
ABSTRACT
Resistance remains an obstacle to anti-programmed cell death protein 1 (PD-1) therapy in human cancer. One critical resistance mechanism is the lack of T cell chemotaxis in the tumor microenvironment (TME). CXCL10-CXCR3 signaling is required for T cell tumor infiltration and tumor immunotherapy. Oncolytic viruses (OVs), including oncolytic adenoviruses (AdVs), induce effective T cell immunity and tumor infiltration. Thus, arming OV with CXCL10 would be an attractive strategy to overcome resistance to anti-PD1 therapy. Here, we successfully constructed a novel recombinant oncolytic adenovirus encoding murine CXCL10, named Adv-CXCL10. Through intratumoural injection, the continuous expression of the functional chemokine CXCL10 in the TME is realized to recruit more CXCR3+ T cells into the TME to kill tumor cells, and the recombinant adenovirus shows great power to ‘fire up’ the TME and enhance the antitumour efficiency of PD-1 antibodies.
Introduction
Programmed cell death protein 1 (PD-1) is a well-known immune checkpoint that regulates T cell function.Citation1 Although targeting PD-1 exhibits marked effects in treating various tumors in clinical practice, its response in most advanced tumor patients remains unsatisfactory.Citation2 Evidence shows that the efficiency of PD-1 blockade therapy is related to the gene microsatellite status, immune cell infiltration in the tumor microenvironment (TME), and interferon gamma (IFN-γ) signaling.Citation3–6 Considering advanced metastatic colorectal cancer (mCRC) as an example, PD-1 antibody monotherapy is only effective in some patients with high microsatellite instability (MSI-H), while most patients with a microsatellite-stable (MSS) status cannot benefit from this monotherapy.Citation6 Notably, however, ~ 90% of mCRC patients have an MSS genotype,Citation7 indicating that they do not respond well to anti-PD-1 therapy. Therefore, identifying appropriate approaches to enhance the efficacy of PD-1 blockade therapy is crucial.
Accumulating evidence has shown that CXCR3 and its ligands play a positive role in tumor prognosis and anti-PD-1 therapy.Citation8–11 CXCR3 is a chemokine receptor expressed in cytotoxic lymphocytes (CTLs), natural killer cells (NKs), NKT cells, DCs and B cells that can be recruited to specific sites by its ligands CXCL9/10/11.Citation10,Citation12 Our previous work has shown that CXCR3 expression in peripheral T cells can be used as a biomarker to predict the efficacy of PD-1 antibodies, while exogenous supplementation with intratumoural CXCL9/10 can assist PD-1 antibodies in inhibiting tumor growth by increasing the proportion of CXCR3+ T cells in the TME in a mouse melanoma model.Citation13 Furthermore, many other studies have suggested that intratumoural CXCL10 signaling is positively correlated with the efficacy of immune checkpoint blockade (ICB) treatment.Citation11,Citation14,Citation15 Consistent with our former discovery, the reported mechanism of these articles is related to more CXCR3+ T cells infiltrating the TME, making ‘cold’ tumors ‘hot’ and helping PD-1 antibodies achieve better antitumour effects. The evidence mentioned above implies the crucial status of CXCR3-CXCL9/10/11 in ICB, and the key point of this hypothesis was the sustained intratumoural chemokine concentration.
Chemokines have a short half-life in vivo, and CXCL10 has limited potential for direct use as a drug for this reason.Citation16 Therefore, strategies for the long-term expression of chemokines such as CXCL10 in the TME, maintenance of a high concentration gradient and continuous recruitment of CXCR3+ T cells must be developed. In recent years, oncolytic viruses (OVs) therapy has been widely studied as an emerging antitumour biotherapy.Citation17,Citation18 Mechanistically, OVs can directly lyse tumor cells. Additionally, it can induce the release of tumor-associated antigen (TAA) and promote pathogen-associated molecular patterns (PAMPs) and cellular danger-associated molecular patterns (DAMPs) to induce the body to produce natural immunity and adaptive immunity to attack tumor cells. Additionally, OVs serve as an excellent vector for exotic antitumour genes; among them, chemokines are the most common because of their extraordinary ability to recruit immune cells.Citation19–21 For example, the well-known oncolytic virus T-VEC is a type of recombinant herpes simplex virus (HSV) loaded with granulocyte-macrophage colony-stimulating factor (GM-CSF), and the data showed the combination of T-VEC with PD-1 antibody exhibited a 33% complete response rate (CR) in melanoma patients and an effective response in sarcomas.Citation22,Citation23
Here, we demonstrate the combination effect in a mouse colon cancer model by inserting the CXCL10 gene into an oncolytic adenovirus and combining it with PD-1 antibody. Regarding the mechanism, the oncolytic adenovirus (Adv-CXCL10) enhances the PD-1 antibody by increasing the number of CXCR3+ T cells in the TME, and the effect disappears when CXCR3 signaling is blocked. Using this approach, we found an effective strategy to sensitize cancer cells to PD-1 antibodies, with positive clinical translational significance.
Materials and methods
Cell lines
The mouse colon carcinoma cell line MC38, CT26 and human embryonic kidney cell line 293 T were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). MC38-CAR was a gift kindly provided by Professor Wei of Nanjing University. CT26 cells were cultured in RPMI 1640 medium, and the other two cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM). All the media were supplemented with 10% FBS, 100 U/mL of penicillin, and 100 mg/mL of streptomycin. Cells were cultured in a 5% CO2, 37°C incubator.
Recombinant adenovirus construction
The generation of the recombinant replicable adenovirus was described as previously reported.Citation24 The E1A cDNA was obtained from 293 T cells. cDNA encoding murine CXCL10 and EGFP was synthesized by Genescript, Nanjing, China. The adenovirus shuttle plasmid required in our study was generated by PCR amplification and connected by ligation PCR. Then the shuttle plasmid was linearized with Pme I and transferred into competent BJ5183 cells containing the adenoviral backbone plasmid (pAdEasy), which was a gift from professor Wei, to create the total length plasmid of recombinant adenovirus. After linearized with Pac I, the whole plasmid was transfected and amplified in 293 T cells to obtain recombinant adenovirus, and the purification procedure was accomplished in Obio Technology (Shanghai, China) by the CsCl method. Viral titer confirmation was performed by infecting 293 T cells using a concentration gradient dilution method and was calculated as follows: TCID50 = 102+ (S/N−0.5) /mL, PFU/mL = 0.7 × TCID50/mL (TCID50: 50% tissue culture infective dose; S: total number of EGFP positive wells; N: replicate number of the same concentration; PFU: plaque-forming units).
Replication of the recombinant adenovirus in tumor cells
MC38-CAR cells were infected with recombinant adenovirus at a multiplicity of infection (MOI) of 10, and the samples were collected 12, 24, 48, 72, 96 and 120 hours after virus infection. Two methods were used for confirmation the replication capacity of the recombinant virus. One was the TCID50 methods described as above, and the other was as follows: viral DNA was collected after 12, 24, 48, 72, 96 and 120 hours using a DNA extraction kit according to the manufacturer’s instructions (19321ES50; YEASEN, Shanghai, China). Next, quantitative PCR was performed, and viral copies were calculated using the comparative threshold cycle method and standardized using the copies of 12 hours.
Enzyme-linked immunosorbent assay (ELISA)
CXCL10 in the cellular supernatant, serum and tumor tissues and IFN-γ and granzyme B in tumor tissues were measured by ELISA following the manufacturers’ instructions. The CXCL10 and Granzyme B ELISA kits were purchased from MultiSciences Biotech (EK268/2; EK2173; Hangzhou, China), and the IFN-γ ELISA kit was obtained from Biolegend (430804; San Diego, CA, USA).
Crystal violet staining and the CCK8 assay
For the crystal violet assay, five thousand MC38-CAR cells were plated in each well of a 96-well plate, and recombinant adenoviruses were added to the wells using different MOIs (MOIs = 0, 5, 10, 20, 40 and 80). The cells were harvested at 48, 72 and 96 hours, fixed with paraformaldehyde for 30 minutes and stained with crystal violet solution for 1 hour. Finally, the crystal violet solution was removed, the wells were washed with PBS 3 times, and photos were taken. The CCK-8 assay was performed similarly to the crystal violet assay. After 48 hours of virus infection, 20 µL of CCK-8 solution was added to each well containing 200 µL of culture medium, and the cells were incubated continuously until the absorbance (A) was measured at 450 nm using a microplate reader within 4 hours. The cell survival relative to control was calculated as follows: cell survival rate (%) = (AMOI=5, 10, 20, 40, 80-Ablank)/(AMOI=0-Ablank) ×100%.
Isolation and culture of murine lymphocytes and the in vitro chemotaxis assay
Lymph nodes were isolated from C57BL/6 mice and carefully ground into single cells and then were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/mL of penicillin, 100 mg/mL of streptomycin, soluble anti-CD3 (5 μg mL−1; 16–0031-86; eBioscience/Thermo Fisher Scientific, Waltham, MA), anti-CD28 (5 μg mL−1; 16–0281-86; eBioscience) and IL-2 (200 U mL−1; 212–12; Peprotech, NJ, USA) for 3 days.Citation25,Citation26 The chemotaxis assay was then performed in 24-well plates with Transwell permeable supports using a 5 μm polycarbonate membrane (3421; Corning Life Sciences, Tewksbury, MA, USA). Briefly, 500 µL of 1640 alone, the supernatant of control adenovirus (Adv-Ctrl)- or Adv-CXCL10-infected MC38-CAR or 1640 with 200 pg/mL of recombinant murine CXCL10 (rCXCL10; 250–16; Peprotech), all of which were supplemented with 10% FBS, were added to the lower well. The activated lymphocytes were washed and resuspended at 6 × 107 per mL in 1640 containing 0.3% FBS, among which 100 μL was loaded into each upper chamber, and the plates were incubated for 2 hours in a 5% CO2 humidified incubator at 37°C. After the incubation period, photos of the cells that migrated into the lower wells were taken under a microscope, and the number of cells was counted using a cell counter (Life Technology, USA). All the conditions were tested in triplicate.
Animals
Six- to eight-week-old female C57BL/6 and BALB/c mice were purchased from Tande Biotechnology (Nanjing, China). They were housed in plastic cages with free access to pellet food and water at 21 ± 2°C and kept on a 12-hour light/dark rhythm. Animal welfare and experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and the ethical regulations of our university (Animal ethical approval number: IACUC2011008). All efforts were made to reduce the number of animals used and minimize animal suffering.
Syngeneic model
5 × 105 MC38-CAR cells were inoculated subcutaneously into the right flank of the C57BL/6 mice. After the tumor volume reached approximately 50–100 mm3, the mice were randomly assigned to different intervention groups. (1) For adenovirus monotherapy experiments, the mice received intratumoural injection (i.t.) of Adv-Ctrl, Adv-CXCL10 or PBS every 2 days for 3 times. The dose of recombinant adenovirus was 3 × 108 PFU per mouse each time. Samples were collected 2 days after the last injection (on day 11). (2) For adenovirus and anti-PD-1 combination therapy, adenovirus was applied to the mice as described above, and mice were injected intraperitoneally (i.p.) with PD-1 antibody at a dose of 5 mg/kg every 2 days for 5 times. Samples were collected 6 days after the last injection (on day 19). Additionally, the survival of mice was observed until the tumor volume of the mouse approached 2000 mm3 or at the indicated time. (3) Regarding the anti-CXCR3 experiment, in addition to the previous intervention, the mice were treated with either the CXCR3 antibody (200 μg per mouse; 126537; Biolegend) or the isotype control (200 μg per mouse; BE0260; Bio X cell) every 7 days. Samples were collected 2 days after the last injection (on day 18). In all the animal experiments, the mice’s body weight was recorded every 2 days, and the tumor volume was measured by assessing the length and width of the tumor using a digital caliper. The volume was calculated according to the following formula: Volume = (long axis × short axis2)/2. The tumor weight was measured after euthanasia of the mice.
Isolation of tumor-infiltrating lymphocytes (TILs) and CD8+ TILs
For TILs isolation, the tumor tissues were mechanically and enzymatically dissociated using a mouse tumor dissociation kit provided by Miltenyi Biotec (130–0960-730; Bergisch Gladbach, Germany). The single-cell suspension was then filtered using a 70 μm mesh. Afterward, the TILs were obtained by density gradient separation using a Percoll gradient (40% – 70%) and centrifugation at 700 × g for 20 min. Finally, this portion of the cells was resuspended in 1640 with 10% FBS for further application. CD8+ TILs were enriched with CD8 (TIL) MicroBeads kit (130–116-478, Miltenyi Biotec) according to the instruction. The CD8+ TILs were cultured in RPMI 1640 medium supplemented with 10% FBS, 200 U/mL of penicillin, 200 mg/mL of streptomycin and IL-2 (200 U mL−1; Peprotech) for further experimentation.
IFN-γ ELISpot assay
The Immune activity of CD8+ TILs was confirmed by IFN-γ ELISpot kit (3321–4AST-2, Mabtech AB, Sweden), following the kit protocol. CD8+ TILs isolated from tumor tissue were seeded into the IFN-γ capture antibody pro-coated 96-well plates along with MC38-CAR or CT26 at an E:T ratio of 10:1 (effector CD8+ TILs: target tumor cells = 104: 103) and cultured in 37°C, 5%CO2 incubator for 36 h. Then cells were removed and the wells were washed with PBS for 4 times. Detection antibody was added into each well for a 2-hour incubation at room temperature (RT), followed by a 4-time rinse with PBS, and wells were added with the streptavidin-alkaline phosphatase (ALP) and went through an hour incubation at RT, followed by the addition of substrate. Finally, the reaction was stopped by washing the wells with tap water once spots appeared. Photos were taken using a dissecting microscope, and the spots number was counted.
Flow cytometry
Single cell suspensions of TILs were stained with different antibodies: Zombie NIRTM (423105; Biolegend) for dead cell preclusion, TruStain FcXTM (anti-mouse CD16/32; Biolegend) for the removal of nonspecific adsorption, CD45-BV421 (103134; Biolegend), CD45-BV510 (103138; Biolegend), CD4-AF488 (557667; BD Biosciences, San Jose, CA), CD4-PE/DazzleTM 594 (100456; Biolegend), CD8-FITC (100706; Biolegend), CD8-BV421 (100738; Biolegend) and CXCR3-PE (155904; Biolegend) to mark different TILs.
Reverse transcription-PCR and quantitative PCR
For the adenovirus replication assay, quantitative PCR was performed as previously reported.Citation27 For tumor tissue mRNA analyses, total RNA was first isolated using TRIzol reagent (Takara Bio, Beijing, China), and cDNA was synthesized using PrimeScript RT Reagent Kits (Takara) according to the manufacturer’s recommendation. Quantitative RT–PCR was performed using SYBR Premix Ex Taq (Takara) and a CFX96 Real-time system (Bio–Rad Laboratories). The amplification program was as follows: one cycle of 95°C for 2 min, followed by 40 cycles of 95°C for 10s, 60°C for 30s, and 95°C for 10s.Citation28 The primer sequences were as follows: viral skeleton protein gene (Hexon), forward 5’-ACCGTGAGGATACTGCGTAC-3’, reverse 5’-TTGCTCGTCTACTTCGTCTT-3’; mouse IFN-γ, forward 5’-ACAGCAAGGCGAAAAAGGATG-3’, reverse 5’-TGGTGGACCACTCGGATGA-3’; mouse Granzyme B, forward 5’-CCACTCTCGACCCTACATGG-3’, reverse 5’-GGCCCCCAAAGTGACATTTATT-3’ and mouse β-actin, forward 5’-GTGACGTTGACATCCGTAAAGA-3’, reverse 5’-GCCGGACTCATCGTACTCC-3’.
Hematoxylin and eosin (H&E) staining, TUNEL assay, immunohistochemistry (IHC) and immunofluorescence (IF)
Tumor tissues were stained with H&E as per standard protocols and analyzed under a light microscope (Olympus). For all staining protocols, the tissue sections were deparaffinized, rehydrated, and washed in 1% PBS–Tween 20. For IHC, the sections were soaked into 3% hydrogen peroxide to block endogenous peroxidases, blocked with 5% goat serum, and incubated with the primary antibodies overnight at 4°C. The sections were then incubated with streptavidin-HRP for 1 hour, stained with diaminobenzidine substrate, and counterstained with hematoxylin. For IF, the slides were stained with primary antibodies overnight at 4°C, incubated with fluorescently labeled secondary antibody for 1 hour the next day, and then counterstained with DAPI for 5 min. The TUNEL assay was performed according to the manufacturer’s protocol using a TMR (red) TUNEL Cell Apoptosis or FITC TUNEL Cell Apoptosis Detection Kit (G1501-50, G1502-50; Servicebio Technology, Wuhan, China). Images were acquired by fluorescence microscopy (Olympus). The antibodies used here included anti-PCNA (sc-56; Santa Cruz, CA, USA), anti-CD4 (sc-13573; Santa Cruz), anti-CD8 (sc-18913; Santa Cruz), anti-granzyme B (sc-8022; Santa Cruz), and anti-IFN-γ (15365-1-AP; Proteintech, USA). The secondary antibodies used in IF were purchased from Thermo Fisher Scientific (A11037, A21135, and A21209).
Statistical analyses
The data were described as mean ± SD. Student’s test and ANOVA were used for analyses. P values < .05 were considered statistically significant. Statistical analyses were conducted using GraphPad Prism Software (Version 7.0; La Jolla, CA).
Materials and Methods of Supplementary data were presented in Supplementary Materials.
Results
Generation of a recombinant oncolytic adenovirus expressing murine CXCL10
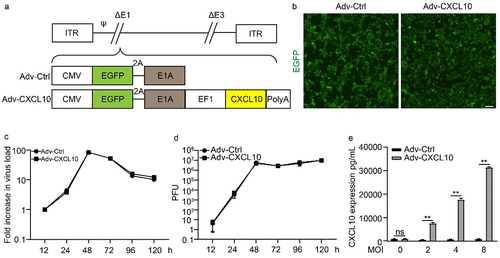
The construction diagram of recombinant oncolytic adenovirus encoding murine CXCL10, here named as Adv-CXCL10, is shown in . After the recombination procedure, EGFP expression was observed on 293 T cells (), confirming the successful packaging of both control adenovirus (Adv-Ctrl) and Adv-CXCL10. To determine whether the insertion of CXCL10 affected the replication capacity of oncolytic adenovirus, we infected the mouse colon cancer cell MC38-CAR with different MOIs of virus. Compared with Adv-Ctrl, Adv-CXCL10 showed equivalent replication ability on MC38-CAR cells (). Furthermore, ELISA confirmed that CXCL10 was expressed by Adv-CXCL10 and secreted into the cell supernatant (). The above results suggest that we successfully constructed a novel recombinant adenovirus encoding and expressing murine CXCL10 without changing its replication ability.
Figure 1. Generation of an oncolytic adenovirus expressing murine CXCL10. (a) Schematic diagram of oncolytic adenovirus construction. Adv-Ctrl, control recombinant adenovirus; Adv-CXCL10, recombinant adenovirus encoding murine CXCL10. (b) Fluorescent images of 293 T cells after 24-hour infection with Adv-Ctrl or Adv-CXCL10. Scale bar, 100 μm. (c-d) MC38-CAR cells were infected with recombinant adenovirus at an MOI of 10 and harvested at the indicated time points. (c) Viral DNA was extracted, and the viral copy number was quantified by quantitative PCR. The figure represents one of three replicative experiments. (d) Viral replication capacity determined by titer test using TCID50 method. The figure represents one of three replicative experiments. (e) CXCL10 concentration in different cell supernatants. MC38-CAR cells were infected with adenovirus at different MOIs. The supernatants were collected 48 hours after virus infection. Three replicates were performed for each sample. (mean ± SD; * P < .05; ** P < .01; ns, not significant)
Adv-CXCL10 not only kills tumor cells but also recruits lymphocytes in vitro
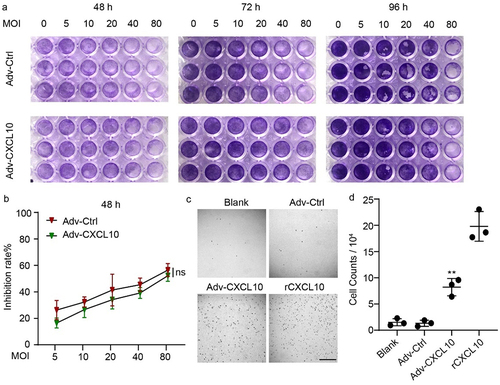
To evaluate the oncolytic ability of the recombinant adenovirus, we infected MC38-CAR cells with adenovirus at increasing MOIs. After 48, 72 and 92 hours, the oncolytic capacity was confirmed by crystal violet staining (). The cell survival rate relative to control at 48 hours was determined by the CCK-8 assay (). These results illustrated time- and MOI-dependent lytic activity against colon cells. Although the violet color of 96 h in seemed deeper than that of 48 or 72 h, it may result from the increasing cell numbers after a longer period of growth, and the obvious stronger killing capacity at 96 hours could still be observed on cells infected with higher MOIs of virus, especially when the MOIs reached to 40 or 80. On the other hand, although Adv-Ctrl seemed to show a relatively stronger oncolytic capacity than Adv-CXCL10, no significant difference was found between them. Next, we examined the chemotactic function of CXCL10 expressed by recombinant adenovirus. Transwell assay showed that more murine primary lymphocytes were attracted to the lower chamber with supernatant containing CXCL10 than to the blank well or supernatant produced by Adv-Ctrl (). In conclusion, the recombinant adenovirus Adv-CXCL10 can kill colon cancer cells similar to the control adenovirus and also produces functional murine CXCL10.
Figure 2. Adv-CXCL10 not only kills tumor cells but also recruits lymphocytes in vitro. (a) MC38-CAR cells were plated into 96-well plates and infected with Advs at the corresponding MOIs for 48, 72 and 96 hours. The oncolytic ability was tested by crystal violet staining. (b) MC38-CAR cells were incubated in 96-well plates and infected with Advs at different MOIs for 48 hours. The cell survival rate relative to control was calculated by the CCK-8 assay. (c&d) Chemotaxis assay of murine primary lymphocytes. (c) Microscope images of lymphocytes migrating to the lower well attracted by different conditional media in the lower chambers. Scale bar, 200 μm. (d) Counts of lymphocytes recruited into the lower chambers by the medium. (The data all showed one of three independent experiments. mean ± SD; * P < .05; ** P < .01; ns, not significant)
Adv-CXCL10 promotes T cell infiltration in MC38-CAR allograft tumors in vivo
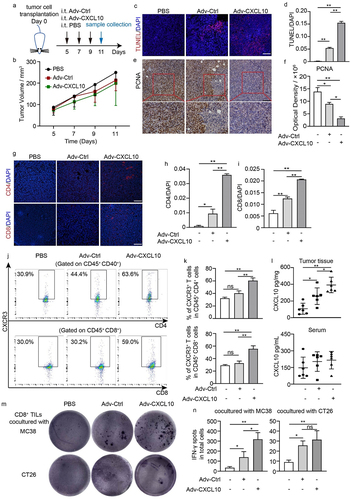
Based on the tumor cell-killing performance in vitro, we next explored the in vivo function of Adv-CXCL10 in an MC38-CAR mouse colon cancer model. The administration of adenovirus is shown in , and the mice were sacrificed and the samples were harvested on Day 11. There was a tendency that when using the recombinant adenovirus monotherapy, the tumor grew slower, although no significant difference was seen (). Notably, the oncolytic adenovirus indeed increased tumor apoptosis and inhibited tumor proliferation (). Because oncolytic adenovirus induced an immune response in vivo and because CXCL10 itself is a chemokine that activates and attracts specific immune cells, we next explored the immune infiltration status in the TME. The adenovirus-treated groups presented a better immune-killing TME, with more CD4+ T cells and CD8+ T cells (, Supplementary Figure S1). Furthermore, the population of CXCR3+ cells was increased in both CD4+ T and CD8+ T cells (gating strategy shown in Supplementary Figure S2), particularly in the Adv-CXCL10 group (), contributing to the higher concentration gradient of CXCL10 in the TME than in other areas (). To further confirm that the T cells attracted into the TME by adenovirus have the antitumour properties, we cocultured them with either MC38 or CT26 colon cells in vitro, and found that these CD8+ TILs (The enrichment efficiency of the CD8+ TILs was tested by flow cytometry, shown in Supplementary Figure S3) had tumor-specific recognition capacity, because they produced more IFN-γ when stimulated by MC38, but not by CT26 (). These phenomena were more obvious in the Adv-CXCL10 treated group.
Figure 3. Adv-CXCL10 promotes T cell infiltration in MC38-CAR allograft tumors in vivo. (a) Brief process of MC38-CAR allograft model construction. Samples were collected on Day 11. (b) Tumor volume changes assessed every 2 days once the treatment began. (c&d) TUNEL staining (c) and quantification (assessed by 3 random areas in (c)) of TUNEL-positive areas in tumor tissues. Scale bar, 100 μm. (e&f) Images of PCNA-positive cells (e) and quantification of PCNA-positive areas (f). Scale bar, 100 μm. (g) Expression of CD4 and CD8 in the tumor tissues of each group was tested by IF. Scale bar, 100 μm. (h&i) Quantitative analysis of CD4 and CD8 positive areas in (g). Three random fields of view per mouse of different groups were calculated. (j&k) Cytometric analysis of CXCR3+ T cells in the TILs of each group. (j) Representative image of 3 replications in each group. (k) Statistical analysis of CXCR3+ T cells in different groups. n = 3. (l) CXCL10 expression in mouse peripheral blood serum and tumor tissues, assessed by ELISA. n = 6. (m-n) IFN-γ ELISpot assay. CD8+ TILs from adenovirus monotherapy MC38-CAR allograft model and MC38 or CT26 were cocultured for 36 h at a 10:1 E/T ratio before harvest. (m) Picture shows the representative image of 3 replications in each group. (n) IFN-γ spots number of different intervention groups in (m). (The data are shown as mean ± SD. * P < .05; ** P < .01; ns, not significant)
Adv-CXCL10 strongly improves the efficiency of anti-PD-1 therapy
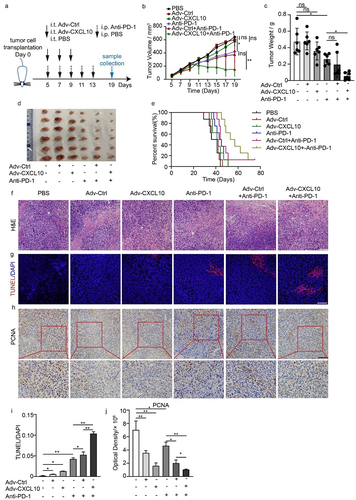
We found an increasing expression of PD-L1 on the MC38-CAR cells when they treated with oncolytic adenovirus. Also, we observed the same tendency during the oncolytic adenovirus therapy in vivo (Supplementary Figure S4). This suggested the treatment of oncolytic adenovirus enabled the TME to turn into a ‘hotter’ position, so we next wondered whether it could further enhance the anti-PD-1 effect in a colon cancer model. A brief experimental procedure is shown in . Consistent with our previous discovery, Adv-Ctrl- or Adv-CXCL10-treated mice showed a lower tumor volume than control mice. Surprisingly, Adv-CXCL10 strongly enhanced the therapeutic effect of the PD-1 antibody, and the tumor volume of the corresponding group even remained at a lower plateau for several days (). Notably, none of the interventions influenced the mouse’s body weight, indicating the safety of these treatments (Supplementary Figure S5). We further observed the survival rate of different groups and found that the mice in the Adv-CXCL10+ anti-PD-1 group lived longer than others (). H&E and TUNEL assays showed the best apoptosis-promoting effect in combination with Adv-CXCL10 and anti-PD-1 therapy (), and this combination also inhibited tumor proliferation compared with other regimens ().
Figure 4. Adv-CXCL10 strongly improves the efficiency of anti-PD-1 therapy. (a) Schematic diagram of the experimental setup for the combination therapy of recombinant adenovirus and PD-1 antibody in MC38-CAR allograft model. Samples were collected on Day 19. (b) Tumor volume changes assessed every 2 days once the treatment began. (c) Analysis of solid tumor weight in 6 different intervention groups. (d) Photos of solid tumors taken after the sacrifice of mice in six groups. (e) Survival curves of mice in different intervention groups. n = 8 for every group. (f) H&E staining of tumor tissues. Scale bar, 100 μm. (g) Apoptosis analysis of tumor tissues assessed by TUNEL staining. Scale bar, 100 μm. (h) Proliferation assay analyzed by PCNA IHC staining. Scale bar, 100 μm. (i&j) Quantification of TUNEL staining and PCNA staining in three different areas in (g) and (h). (n = 6 mice per group in a-d, f-j. The data are shown as mean ± SD. * P < .05; ** P < .01 ns, not significant.)
Adv-CXCL10 enhances anti-PD-1 therapy by remodeling an antitumour immune microenvironment
Immunofluorescence and flow cytometry were used to prove the capacity of the combination therapy to recruit immune cells. CD4+ T and CD8+ T cells of the Adv-CXCL10 and PD-1 antibody combination regimen significantly increased compared with those in other groups (), and the percentage of CXCR3+ T cells presented a synchronous increase in the combination group (). IFN-γ plays an essential role in the CXCL10-CXCR3 axis and is a tumor-suppressive cytokine, and we confirmed an increase in IFN-γ in Adv-CXCL10 and anti-PD-1 combination therapy (). Consistent with this finding, granzyme B showed the same variation (), further confirming the function of Adv-CXCL10 in enhancing the antitumour efficiency of the PD-1 antibody.
Figure 5. Adv-CXCL10 enhanced anti-PD-1 therapy by recruiting more CXCR3+ T cell into TME. Samples were from MC38-CAR allograft model, collected on Day 19. (a&c) Images of CD4 and CD8 positive areas in tumor tissues assessed by IF. Scale bar, 100 μm. (b&d) Quantification of CD4 and CD8-positive areas in (a) and (c). Three random fields of view per mouse of different groups were calculated. (e&g) The percentage of CD4+CXCR3+ T and CD8+CXCR3+ T cells in TILs shown by flow cytometry. Pictures presented typical examples of 3 samples in each group. (f&h) Quantification of CXCR3+ T cell percentages in different intervention groups. (The data are shown as mean ± SD. * P < .05; ** P < .01; ns, not significant)
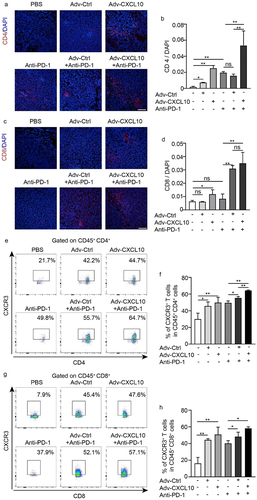
Figure 6. Adv-CXCL10 enhances anti-PD-1 therapy by remodeling an antitumour immune microenvironment. Samples were from MC38-CAR allograft model, collected on Day 19. (a&b) IFN-γ expression in tumor tissues presented by IF (a) and quantification analysis (b). Scale bar, 100 μm. (c&d) mRNA (tested by qPCR) and protein quantification (tested by ELISA) of IFN-γ expression in tumor tissues. (e&f) Granzyme B expression in tumor tissues presented by IF (e) and quantification analysis (f). Scale bar, 100 μm. (g&h) mRNA (tested by qPCR) and protein quantification (tested by ELISA) of granzyme B expression in tumor tissues. (n = 6 mice per group. The data are shown as mean ± SD, * P < .05; ** P < .0001; ns, not significant.)
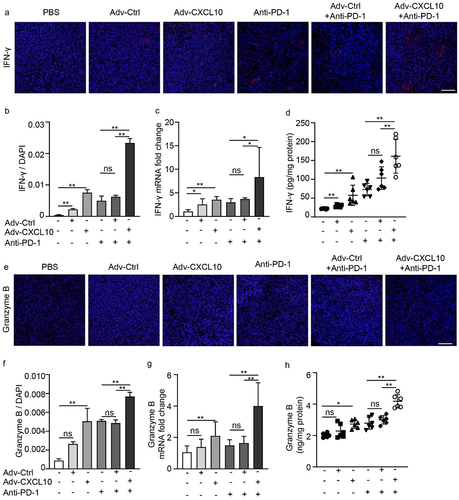
The in vivo Adv-CXCL10 combination effect with PD-1 antibody depends on CXCR3+ cells
The discovery above suggests the important role of CXCR3+ cells in the combination regimen. To confirm our findings, a CXCR3 blockade experiment was performed when mice were administered a combination of Adv-CXCL10 and PD-1 antibody (). CXCR3+ T cells were eliminated 1 day after administering CXCR3 antibody, and the blocking effect was observed even for 7 days (Supplementary Figure S6). Consequently, the antitumour effect was abrogated in both Adv-Ctrl and Adv-CXCL10 with PD-1 antibody combination therapy (). Taken together, the results indicate that CXCR3 signaling is vital in combination therapy.
Figure 7. The in vivo Adv-CXCL10 combination effect with PD-1 antibody depends on CXCR3+ cells. (a) Brief introduction of CXCR3 blockade experiments of MC38-CAR allograft model. (b) Photos of solid tumors taken after the sacrifice of mice in separate groups. (c) Tumor volume changes assessed every 2 days once the intervention began. (d) Analysis of solid tumor weight in 5 different intervention groups. (n = 6 mice per group, data are shown as the mean ± SD, * P < .05, ** P < .01, ns, not significant.)
Discussion
Although ICB has achieved positive clinical results in some tumor types, a large proportion of patients cannot benefit from ICB monotherapy. The reason may be related to the ineffective infiltration of immune cells in the TME according to numerous studies.Citation29–32 Tumors can be divided into 3 different types based on the infiltration of immune cells in the TME: inflammatory, immune excluded and immune desert phenotypes.Citation33 Evidence shows that immune cells in the TME and intratumoural IFN-γ signaling are more important for tumor prognosis than the TNM staging method in some solid tumors.Citation34 For such ‘cold tumours’, different strategies, such as promoting T cell infiltration and activating tumor immunogenicity, can be used to increase the inhibition capacity of ICB.Citation35 The presence of specific chemokines, such as CXCL10, is associated with different immune cell subsets and a high density of T cell subsets in specific tumor regions.Citation9 Multiple reported internal mechanisms of different combination strategies to increase the efficacy of PD-1 antibody also point to the CXCR3-CXCL9/10 axis.Citation14,Citation15,Citation36,Citation37 We confirmed the same conception in both CT 26 and MC38 mouse colon cancer models because adding rCXCL10 to PD-1 antibody for treatment has a better response than using PD-1 antibody alone (Supplementary Figure S7). This finding suggests that this signal plays a crucial role in ICB combination therapy. Previous reports, our previous studies and CXCR3 blocking experiments in the present study all confirmed that the antitumour effect of PD-1 blocking therapy was significantly weakened after the CXCR3 signaling was lost.Citation13,Citation38
As an approach to biotherapy for cancer, OVs has attracted researchers’ attention for several decades.Citation18,Citation39 OVs can attack the tumor area in multiple ways: they can infect and lyse the cancer cell directly, and also have the ability to cause collateral destruction within the TME. For instance, they can lead to bystander effect after entering the TME, cause hypoxia environment and develop growth arrest or death in uninfected cells.Citation20,Citation21 That may partially explain why the uninfected areas (EGFP negative areas) also showed low proliferation ability (Supplementary Figure S8). However, many technical challenges exist concerning their application because of toxicity, side effects, and nonspecific replication, among others. In recent years, scientists have tried to address these technical barriers in different ways, such as engineering viruses to specifically infect tumor cells without harming normal cells.Citation18 In our experiment, attenuated type 5 adenovirus (Adv5) lacking the E1B gene was used. Deletion of the E1B gene of Adv5 disabled the ability of Adv5 to downregulate p53 activity, ensuring its specific replication in tumor cells (because most tumors have highly dysfunctional p53 activity) without causing toxicity to normal cells.Citation20,Citation27,Citation40,Citation41 In 2006, China officially approved a modified oncolytic type 5 adenovirus – H101 – to treat head and neck squamous cell carcinoma, also demonstrating the safety of this type of oncolytic virus.Citation27 Notably, Adv5 infects cells by binding coxsackievirus and adenovirus receptor (CAR) of target cells.Citation42 Thus, human cells are susceptible to Adv5 as permissive cells. However, this receptor is absent in most established mouse cell lines, limiting the virus’s ability to infect murine cell lines.Citation43 Therefore, we used the stably transfected mouse colon cancer cell line MC38-CAR to better simulate recombinant oncolytic adenovirus in the human body. Consequently, MC8-CAR cells exhibited significant fluorescence when infected with recombinant adenovirus at an MOI of 10 compared with MC38 wild type (MC38 WT) in vitro (Supplementary Figure S9), and the addition of the CAR (mouse origin) didn’t influence the growth ability of MC38 in vitro, and didn’t show immunogenic response in vivo (Supplementary Figure S10).
As mentioned previously, OVs can be used as a powerful tool to carry exogenous genes so that some immunoregulatory genes can be expressed in the TME to enhance the antitumour effect.Citation19 Based on the size of the genome, different OVs have different capacities for exogenous gene insertion, and larger viruses can accommodate larger transgenes.Citation19 For example, vaccinia virus (VV) inserts large (25–40 kb) exogenous gene fragments because of its more than 180 kb genome,Citation44 while adenovirus has a moderate genome (32 kb), so it has a relative weaker transgene capacity than VV.Citation19 As immune regulators, chemokines are highly suitable to integrate into OVs, and they can inhibit tumor growth along with OVs.Citation45 In our study, the coding sequence of CXCL10 (297 bp) from NBCI database with gene ID: 15945 was cloned into the adenovirus. And the inserting CXCL10 into adenovirus did not affect the replication capacity and oncolytic ability of the recombinant virus ().
Different studies have inserted various cytokines and chemokines into the OVs for different purpose. For example, RANTES (regulated upon activation, normally T expressed, and presumably secreted, also known as CCL5) is inserted into OV to increase the infiltrations of DCs in the TME, and thus triggering antitumour properties of cytotoxic T cells.Citation46,Citation47 However, this is an indirect way and ultimately the involvement of CD8+ T cells is required to inhibit tumor progression. On the other hand, one of the RANTES’ receptors, CCR5, is reported to be expressed on regulatory T cells,Citation46 while other studies point out that RANTES/CCR5 signaling may stimulate tumor progression and metastasis,Citation48 so the application of RANTES in antitumour treatment may be restricted. IL-15 is another potent cytokine which is widely utilized in antitumour research. IL-15 can promote NK, NK T cell and T cell proliferation and activation, and sustain the survival of these cells.Citation49 For this reason, researchers constructed IL-15 contained OV to maintain the survival of CAR-T in solid tumor, and received a better response.Citation50 Different from IL-15, we constructed a recombinant adenovirus armed with CXCL10, which is not only able to activate T cells, but also able to attract more activated CXCR3+ T cells into the TME, thus enhance the PD-1 antibody therapy. CXCR3 has three ligands, including CXCL9, CXCL10 and CXCL11, and all of them can function as CXCR3+ T cell trafficker.Citation12 Several studies have explored the antitumour efficiency of CXCL9 or CXCL11-armmed OVs. For example, CXCL11 armed oncolytic poxvirus elicits immune response in mouse melanoma model and colon cancer model, and the authors also mentioned in their article that OVs expressing CXCR3 ligands could be an effective antitumour strategy,Citation51 which further confirmed the effectiveness of our recombinant adenovirus. Among these three ligands, it is hard to say which one is better, while based on our former discovery, we confirmed that our recombinant adenovirus can lyse tumor cells directly, and more importantly, we realized the goal of stably expressing and building a higher concentration gradient of CXCL10 in the TME than in serum, thus increasing CXCR3+ T cells infiltration. However, we did not observe a significant inhibition of mouse subcutaneous colon cancer when using the recombinant adenovirus alone, although there was a tendency for that effect (). The reason may be related to the amount and frequency of the virus administered. Additionally, based on the insensitivity of PD-1 antibody monotherapy and multiple characteristics of OVs mentioned above, we found that the combination of these two strategies produced a significant tumor suppression effect lasting a long time. We demonstrated increased CXCR3+ T cell infiltration and increased IFN-γ and granzyme B production in the TME, which are common killing factors for antitumour immunity.
In summary, we constructed a recombinant oncolytic adenovirus encoding the CXCL10 gene to fire up and reshape the TME by multiple means to make tumors more sensitive to anti-PD-1 therapy. Our work suggests that oncolytic viruses can be used as a safe and effective adjuvant tool to carry exogenous immune regulatory factors, such as CXCL10, activate the immune landscape within the TME and cooperate with PD-1 antibodies to achieve antitumour effects through multiple mechanism. Our experiment is based on a murine system, and further validation in the human system is required to prove this combination regimen in the future.
Author contributions
XF.L., MJ.L., XX.W., YH.G. and J.D. designed the study. XF.L., MJ.L., MM.Y., J.Y., W.Z., LY.X., XH.W., BQ.H., YC.Y., B.W., CL.G., and M.W. performed experiments and analyzed data. XF.L., MJ.L., and XX.W. interpreted data and contributed to the discussion. XF.L., MJ.L., XX.W., YH.G. and J.D. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Supplemental Material
Download MS Word (2.7 MB)Acknowledgments
We thank Mrs. Jie Zhu and Mr. Dianhua Chen (Nanjing University) for the help of Flow Cytometry.
Disclosure statement
All authors declare that they have no conflict of interests.
Data availability statement
We confirm we understand the terms of the share upon reasonable request data policy and we will make the data freely available upon request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2118210
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12: 252–13. doi:10.1038/nrc3239.
- Li X, Song W, Shao C, Shi Y, Han W. Emerging predictors of the response to the blockade of immune checkpoints in cancer therapy. Cell Mol Immunol. 2019;16: 28–39. doi:10.1038/s41423-018-0086-z.
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375: 819–829. doi:10.1056/NEJMoa1604958.
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127: 2930–2940. doi:10.1172/JCI91190.
- Kikuchi T, Mimura K, Okayama H, Nakayama Y, Saito K, Yamada L, Endo E, Sakamoto W, Fujita S, Endo H, et al. A subset of patients with MSS/MSI-low-colorectal cancer showed increased CD8(+) TILs together with up-regulated IFN-gamma. Oncol Lett. 2019;18: 5977–5985. doi:10.3892/ol.2019.10953.
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372: 2509–2520. doi:10.1056/NEJMoa1500596.
- Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394: 1467–1480. doi:10.1016/S0140-6736(19)32319-0.
- Wang H, Li S, Wang Q, Jin Z, Shao W, Gao Y, Li L, Lin K, Zhu L, Wang H, et al. Tumor immunological phenotype signature-based high-throughput screening for the discovery of combination immunotherapy compounds. Sci Adv. 2021;(7). doi:10.1126/sciadv.abd7851.
- Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, Camus M, Gillard M, Bruneval P, Fridman W, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138: 1429–1440. doi:10.1053/j.gastro.2009.10.057.
- Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity. 2021;54: 859–874. doi:10.1016/j.immuni.2021.01.012.
- Shi Z, Shen J, Qiu J, Zhao Q, Hua K, Wang H. CXCL10 potentiates immune checkpoint blockade therapy in homologous recombination-deficient tumors. Theranostics. 2021;11: 7175–7187. doi:10.7150/thno.59056.
- Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, Lenz HJ. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev. 2018;63: 40–47. doi:10.1016/j.ctrv.2017.11.007.
- Han X, Wang Y, Sun J, Tan T, Cai X, Lin P, Tan Y, Zheng B, Wang B, Wang J, et al. Role of CXCR3 signaling in response to anti-PD-1 therapy. EBioMedicine. 2019;48: 169–177. doi:10.1016/j.ebiom.2019.08.067.
- Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Arakelian T, Viklund J, De Milito A, Hasmim M, Andersson M, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv. 2020;6: eaax7881. doi:10.1126/sciadv.aax7881.
- Janji B, Hasmim M, Parpal S, De Milito A, Berchem G, Noman MZ. Lighting up the fire in cold tumors to improve cancer immunotherapy by blocking the activity of the autophagy-related protein PIK3C3/VPS34. Autophagy. 2020;16: 2110–2111. doi:10.1080/15548627.2020.1815439.
- Barash U, Zohar Y, Wildbaum G, Beider K, Nagler A, Karin N, Ilan N, Vlodavsky I. Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia. 2014;28: 2178–2187. doi:10.1038/leu.2014.121.
- Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer. 2020;8(2):e001486. doi:10.1136/jitc-2020-001486.
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30: 658–670. doi:10.1038/nbt.2287.
- Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18: 498–513. doi:10.1038/s41577-018-0014-6.
- Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14: 642–662. doi:10.1038/nrd4663.
- Martin NT, Bell JC. Oncolytic virus combination therapy: killing one bird with two stones. Mol Ther. 2018;26: 1414–1422. doi:10.1016/j.ymthe.2018.04.001.
- Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves Anti-PD-1 immunotherapy. Cell. 2017;170(1109–1119):e1110. doi:10.1016/j.cell.2017.08.027.
- Talimogene laherparepvec plus pembrolizumab is effective in sarcomas. Cancer Discov. 2020;10(3):340. doi:10.1158/2159-8290.CD-RW2020-018.
- Zhang Y, Zhang H, Wei M, Mou T, Shi T, Ma Y, Cai X, Li Y, Dong J, Wei J, et al. Recombinant adenovirus expressing a soluble fusion protein PD-1/CD137L subverts the suppression of CD8(+) T Cells in HCC. Mol Ther. 2019;27: 1906–1918. doi:10.1016/j.ymthe.2019.07.019.
- Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184: 963–969. doi:10.1084/jem.184.3.963.
- Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101: 746–754. doi:10.1172/JCI1422.
- Zhang H, Zhang Y, Dong J, Li B, Xu C, Wei M, Wu J, Wei J. Recombinant oncolytic adenovirus expressing a soluble PVR elicits long-term antitumor immune surveillance. Mol Ther Oncolytics. 2021;20: 12–22. doi:10.1016/j.omto.2020.11.001.
- Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, Wu X, Wu X, Shen Y, Shu Y, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10: 972–985. doi:10.4161/auto.28374.
- Camus M, Tosolini M, Mlecnik B, Pages F, Kirilovsky A, Berger A, Costes A, Bindea G, Charoentong P, Bruneval P, et al. Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res. 2009;69: 2685–2693. doi:10.1158/0008-5472.CAN-08-2654.
- Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, Rosenblum M, Harview CL, Taube JM, Handley N, et al. Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res. 2017;5: 417–424. doi:10.1158/2326-6066.CIR-16-0325.
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515: 568–571. doi:10.1038/nature13954.
- Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27: 1492–1504. doi:10.1093/annonc/mdw217.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541: 321–330. doi:10.1038/nature21349.
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313: 1960–1964. doi:10.1126/science.1129139.
- Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18: 197–218. doi:10.1038/s41573-018-0007-y.
- Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, Yuan J, Rana TM. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39: e104514. doi:10.15252/embj.2020104514
- Luo R, Firat E, Gaedicke S, Guffart E, Watanabe T, Niedermann G. Cisplatin facilitates radiation-induced abscopal effects in conjunction with PD-1 checkpoint blockade through CXCR3/CXCL10-mediated T-cell recruitment. Clin Cancer Res. 2019;25: 7243–7255. doi:10.1158/1078-0432.CCR-19-1344.
- Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, Luster AD. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of Anti-PD-1 therapy. Immunity. 2019;50(1498–1512):e1495. doi:10.1016/j.immuni.2019.04.010.
- Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13: 84. doi:10.1186/s13045-020-00922-1.
- Hoeben RC, Uil TG. Adenovirus DNA replication. Cold Spring Harb Perspect Biol. 2013;5: a013003–a013003. doi:10.1101/cshperspect.a013003.
- Zhang Y, Wu J, Zhang H, Wei J, Extracellular Vesicles-Mimetic WJ. Encapsulation improves oncolytic viro-immunotherapy in tumors with low coxsackie and adenovirus receptor. Front Bioeng Biotechnol. 2020;8: 574007. doi:10.3389/fbioe.2020.574007.
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275: 1320–1323. doi:10.1126/science.275.5304.1320.
- Howitt J, Anderson CW, Freimuth P. Adenovirus interaction with its cellular receptor CAR. Curr Top Microbiol Immunol. 2003;272: 331–364. doi:10.1007/978-3-662-05597-7_11.
- Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, Liu W, Storkus WJ, He Y, Liu Z, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer. 2019;7(6). doi:10.1186/s40425-018-0495-7
- Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7(5):a016303. doi:10.1101/cshperspect.a016303.
- Li J, O’Malley M, Urban J, Sampath P, Guo ZS, Kalinski P, Thorne SH, Bartlett DL. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. 2011;19: 650–657. doi:10.1038/mt.2010.312.
- Lapteva N, Aldrich M, Weksberg D, Rollins L, Goltsova T, Chen SY, Huang XF. Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J Immunother. 2009;32: 145–156. doi:10.1097/CJI.0b013e318193d31e.
- de Oliveira CE, Oda JM, Losi Guembarovski R, de Oliveira KB, Ariza CB, Neto JS, Banin Hirata BK, Watanabe MA. CC chemokine receptor 5: the interface of host immunity and cancer. Dis Markers. 2014;2014(126954):1–8. doi:10.1155/2014/126954.
- Budagian V, Bulanova E, Paus R, Bulfone-Paus S. IL-15/IL-15 receptor biology: a guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006;17: 259–280. doi:10.1016/j.cytogfr.2006.05.001.
- Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4: e988098. doi:10.4161/21505594.2014.988098.
- Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology. 2016;5: e1091554. doi:10.1080/2162402X.2015.1091554.