL-1α is increased in many inflammatory diseases, including acute liver inflammation,Citation39 acute viral hepatitisCitation40 and drug-induced liver injury.Citation41 Moreover, in hypercholesterolaemic mice, IL-1α deficiency in kupffer cells decreases inflammatory cytokines expression and attenuates liver inflammation.Citation42 Transformation of steatosis to steatohepatitis and liver fibrosis is significantly reduced in IL-1α deficient mice.Citation43 In diethylnitrosamine-induced liver carcinogenesis model, release of IL-1α from necrotic hepatocytes can initiate inflammation and promote carcinogen-induced compensatory proliferation and carcinogenesis.Citation44 Our previous study demonstrated that membrane IL-1α promotes the activation of T and NK cells to inhibit the HCC development,Citation6 while passive release of IL-1α from necrotic hepatocytes may not be the major mode of IL-1α release during tumor development. IL-1α could be cleaved by intracellular calpains and actively secreted to regulate anti-tumor immune responses. In the present study, we demonstrated, for the first time, that tumoral-secreted IL-1α significantly promoted tumor development, while systemic IL-1α administration inhibited tumor growth in the same murine HCC model. Tumoral-secreted IL-1α suppressed T cell functions via promoting MDSCs in the tumor microenvironment, while systemic IL-1α may directly enhance T cell responses. Further studies showed that calpain 1 deficiency could reduce tumoral IL-1α secretion and tumor growth. Thus, calpain 1 expression at the tumor site may be targeted to specifically suppress tumoral IL-1α secretion.
It remains as a fundamental scientific question how the leaderless cytokines can be actively transported out of the cells. For a long period of time, the release of IL-1α was believed as passive due to the loss of cell membrane integrity, such as necrosis or physical damage.Citation10 However, recent evidence has implicated an active transport route for IL-1α secretion. Zhang et al. reported a universal unconventional protein secretion (UPS) route for leaderless protein, including the mature form of IL-1α, IL-1β, IL-36α, IL-36RA, IL-37, IL-38, etc. They revealed that HSP90A binds and unfolds leaderless proteins, then interacts with the TMED10, resulting in the oligomerization of TMED10 to form a protein channel that translocates the leaderless protein into the lumen of the ER-Golgi intermediate compartment (ERGIC) for subsequent secretion out of the cell via a vesicular intermediate.Citation45 Notably, they performed the experiments with mature form of IL-1α, whether this UPS could also transport the full-length IL-1α is unknown. More recently, Tsuchiya et al. discovered a new secretion route for IL-1α.Citation46 They found that IL-1α maturation resulted in more IL-1α release from macrophages upon GSDMD-mediated pore formation. These findings suggested mature IL-1α might be the dominant form that could be actively transported compared with the pro-IL-1α. However, we cannot exclude the possibility that pro-IL-1α could also be actively transported out of the cell via an unknown route. Further studies are needed to address this question.
In the in vitro experiments, we demonstrated that both tumoral-secreted IL-1α and recombinant IL-1α protein could promote T cell activation. However, tumoral-secreted IL-1α inhibited T cell activation to promote tumor development in vivo. IL-1α signaling can drive myelopoiesis in the bone marrow.Citation47 In the tumor microenvironment, released IL-1α is an upstream and alarm cytokine which triggers the production of other pro-inflammatory cytokines, chemokines and growth factors to initiate inflammatory responses. Our previous studies have demonstrated that secreted IL-1α from hepatocyte can promote the recruitment and expansion of MDSCs in the liver during acute liver inflammation through the induction of CCL2, CXCL5 and IL-6 expressions.Citation33 Here, we also demonstrated tumoral-released IL-1α could recruit MDSCs to the tumor microenvironment in a CXCR2-mediated chemotaxis manner. MDSCs consisting of immature myeloid cells and myeloid progenitor cells suppress immune responses and promote tumor development.Citation48 They have been found to accumulate in human HCC and inhibit anti-tumor immune response.Citation49 The depletion of MDSCs dramatically inhibited the tumor growth induced by tumoral-secreted IL-1α, suggesting the T cell inhibition by tumoral-secreted IL-1α was mediated by MDSCs. On the other hand, systemic IL-1α expression does not specifically increase IL-1α levels in the tumor microenvironment to induce the cytokine and chemokine gradient that can promote MDSC proliferation and recruitment in the liver. On the contrary, systemic IL-1α can promote T cell activation in the peripheral and secondary lymphoid organs. The current therapeutic strategies in clinical trials targeting IL-1α can reduce overall IL-1α levels or systemically block its function, which may achieve some clinical benefits, but are not optimal.
Pro-IL-1α has been well characterized to be cleaved by the calcium-dependent proteases: calpain family. Though pro-IL-1α is biological active, calpain cleaved secreted (mature) IL-1α results in seven-fold increase in bioactivity.Citation50 Besides its cleavage activity, calpain has also been proved to be involved in IL-1α secretion.Citation51 So far, more than 10 calpain family members have been identified. Among them, calpain 1 and calpain 2 are the most studied and ubiquitously expressed. Calpain 1 expression has been found to be increased in schwannomas, meningiomas and renal cell carcinoma.Citation52 Calpain 1 could affect tumor development in many aspects: cell migration, proliferation, death and autophagy.Citation53 However, the detailed mechanism is still unclear. Here, for the first time, we demonstrated that calpain 1 is required for tumoral secreted IL-1α-mediated tumor progression in HCC. Interestingly, Calpain 1 KO in parental hepa1-6 cells exhibited enhanced tumor development in vivo with no impact on MDSCs recruitment in the absence of IL-1α expression. These results suggest a dual role of Calpain 1 in cancer development. Calpain 1 could act as a tumor suppressor in tumors without IL-1α expression which is likely the case for tumors without the involvement of inflammation, while it could promote tumor growth in the IL-1α enriched settings. Therefore, cautions need to be taken when targeting Calpain 1 for cancer therapy and evaluation of inflammation status and IL-1α expression is required. Calpain 2 knock out in hepa1-6 cells did not reduce the level of IL-1α secretion (data not shown). Thus, calpain 1 could be a potential therapeutic target for specifically reducing tumoral IL-1α secretion and inhibiting tumor development. Over the years, more than 40 calpain inhibitors have been developed and some of them have been trialed to treat diseases such as Alzheimer disease and atopic dermatitis.Citation54 However, most of the calpain inhibitors have been abandoned due to low efficiency, high cost or severe side effect. Therefore, more targeted and specific calpain inhibition strategies are needed in the future.
In summary, our studies demonstrated that the location of released IL-1α is critical for its role in tumor development. Tumoral-secreted IL-1α has a tumor-promoting function and suppresses the anti-tumor immune responses through MDSC recruitment to the tumor microenvironment. In contrast, systemic IL-1α administration leads to the reduction of tumor development by directly activating T cells. Therefore, understanding the differential functions of IL-1α release and distribution may facilitate the discovery of novel strategies for more targeted and effective immunotherapy.
Availability of data and material
All data that support the findings of this study are available from the corresponding authors upon reasonable requests.
Supplemental Material
Download MS Word (1.9 MB)Acknowledgments
We thank Dr. P. Hutchinson and Mr. G. Teo (NUS Immunology Program Flow Cytometry Core Facility) for assistance with FACS analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2088467
Additional information
Funding
References
- Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–14. doi:10.1111/imr.12621.
- Chiu JW, Binte Hanafi Z, Chew LCY, Mei Y, Liu H. IL-1alpha processing, signaling and its role in cancer progression. Cells. 2021;10(1):92. doi:10.3390/cells10010092.
- Buryskova M, Pospisek M, Grothey A, Simmet T, Burysek L. Intracellular interleukin-1alpha functionally interacts with histone acetyltransferase complexes. J Biol Chem. 2004;279(6):4017–4026. doi:10.1074/jbc.M306342200.
- Yano S, Banno T, Walsh R, Blumenberg M. Transcriptional responses of human epidermal keratinocytes to cytokine interleukin-1. J Cell Physiol. 2008;214(1):1–13. doi:10.1002/jcp.21300.
- Brody DT, Durum SK. Membrane IL-1: IL-1 alpha precursor binds to the plasma membrane via a lectin-like interaction. J Immunol. 1989;143:1183–1187.
- Lin D, Lei L, Liu Y, Zhang Y, Hu B, Bao G, Song Y, Jin Z, Liu C, Mei Y, et al. Membrane IL1alpha inhibits the development of hepatocellular carcinoma via promoting T- and NK-cell activation. Cancer Res. 2016;76(11):3179–3188. doi:10.1158/0008-5472.CAN-15-2658.
- Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Luthi AU, Reeves EP, McElvaney NG, Medema JP, Lavelle EC, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell. 2011;44(2):265–278. doi:10.1016/j.molcel.2011.07.037.
- Xu D, Mu R, Wei X. The roles of IL-1 family cytokines in the pathogenesis of systemic sclerosis. Front Immunol. 2019;10:2025. doi:10.3389/fimmu.2019.02025.
- Rider P, Kaplanov I, Romzova M, Bernardis L, Braiman A, Voronov E, Apte RN. The transcription of the alarmin cytokine interleukin-1 alpha is controlled by hypoxia inducible factors 1 and 2 alpha in hypoxic cells. Front Immunol. 2012;3:290. doi:10.3389/fimmu.2012.00290.
- Di Paolo NC, Shayakhmetov DM. Interleukin 1 alpha and the inflammatory process. Nat Immunol. 2016;17(8):906–913. doi:10.1038/ni.3503.
- Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood. 2010;115(17):3632–3639. doi:10.1182/blood-2009-11-252643.
- Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, Libby P. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139:1902–1910.
- Menghini P, Corridoni D, Butto LF, Osme A, Shivaswamy S, Lam M, Bamias G, Pizarro TT, Rodriguez-Palacios A, Dinarello CA, et al. 2019. Neutralization of IL-1alpha ameliorates Crohn’s disease-like ileitis by functional alterations of the gut microbiome. Proc Natl Acad Sci U S A. 116(52): 26717–26726.
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6(4):232–241. doi:10.1038/nrrheum.2010.4.
- Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51(1):27–41. doi:10.1016/j.immuni.2019.06.025.
- Xiao Z, Singh S, Singh M. Improving cancer immunotherapy by targeting IL-1. Oncoimmunology. 2021;10(1):2008111. doi:10.1080/2162402X.2021.2008111.
- Leon X, Bothe C, Garcia J, Parreno M, Alcolea S, Quer M, Vila L, Camacho M. Expression of IL-1alpha correlates with distant metastasis in patients with head and neck squamous cell carcinoma. Oncotarget. 2015;6(35):37398–37409. doi:10.18632/oncotarget.6054.
- Charbonneau B,MS, Block WR, Bamlet RA, Vierkant KR, Kalli Z, Fogarty DN, Rider TA, Sellers SS, Tworoger E, Poole HA, et al. Risk of ovarian cancer and the NF-κB pathway: genetic association with IL1A and TNFSF10. Cancer Res. 2014;74(3):852–861. doi:10.1158/0008-5472.CAN-13-1051.
- Kuan EL, Ziegler SF. A tumor-myeloid cell axis, mediated via the cytokines IL-1alpha and TSLP, promotes the progression of breast cancer. Nat Immunol. 2018;19(4):366–374. doi:10.1038/s41590-018-0066-6.
- Dvorkin T, Song X, Argov S, White RM, Zoller M, Segal S, Dinarello CA, Voronov E, Apte RN. Immune phenomena involved in the in vivo regression of fibrosarcoma cells expressing cell-associated IL-1alpha. J Leukoc Biol. 2006;80(1):96–106. doi:10.1189/jlb.0905509.
- Song X, Voronov E, Dvorkin T, Fima E, Cagnano E, Benharroch D, Shendler Y, Bjorkdahl O, Segal S, Dinarello CA, et al. Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J Immunol. 2003;171(12):6448–6456. doi:10.4049/jimmunol.171.12.6448.
- Voronov E, Weinstein Y, Benharroch D, Cagnano E, Ofir R, Dobkin M, White RM, Zoller M, Barak V, Segal S, et al. Antitumor and immunotherapeutic effects of activated invasive T lymphoma cells that display short-term interleukin 1alpha expression. Cancer Res. 1999;59:1029–1035.
- Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, Caucheteux S, Ratner-Hurevich M, Berzofsky JA, Nir-Paz R, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med. 2013;210(3):491–502. doi:10.1084/jem.20122006.
- Abdel-Wahab N, Safa H, Abudayyeh A, Johnson DH, Trinh VA, Zobniw CM, Lin H, Wong MK, Abdelrahim M, Gaber AO, et al. Checkpoint inhibitor therapy for cancer in solid organ transplantation recipients: an institutional experience and a systematic review of the literature. J Immunother Cancer. 2019;7(1):106. doi:10.1186/s40425-019-0585-1.
- Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res. 2012;96(1):32–37. doi:10.1093/cvr/cvs163.
- Starling S. Innate immunity: revealing the secrets of IL-1 secretion. Nat Rev Immunol. 2017;18(1):2–3. doi:10.1038/nri.2017.155.
- Hickish T, Andre T, Wyrwicz L, Saunders M, Sarosiek T, Kocsis J, Nemecek R, Rogowski W, Lesniewski-Kmak K, Petruzelka L, et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2017;18(2):192–201. doi:10.1016/S1470-2045(17)30006-2.
- Liu Y, Song Y, Lin D, Lei L, Mei Y, Jin Z, Gong H, Zhu Y, Hu B, Zhang Y, et al. NCR(-) group 3 innate lymphoid cells orchestrate IL-23/IL-17 axis to promote hepatocellular carcinoma development. EBioMedicine. 2019;41:333–344. doi:10.1016/j.ebiom.2019.02.050.
- Hu B, Bao G, Zhang Y, Lin D, Wu Y, Wu D, Liu H. Donor NK Cells and IL-15 promoted engraftment in nonmyeloablative allogeneic bone marrow transplantation. The Journal of Immunology. 2012;189(4):1661–1670. doi:10.4049/jimmunol.1103199.
- Lee CR, Kwak Y, Yang T, Han JH, Park SH, Ye MB, Lee W, Sim KY, Kang JA, Kim YC, et al. Myeloid-Derived suppressor cells are controlled by regulatory T cells via TGF-beta during murine colitis. Cell Rep. 2016;17(12):3219–3232. doi:10.1016/j.celrep.2016.11.062.
- Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1 + /CD11b + myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–6721. doi:10.1158/1078-0432.CCR-05-0883.
- Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485–498. doi:10.1038/s41577-020-00490-y.
- Lin D, Lei L, Zhang Y, Hu B, Bao G, Liu Y, Song Y, Liu C, Wu Y, Zhao L, et al. Secreted IL-1α promotes T-cell activation and expansion of CD11b + Gr1 + cells in carbon tetrachloride-induced liver injury in mice. Eur J Immunol. 2015;45(7):2084–2098. doi:10.1002/eji.201445195.
- Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi:10.1038/s41587-019-0114-2.
- Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev. 2018;281(1):57–61. doi:10.1111/imr.12614.
- Keenan BP, Fong L, Kelley RK. Immunotherapy in hepatocellular carcinoma: the complex interface between inflammation, fibrosis, and the immune response. J ImmunoTher Cancer. 2019;7(1):267. doi:10.1186/s40425-019-0749-z.
- Refolo MG, Messa C, Guerra V, Carr BI, D’Alessandro R. Inflammatory mechanisms of HCC development. Cancers. 2020;12(3):641. doi:10.3390/cancers12030641.
- Apte RN, Voronov E. Immunotherapeutic approaches of IL-1 neutralization in the tumor microenvironment. J Leukoc Biol. 2017;102(2):293–306. doi:10.1189/jlb.3MR1216-523R.
- Gehrke N, Hovelmeyer N, Waisman A, Straub BK, Weinmann-Menke J, Worns MA, Galle PR, Schattenberg JM. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J Hepatol. 2018;68(5):986–995. doi:10.1016/j.jhep.2018.01.008.
- Kasprzak A, Zabel M, Wysocki J, Seidel J, Surdyk-Zasada J, Filipiak B. Expression of mRNA for cytokines (TNF-alpha and IL-1alpha) in human cytomegalovirus (HCMV) and hepatitis B virus (HBV) infections. Folia Histochemica Et Cytobiologica. 2002;40:63–68.
- Steuerwald NM, Foureau DM, Norton HJ, Zhou J, Parsons JC, Chalasani N, Fontana RJ, Watkins PB, Lee WM, Reddy KR, et al. Profiles of serum cytokines in acute drug-induced liver injury and their prognostic significance. PloS one. 2013;8:e81974.
- Olteanu S, Kandel-Kfir M, Shaish A, Almog T, Shemesh S, Barshack I, Apte RN, Harats D, Kamari Y. Lack of interleukin-1alpha in Kupffer cells attenuates liver inflammation and expression of inflammatory cytokines in hypercholesterolaemic mice. Digestive and Liver Disease: Official Journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2014;46(5):433–439. doi:10.1016/j.dld.2014.01.156.
- Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. 2011;55(5):1086–1094. doi:10.1016/j.jhep.2011.01.048.
- Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14(2):156–165. doi:10.1016/j.ccr.2008.06.016.
- Zhang M, Liu L, Lin X, Wang Y, Li Y, Guo Q, Li S, Sun Y, Tao X, Zhang D, et al. A Translocation Pathway for Vesicle-Mediated Unconventional Protein Secretion. Cell. 2020;181(3):637–652 e615. doi:10.1016/j.cell.2020.03.031.
- Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR, Kinoshita T, Suda T. Gasdermin D mediates the maturation and release of IL-1alpha downstream of inflammasomes. Cell Rep. 2021;34(12):108887. doi:10.1016/j.celrep.2021.108887.
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18(6):607–618. doi:10.1038/ncb3346.
- Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-Derived suppressor cells as a therapeutic target for cancer. Cells. 2020;9(3):561. doi:10.3390/cells9030561.
- Lu LC, Chang CJ, Hsu CH. Targeting myeloid-derived suppressor cells in the treatment of hepatocellular carcinoma: current state and future perspectives. J Hepatocellular Carcinoma. 2019;6:71–84. doi:10.2147/JHC.S159693.
- Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015;42(6):991–1004. doi:10.1016/j.immuni.2015.06.003.
- England H, Summersgill HR, Edye ME, Rothwell NJ, Brough D. Release of interleukin-1alpha or interleukin-1beta depends on mechanism of cell death. J Biol Chem. 2014;289(23):15942–15950. doi:10.1074/jbc.M114.557561.
- Kimura Y, Koga H, Araki N, Mugita N, Fujita N, Takeshima H, Nishi T, Yamashima T, Saido TC, Yamasaki T, et al. The involvement of calpain-dependent proteolysis of the tumor suppressor NF2 (merlin) in schwannomas and meningiomas. Nat Med. 1998;4(8):915–922. doi:10.1038/nm0898-915.
- Shapovalov I, Harper D, Greer PA. Calpain as a therapeutic target in cancer. Expert Opin Ther Targets. 2022;26(3):217–231. doi:10.1080/14728222.2022.2047178.
- Ono Y, Saido TC, Sorimachi H. Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov. 2016;15(12):854–876. doi:10.1038/nrd.2016.212.