
ABSTRACT
Diffuse midline gliomas (DMG) are a highly aggressive and universally fatal subgroup of pediatric tumors responsible for the majority of childhood brain tumor deaths. Median overall survival is less than 12 months with a 90% mortality rate at 2 years from diagnosis. Research into the underlying tumor biology and numerous clinical trials have done little to change the invariably poor prognosis. Continued development of novel, efficacious therapeutic options for DMGs remains a critically important area of active investigation. Given that DMGs are not amenable to surgical resection, have only limited response to radiation, and are refractory to traditional chemotherapy, immunotherapy has emerged as a promising alternative treatment modality. This review summarizes the various immunotherapy-based treatments for DMG as well as their specific limitations. We explore the use of cell-based therapies, oncolytic virotherapy or immunovirotherapy, immune checkpoint inhibition, and immunomodulatory vaccination strategies, and highlight the recent clinical success of anti-GD2 CAR-T therapy in diffuse intrinsic pontine glioma (DIPG) patients. Finally, we address the challenges faced in translating preclinical and early phase clinical trial data into effective standardized treatment for DMG patients.
Introduction
Central nervous system (CNS) tumors are the most common solid malignancy in children and are the primary cause of pediatric cancer-associated mortality.Citation1 Pediatric high-grade gliomas (pHGGs) are aggressive neoplasms that originate from glial lineages in the developing CNS. While many types of pHGGs commonly occur in the cerebral hemispheres, including pediatric glioblastoma (GBM) and anaplastic astrocytomas, some of the most lethal subtypes arise in the thalamus, spinal cord, and/or brainstem, where they are subclassified as diffuse midline gliomas (DMG). Up to 80% of DMGs arise in the pons, where they are also referred to as diffuse intrinsic pontine glioma (DIPG).Citation2,Citation3 Pathogenetically, the 2021 World Health Organization (WHO) classification of CNS tumors distinguishes isocitrate dehydrogenase (IDH)- and histone 3 (H3)-wildtype pHGGs, which may sometimes arise in midline structures, from H3 lysine 27 (K27)-altered DMG.Citation4 Irrespective of the location and pathogenetic molecular alteration, H3K27-altered DMGs are classified as WHO grade 4 tumors and bear dismal prognoses.Citation4
In the United States, there are approximately 400 new diagnoses of DIPG per year,Citation2 with a median age at presentation of 6.8 years and a median overall survival of 11 months (interquartile range, 7.5 to 16 months).Citation3 Histologic analysis often reveals a high-grade astrocytoma, though interestingly tumor grade does not correlate with either the rate of tumor progression nor clinical prognosis. DIPGs often have a rapid local infiltration, and approximately 20% of DIPG patients develop neuraxis metastases.Citation5 Decades of research have thus far failed to produce a therapeutic intervention with any meaningful survival benefit, as significant barriers hinder the successful study and treatment of DIPG. Apart from the aggressive nature of the disease, its anatomically challenging location in the brainstem and immunological senescence further complicate therapeutic angles. DIPG arises in the pons, a midline structure that is a crucial regulator of vital functions such as respiration, blood pressure, cardiac rhythm, and sleep-wake cycles. The pons also contains critical interneuron tracks that connect upper and lower motor neurons and is the site from which numerous cranial nerves emerge; accordingly, surgical resection in this area is contraindicated as damage can result in significant morbidity, including autonomic dysregulation, diplopia, hemiparesthesia, dysarthria, gait disturbance, and hemiparesis/hemiplegia.Citation6,Citation7 Historically, biopsy was generally not performed unless the clinical picture or imaging findings were highly atypical. However, the universal failure of early phase clinical trials has underscored the importance of understanding the molecular biology of these tumors, and studies published in recent years have revealed that surgical biopsy can be performed safely and with high diagnostic yield, resulting in a trend toward tissue diagnosis for many patients with radiographic evidence of DIPG.Citation8–10
Focal radiation therapy remains the standard of care for DIPG, primarily due to the failure of surgery and available chemotherapeutic agents to provide any clinical benefit. Radiation therapy provides temporary symptomatic relief in 70–80% of patients and is thus largely considered a palliative measure.Citation11 For DIPG patients that respond to upfront radiation, there is some evidence to suggest re-irradiation after first relapse may lead to meaningful clinical improvement and radiologic response with minimal risk of acute toxicity; however, re-irradiation still does not impact overall survival and more data is needed to identify patients most likely to benefit from this strategy.Citation12–14
Another major obstacle to therapeutic development and advancement is the heterogeneous epigenetic and genetic landscapes identified in these aggressive tumors to date. The identification of lysine 27 to methionine gene mutations in histone H3.1 and H3.3 (H3.1K27M and H3.3K27M mutations), which are present in upwards of 85% of DIPG tumors, resulted in the revision of WHO CNS tumor classification guidelines and motivated studies interrogating the epigenetic landscape and global transcriptome dysregulation of DIPG tumors.Citation15 As targeting these histone mutations directly has not yet proven to be a viable strategy, current focus has shifted to other epigenetic aberrations associated with these tumors, such as hypomethylation and increased acetylation of H3K27 by EZH2 and histone deacetylases, respectively.Citation16 Significant inter- and intra-tumoral heterogeneity is further evidenced by integrated molecular profiling and genomic analyses of DIPG patient samples. For instance, several chromosomal and genetic aberrations, notably gains of chromosome 1q along with losses of 11p, 13q, and 14q, can be used to differentiate DIPG from other pediatric high-grade DMGs. Numerous studies have also revealed genetic alterations in PDGFRA, TP53, MYC, PVT-1/MYC, RB1, and PTEN.Citation15,Citation17,Citation18 Concomitant copy number alterations of TP53, PPM1D, and ACVR1 further connote a complex interplay between canonical histone aberrations and methylation derangements that may accelerate tumorigenesis.Citation17 Genetic or epigenetic-based monotherapy or combination approaches have not demonstrated curative potential in clinical trials to date, highlighting the necessity to explore alternative treatment modalities.Citation19
Finally, the etiology underlying tumor recurrence remains largely unknown, and most children die shortly after first relapse. While this phenomenon remains an active area of research, deficiencies in DNA repair mechanisms have been posited as a potential cause. Most DIPG tumors overexpress poly(ADP-ribose) polymerase (PARP1), a protein essential for repairing single-stranded DNA breaks that has been implicated in resistance to chemotherapy and is widely dysregulated in a number of solid malignancies.Citation18 Further insight into DIPG/DMG mechanisms of resistance to treatment are needed to create and refine targeted treatment approaches.
Immunotherapeutic challenges
Immunotherapy has emerged as a novel treatment modality for both solid and hematologic malignancies and has been incorporated into the standard of care for many adult and pediatric cancers.Citation20 However, the applicability and efficacy of immunomodulatory treatment methods for DMG patients have not yet been established. The advancement of next-generation sequencing technology and more robust in vitro and in vivo disease models has broadened our knowledge of the molecular and genetic heterogeneity of DMG tumors and enabled identification of antigenic regions specific to mutated tumor cells that may ultimately serve as therapeutic targets. One notable risk inherent to immunotherapy is that of sequelae from widespread immunological activation and the induction of a pro-inflammatory state that could cause significant edema and extravasation of fluid within midline structures such as the brainstem, thus worsening tumor-associated symptoms. Activating an effective immune response while minimizing potentially devastating side effects of inflammation is a central consideration in designing and implementing safe immunotherapies for DIPG/DMG.Citation21,Citation22
Another physical obstacle to treatment of DMG is the blood–brain barrier (BBB), which limits the distribution of systemically administered therapeutic agents.Citation23 While some small and lipophilic molecules delivered systemically can enter the brain by crossing the BBB, high doses are typically needed to achieve therapeutic levels in target tissues that can lead to substantial toxicity.Citation23,Citation24
In addition to anatomic considerations, DIPG has been shown to be remarkably immunologically senescent, even in comparison to other “immune-cold” tumors like adult GBM. For instance, DIPG samples contain a smaller absolute number of glioma-associated microglia/macrophages (GAMs) as compared to adult GBM tissue.Citation25,Citation26 In turn, DIPG-GAMs secrete markedly fewer chemokines/cytokines and express significantly lower levels of inflammatory markers such as IL6, IL-1α, IL-1β, CCL3, and CCL4.Citation27 The paucity of chemotactic cues and soluble mediators of inflammation in the DIPG microenvironment is mirrored by an expected decrease in the magnitude of infiltrating CD3+ T-lymphocytes. Bulk and single-cell sequencing analyses have also shown transforming growth factor beta 1 (TGFβ1), a known immunosuppressive growth factor, to be upregulated in DIPG, suggesting that TGFβ1 may prevent appropriate T-lymphocyte activation against DIPG.Citation27 However, DIPG was found to have a greater inflammatory milieu compared to hemispheric pHGGs; although the relatively “colder” immune signature of DIPG presents unique difficulties in developing effective immunomodulatory treatment methods, the ability to manipulate the immunosuppressive microenvironment of this tumor may represent an alternative and complementary avenue for treatment.
Immunotherapy approaches
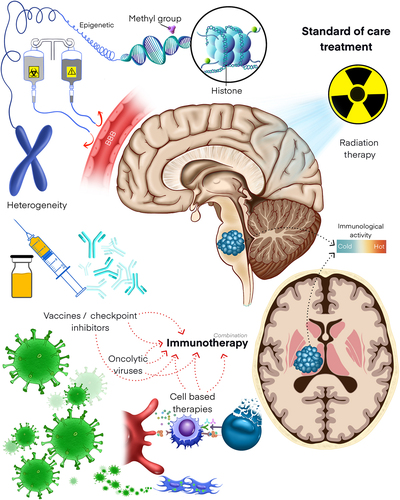
The current literature on immunotherapy in DIPG/DMG is somewhat limited; however, there is a growing body of preclinical and clinical data on the use of immunotherapy in other brain tumor types that may inform therapeutic design. The basis of inducing an immune response to a tumor involves first increasing the antigenicity of the tumor, and second manipulating the immune system to attack the cancerous tissue. This review will highlight adoptive cell transfer therapies, oncolytic viruses, immune checkpoint inhibition, tumor vaccines, and immune cell engineering approaches as promising modalities of immunotherapy for DMGs ().
Figure 1. Immunotherapeutic and combination therapy modalities in DMG. DMGs are highly aggressive and often fatal tumors of the pediatric central nervous system. As the anatomical location of these tumors precludes total surgical resection, chemo- and radiotherapies comprise the current mainstays of treatment. Unfortunately, these treatment modalities have not significantly improved the dismal prognoses of DMGs, underscoring the urgency of identifying efficacious alternatives. Obstacles to therapeutic development include addressing intra- and intertumoral heterogeneity, overcoming blood-brain barrier penetrance, and modulating the relatively “cold” immune environment of DMGs. Despite these obstacles, emerging evidence demonstrates strong potential for immune checkpoint blockade, adoptive cell transfer, oncolytic viral therapies, and tumor vaccines as novel therapies for DMG. Pre-clinical studies and clinical trials are also interrogating the synergistic effects of these immunotherapies with chemotherapy and radiotherapy.
Adoptive cell transfer
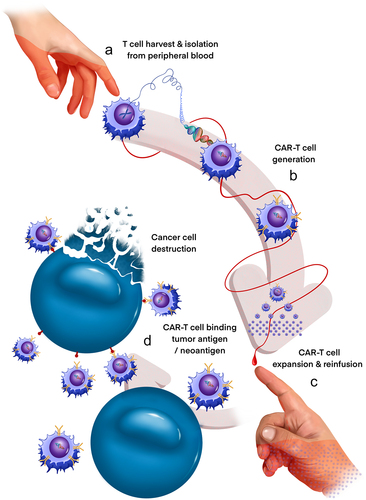
Adoptive cell transfer (ACT) is a form of immunotherapy in which immune cells are isolated from a patient, modified, expanded ex vivo, and then transferred back into the patient ().Citation27 T-cells genetically modified to express chimeric antigen receptor (CAR-T) have shown remarkable rates of clinical response and remission in the setting of various hematologic malignancies and have since become the first FDA-approved ACT for the treatment of acute lymphoblastic leukemia and diffuse large B-cell lymphoma.Citation28 CAR-T cells are engineered to specifically target tumor-associated antigens and are composed of an extracellular Fc domain, a transmembrane domain, and an intracellular domain that allow for potent cytotoxicity completely independent of major histocompatibility complex (MHC) activation.Citation29 There are now several generations of signaling domains that have been utilized, with recent advances leading to significantly enhanced and sustained T-cell response.Citation30
Figure 2. Generation of CAR-T cells for anti-DMG therapy. CAR-T cells present a powerful new approach to precision immunotherapy in DMG. CAR-T cells are generated from a DMG patient’s own T cells (a) and directed against tumor-specific antigens/neoantigens by genetic introduction of a chimeric antigen receptor (CAR) gene (b). Clonally expanded CAR T cells (c) are then reinfused into the originating patient and are activated to promote enhanced tumor cell-specific destruction (d). Treatment of 4 DMG patients with GD2-directed CAR-T cells recently demonstrated both radiologic and clinical benefit, evidencing the transformative potential of this novel therapy.
To date, four clinical trials have been conducted using CAR-T therapy in adult GBM with preliminary results showing no dose-limiting toxicities and evidence of an antitumor response in a small number of patients.Citation31 The antigens targeted – epidermal growth factor receptor variant III (EGFRvIII), human epidermal growth factor receptor 2 (HER2), and interleukin 13 receptor subunit alpha 2 (IL-13Rα2) – are also expressed in a subset of pediatric HGGs.Citation30,Citation32–34 While previous trials in DIPG based on data extrapolated from adult GBM trials have uniformly failed, it is possible that this targeted approach may extend to DIPG given the overlap in antigen profile. These targets are being explored in the ongoing BrainChild-01 (HER2; NCT03500991) and BrainChild-02 (EGFR; NCT03638167) pediatric clinical trials. Of note, CAR-T cell infusion has been trialed both intracraniallyCitation35 and intravenously (IV) in other disordersCitation36,Citation37 with imaging and pathology data demonstrating appropriate infiltration of inflammatory cells and the presence of CAR-T cells via both routes of administration.Citation38 The ability to deliver CAR-T cells via IV administration for the treatment of brainstem tumors would bypass the risk and difficulty associated with direct intra-tumoral CAR-T injection, thereby transforming this therapeutic approach.
Another significant recent advancement in CAR-T therapy for DIPG was the development and assessment of anti-disialoganglioside 2 (GD2) CAR T-cells to target DIPG and other DMGs.Citation39 Gangliosides are glycosphingolipids that are commonly found on the surface of cells in the mammalian nervous system.Citation40 While the majority of gangliosides are poor therapeutic targets given their near ubiquitous expression across a variety of normal tissue types, GD2 is unique given its differential overexpression in many solid tumor types compared to surrounding healthy tissue, including gliomas, neuroblastoma, and osteosarcoma.Citation41 The anti-GD2 antibody dinutuximab was approved by the U.S. Food and Drug Administration (FDA) for the treatment of high-risk pediatric neuroblastoma in 2015, motivating further translational and clinical investigation into anti-GD2 therapies.Citation42 High expression of GD2 was identified in four independent patient-derived DIPG cultures bearing the H3K27M mutation, providing evidence for targeting GD2 in these high-grade tumors. Given that anti-GD2 antibodies had limited penetrance of the BBB, GD2-CART cells, which readily cross the BBB, proved an attractive alternative.Citation41
Initial results from the first dose level of a pediatric phase I clinical trial employing GD2-CAR T cell therapy for DIPG and other H3K27M-mutated DMGs have demonstrated a tolerable safety profile and clear signs of T cell expansion and activity including clinical responses.Citation22,Citation43,Citation44 In the largest clinical series of CAR-T in DIPG to date, Majzner et al. described the treatment of 4 pediatric or young adult patients with H3K27M-mutated DIPG or DMG with GD2-directed CAR-T cells.Citation22 Three of the 4 reported patients experienced clinical and radiographic improvement following the initial IV infusion of GD2-CAR T cells; 1 patient experienced a 90% reduction in her spinal DMG, while another demonstrated improved motor function and decreased midbrain tumor signal by MRI.Citation22 All 3 of these patients received at least one intracranial infusion of GD2-CAR T cells in the months following their initial IV therapy due to stagnation or worsening of their clinical or radiographic disease, with one patient receiving five total GD2-CAR T cell treatments before passing away.Citation22 In the sole patient who did not respond to IV GD2-CAR T cell therapy, quantitative polymerase chain reaction analysis of autopsy brain tissue revealed evidence of GD2-CAR T cell infiltration specifically into her tumor with sparing of normal cortex,Citation22
In April of 2022, Majzner et al. presented preliminary findings for the 3 + 3 phase I dose escalation trial of GD2-CAR T in H3K27M DMGs, which included 11 treated patients out of 13 total enrollees.Citation45 Nine of the 10 follow-up patients experienced clinical and radiographic improvement after initial IV infusion, similar to the previously reported patients; all 9 also received at least one subsequent intracranial infusion.Citation45 Two patients experienced dramatic reductions in tumor volume (over 95% and 98% respectively), and 4 patients were still receiving infusions at the time of data release.Citation45 These promising phase I studies therefore not only portend an important role for CAR-T cells in the future of DIPG/DMG management, but also highlight the necessity for careful clinical planning and monitoring of these patients during therapy.
Clinical benefit notwithstanding, the side effect profile of CAR-T therapy presents a clinical challenge.Citation46 Cytokine release syndrome (CRS), cytokine release encephalopathy syndrome (CRES), and tumor inflammation-associated neurotoxicity (TIAN) are CAR-T-associated complications that can range from mild inflammatory changes and confusion to fluid overload, respiratory failure, seizures/obtundation, and death.Citation22,Citation46 A significant cause of morbidity in CAR-T-treated patients with hematologic malignancies, these toxicities may become more pronounced in anatomically constrained solid neurologic tumors. The pathogenesis of both CRS, CRES, and TIAN stems from the induction of IL-1 and IL-6 mediated inflammation with sharp upregulation of T-cell activity after engaging with tumor antigens. Tocilizumab and siltuximab, both anti-IL-6 monoclonal antibodies, have been used in conjunction with corticosteroids to treat CRS, CRES, and TIAN, allowing for toxicity abrogation as necessary.Citation46 Anakinra, an IL-1 antagonist, can similarly be used to mediate the neurotoxic inflammatory effects of tumor CAR-T therapy.Citation22 Studies of adult GBM patients infused with CAR-T cells demonstrated tolerable side effect profiles, with none of the patients requiring tocilizumab infusions.Citation47 In the phase I study of GD2-CAR T in DMG, however, all 4 patients required aggressive management of TIAN using tocilizumab and either anakinra or corticosteroids after their initial IV infusion. Two of 3 patients required similar treatment following their subsequent intracranial infusion(s); the spinal DMG patient required a cocktail of anakinra, siltuximab, corticosteroids, and dasatinib to suppress CAR-T activity.Citation22 In general, patients experienced worse cytokine release syndrome after IV infusion of GD2-CAR T cells compared to subsequent intracranial infusions.Citation22 Single-cell RNA-sequencing analysis of CSF samples isolated from patients revealed differential enrichment of pro-inflammatory myeloid cells during peak post-intracranial infusion inflammatory periods compared to peak post-IV inflammatory periods.Citation22 Of the patients from the recently reported 3 + 3 trial (NCT04196413), all experienced TIAN symptoms that were managed with anakinra with additional corticosteroids or CSF drainage as required.Citation45 Together, the data from these monumental clinical investigations demonstrate the power of an immune-based therapeutic approach to DMGs and emphasize opportunities for future investigation in this arena.
As more preclinical and clinical data become available and our understanding of the genetic makeup of DIPG/DMG expands, the use of CAR-T cells to target neo-antigens may prove to be a viable, precision immunotherapeutic option for patients. Further studies assessing the tolerability of neuroinflammatory related side effects in children are needed, and further delineation of neo-antigens along with maturation of administration techniques will be critical.
NK/CAR-NK therapy
While CAR-T therapy remains the predominant ACT approach in DMG and other tumors, recent studies are also investigating novel CAR-natural killer (CAR-NK) cell strategies. In general, CAR-NK approaches have several advantages over CAR-T cell-based therapies. For instance, current clinical protocols often are limited to autologous CAR-T cells to prevent graft-versus-host disease, whereas patients can safely receive allogeneic NK/CAR-NK cell therapies.Citation48,Citation49 Additionally, CAR-NK cells can be derived from a variety of sources, including autologous or non-HLA-matched peripheral blood mononuclear cells, cord blood, induced pluripotent stem cells, and others; most CAR-T cells are generated from patient leukapheresis, although novel allogeneic forms of CAR-T cells are under investigation.Citation49 Finally, preliminary evidence suggests that CAR-NK therapy may have higher anti-tumoral efficacy with lower neurotoxicity and cytokine release syndrome-related sequelae compared to CAR-T cells; the innate immune functions of CAR-NK cells augment the CAR-mediated cytotoxicity of these cells compared to their CAR-T counterparts.Citation49,Citation50
NK cells have demonstrated cytotoxic potential against DIPG cells in vitro, mediated by binding between the NKG2D-activating receptor on NK cells and stress-response ligands upregulated on the DIPG cell surface.Citation51 Investigation of immune cell infiltration and survival in DIPG suggests that increased NK cell infiltration is correlated with better prognosis, supporting the translational study of CAR-NK in this disease.Citation52 Combining NK-based therapy with existing cancer therapies may also augment the resulting anti-tumoral immune response. For instance, current agents undergoing clinical trial for DIPG may further enhance NK-mediated killing. The narrow-spectrum histone deacetylase inhibitor entinostat increases NKG2D expression on NK cells and thus cytotoxic capacity.Citation53 Lysine-specific demethylase-1 (LSD1) inhibitors increased expression of NK cell-activating ligands on DIPG cells in vitro, corresponding to increased NK cell-mediated tumor cell lysis following LSD1 inhibitor therapy.Citation52
Several obstacles exist for the development and clinical implementation of NK cell-based therapies. Critically, CAR-NK cells persist for shorter times in vivo compared to CAR-T cells, which may necessitate larger and/or more frequent infusions to achieve similar tumor infiltration levels.Citation49 Additionally, viral transduction of NK cells is more challenging and less successful than in T cells, requiring investigators to pursue alternative methods of vector integration.Citation50,Citation54 Finally, NK cells proliferate at a much lower rate than T cells, making it more difficult to generate an expanded pool of CAR-NK cells for clinical application.Citation54 While it will likely require years of additional investigation and refinement before NK cell-based immunotherapies are ready for clinical trials in the treatment of DMG, the above studies demonstrate the potential of manipulating innate immunity as a therapeutic approach.
Oncolytic viruses
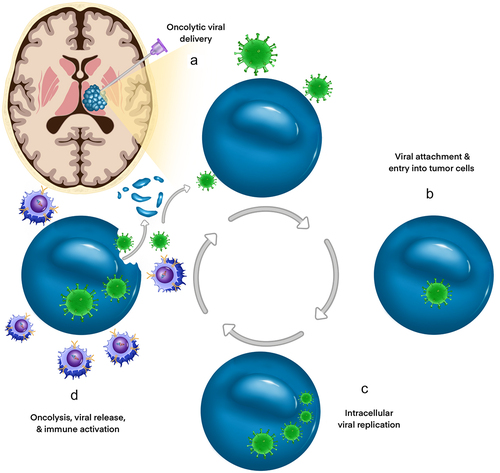
Oncolytic viruses (OVs) are an emerging class of immune-oncologic agents that are used to promote a robust antitumor immune response through selective tumor lysis and induction of anti-tumor immunity.Citation55 OVs have become a promising and evolving modality of therapy in many types of CNS and extracranial solid tumors. A select number of native OVs (Seneca Valley virus, Newcastle virus, and reovirus) demonstrate potent antitumor efficacy while others (oncolytic herpes virus (oHSV), oncolytic adenoviruses, and oncolytic measles virus) have been modified to improve specificity, immunogenicity, and safety.Citation56 The selectivity of OVs for cancer cells is thought to arise from intrinsic abnormalities of cell signaling and antiviral machinery that confer a selective advantage for viral replication.Citation57 Subsequent OV-mediated cytolysis is dependent upon 1) viral entry into cancer cells, 2) viral replication within the tumor, and 3) the secondary immune response to the virus, a coordinated series of events that underlies both the safety and efficacy of OVs ().Citation56,Citation58
Figure 3. Mechanisms of DMG targeting by immunovirotherapy. Oncolytic viruses (OV) are promising novel therapies for DMG as they can be delivered directly via intratumoral injection (a), bypassing the BBB. OV entry (b) and replication (c) within DMG cells induces direct oncolysis and release of new viral particles into the tumor bed, facilitating further inoculation and lysis of surrounding tumor cells (d). Tumor cell debris increases the exposure of the patient’s immune system to both existing and novel tumor antigens, bolstering immune-mediated anti-tumoral effects.
Oncolytic herpes simplex virus (oHSV)
Virus uptake into tumor cells is the first step in being able to target aberrant tissue, and the exploitation of specific surface uptake proteins can enhance viral entry and subsequent oncolysis. For example, nectin-1 (CD111) is an important receptor molecule in promoting oHSV entry and can be used to predict sensitivity to herpes oncolytic therapy in medulloblastoma and HGG pediatric xenograft models.Citation59 Expression of CD111 was significantly higher in pediatric brain tumor xenografts relative to adult GBM, suggesting that pediatric brain tumors may be ideally suited for oHSV virotherapy.Citation60 Critically, a clinical trial utilizing the oHSV G207 in recurrent pediatric cerebellar brain tumors is currently recruiting patients.Citation61 A second phase I study focusing on G207 as a treatment for pHGG (NCT02457845) recently reported safety of the approach and impressive efficacy results with radiographic, neuropathological, and/or clinical responses demonstrated in 11 out of 12 patients.Citation62 The median overall survival was 12.2 months in those patients who received G207 compared to 5.6 months in historical controls. Such work demonstrates the potential of OVs as treatments for DMG and should comprise a central area of future research. Notably, no pediatric or adult patient treated with G207 in multiple phase I trials suffered from virus-associated neurotoxicities such as encephalitis.Citation63–65 Upcoming trials will further elucidate the safety profile for oHSV therapy in DIPG/DMG.
Oncolytic adenovirus
Oncolytic adenoviruses have also been genetically engineered to selectively infect tumor cells by targeting aberrancies in the retinoblastoma tumor suppressor (Rb) signaling pathway that are present in most gliomas.Citation66,Citation67 Wild-type (WT) adenovirus produces early region 1A (E1A) proteins upon cell entry, which bind to Rb and release E2F-family transcription factors from preexisting Rb-E2F cellular complexes. Collectively, this promotes cell cycle progression and transcriptional activation. By introducing a deletion in the Rb binding domain of E1A, Fueyo and colleagues created a tumor-selective adenovirus that showed impressive cytolytic activity in vitro and in vivo.Citation67 Critically, Rb mutations appear in 59% of pHGG cell lines, suggesting this strategy may also be efficacious in treating patients with DMGs.Citation68 Additional alterations of oncolytic adenoviruses may enhance glioma tropism, as evidenced by DNX-2401, a modified oncolytic adenovirus containing an integrin binding RGD-4C motif. A phase I clinical trial completed in adults using DNX-2401 in recurrent malignant glioma (NCT00805376) demonstrated a 3-year survival of 20%.Citation69 Importantly, pathologic analysis showed evidence of DNX-2401 viral replication and lymphocytic infiltration within tumor cells.Citation70 Additional preclinical data demonstrating the oncolytic effect and robust immune response induced by DNX-2401Citation71 motivated additional phase I/II clinical trials (NCT03178032). Of note, the results of using DNX-2401 in combination with radiotherapy for newly diagnosed DIPG have recently been reported with exciting responses to the virus having been noted.Citation72
“Armed” oncolytic viruses
Second-generation oncolytic viruses modified to express immune adjuvants such as cytokines and/or immune checkpoint proteins have demonstrated enhanced antitumor activity and augmented antitumor immune memory in a myriad of models;Citation56,Citation73 as an example such an approach is being evaluated in brain tumors using engineered forms of oHSV with M002 (expressing murine IL-12) and M032 (expressing human IL-12) showing high tumor affinity and robust CD4+, CD8+, and NK cell infiltration.Citation74 Similar findings were reported in primate studies, supporting the safety of intracranial engineered oHSV inoculation for use in phase I trials in adults with GBM.Citation75 Though additional safety and long-term outcomes data are needed, preclinical and early trial results for oncolytic viral therapy with armed viruses in HGG are encouraging and may transform the treatment and outlook of DMGs.
Mesenchymal stem cells (MSC)-carrying oncolytic viruses
The efficacy of OV-mediated immunotherapy for brain cancer is influenced by targeted delivery. The homing capacity of mesenchymal stem cells (MSCs) to tumors makes them excellent carriers of anticancer therapeutics. MSCs can cross the BBB and reach brain tumor tissue following systemic administration.Citation76 A recent preclinical study showed that OV-carrying MSCs delay elimination of the virus by the host immune system,Citation77 and decreases neuroinflammatory response, thereby providing neuroprotection to normal peritumoral brain.Citation78 Endovascular, intra-arterial delivery techniques with perfusion guidance have also shown promising results in other HGGs that may be translatable to DMG.Citation79 The results from the preclinical studies in GBM models led to a phase I clinical trial investigating the safety of MSC carrying OV for treating GBM patients (NCT03072134). MSCs also successfully deliver OV to DIPG patient-derived xenograft (PDX) models and increase survival using an intranasal delivery approach.Citation80 Carceller et al. reported a case of intra-arterial administration of autologous MSCs infected with an oncolytic adenovirus, ICOVIR-5, for the treatment of DIPG in a 9-year-old girl.Citation81
These experimental approaches may ultimately inspire a less invasive therapeutic modality that is capable of facilitating CNS delivery via an IV route (see for a summary of major clinical trials using adoptive cell transfer and oncolytic viruses for DIPG/DMG patients).
Table 1. Major current clinical trials of treatment using adoptive cell transfer and oncolytic virus for DMG patients.
Vaccines
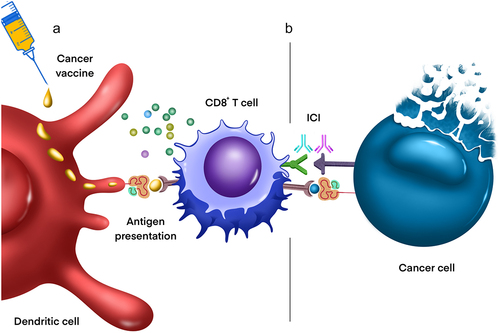
Tumor vaccines are a form of immunotherapy that provokes a T-cell response to tumor-specific antigens. Vaccines are often conjugated to immunostimulatory biological adjuvants, which enhance the potency of the epitope-specific adaptive immune response (). Current tumor vaccines being evaluated for efficacy in the treatment of DIPG/DMGCitation82 include a H3K27M peptide vaccine and imiquimod (INTERCEPT-H3; NCT04808245), H3.3-K27M neoantigen vaccine (ENACTING; NCT04749641), TTRNA-DC vaccine with GM-CSF (TTRNA-xALT; BRAVO; NCT03396575) combined with chemotherapy, K27M peptide with nivolumab (NCT02960230), rHSC-DIPGVax (NCT04943848), adjuvant dendritic cell vaccine (ADDICT-pedGLIO; NCT04911621), and PEP-CMV (NCT05096481) (see ). As our understanding of the genomic landscape of these tumors has expanded, our ability to identify tumor-specific neoantigens to serve as vaccine targets has increased in tandem. Our current understanding of these therapies is based on decades of studies that have investigated the activity of vaccines against a myriad of tumors including breast, lung, melanoma, pancreatic, colorectal, and renal cancers with varying degrees of success.Citation83 Tumor vaccine design must consider and address inter- and intratumoral heterogeneity, which may drive escape variant selection and preclude therapeutic efficacy if a single antigen is utilized.
Table 2. List of current clinical trials utilizing immunotherapy for the treatment of DMG.
Figure 4. Combination vaccination therapy and/or immune checkpoint blockade in DMG. a) Cancer vaccination exposes dendritic cells to tumor-specific antigens which are ultimately presented to T cells in secondary lymphoid organs, activating cytotoxic CD8+ T cells and thereby promoting their migration into the tumor microenvironment. b) Immune checkpoint proteins (e.g., those expressed on DMG cells) bind to receptors on infiltrating lymphocytes, promoting T cell anergy and resistance to immunotherapy. Combining cancer vaccination with immune checkpoint blockade (ICB) inhibits such immunosuppressive interactions, facilitating anti-tumoral inflammation and therefore cancer cell destruction. Current ICB targets in DMG include the PD-1/PD-L1, CD47/SIRPa, and IDO axes.
In addition to appropriate antigen selection, the subsequent immune response to the vaccine is critical in determining its effectiveness. It was initially assumed that antitumor activity would largely be conferred by tumor cytolysis, making an MHC class-I predominant vaccine the optimal choice. However, findings from murine models have shown that significant fractions of non-synonymous tumor mutations are immunogenic and recognized by CD4+ T cells, which help orchestrate and potentiate a systemic antitumor response.Citation84 This work demonstrated that MHC class II-restricted epitopes resulted in a more equal distribution of CD4+ and CD8+ T-cell responses in accordance with marked inhibition of tumor growth in vivo. An approach using tumor exome sequencing to build a poly-neoepitope vaccine may therefore be highly beneficial in DMG.
Criteria that govern antigen selection include 1) differential expression within the tumor cell population, 2) necessity for cellular survival, and 3) immunogenicity. DMG contains several exciting antigen prospects, most notably the H3.3K27M mutation. The exact mechanism this unique mutation plays in cell division has not yet been completely elucidated, but extensive work has detailed the multifaceted role of histones in architecting the epigenetic landscape of oncogenesis.Citation85 Initial attempts to target H3.3K27M support its candidacy as a tumor antigen of interest; for instance, experimental implementation of a peptide vaccine directed against H3.3K27M produced an effective, mutation-specific CD4+ and CD8+ mediated immune response with antigen presentation on both MHC classes I and II.Citation86 They observed tumor regression in vivo using murine DIPG models, though these were not orthotopic. This vaccine epitope is currently being tested in a phase I clinical trial in combination with checkpoint inhibitors (NCT02960230).
In conjunction with antigen-vaccine specificity, the vector construct of the vaccine also plays a critical role in the potency of the immune response. There are multiple classes of vectors used in vaccine construction, including peptides, viral vectors (used in cancers typically associated with viral infections), nucleic acids (both RNA and DNA), and cellular vaccines.Citation87 Single-peptide antigen vaccines have been insufficient in producing robust, clinically beneficial immune responses,Citation88 potentially secondary to the use of short peptide chains (<15 amino acids) that bind effectively to MHC class I molecules but do not require processing by antigen-presenting cells (APCs). Consequently, there is no co-stimulation of immune effector cells required to prevent cytotoxic T cell dysregulation, promoting the development of antigen tolerability. The addition of toll-like receptor (TLR) agonists and synthetic long peptides (SLPs) containing both MHC class I and II epitopes to peptide vaccines elicit a far more potent immune response with a balanced induction of CD8+ and CD4+ T cells.Citation87
Antigen presentation by professional APCs also plays a pivotal role in tumor-specific vaccination; numerous ongoing trials are evaluating the efficacy of APCs transfected with tumor antigens, such as whole tumor cells, peptide extract, or tumor-derived RNA, in inducing tumoral immunity (e.g., NCT04749641). In particular, dendritic cell (DC)-based therapies may prove a promising treatment modality for pediatric brain tumors, including DIPG/DMG. Investigators looked at DC activity against multiple tumor antigens including peptides,Citation89 tumor homogenate,Citation89 and ribonucleic acid (RNA).Citation90 Application of DCs pulsed with EGFRvIII glioma homogenate to a syngeneic TGF-β-secreting murine glioma model demonstrated both safety and tolerability, as well as tumor regression and upregulation of CD4+ and CD8+ cells.Citation91 As techniques to load cells with antigen and subsequently deliver them to patients have improved, the limiting factor in the progression of APC vaccination development is largely attributable to a lack of understanding of the DIPG/DMG neoantigen profile. The discovery of mutant H3.3K27M has inspired multiple studies investigating its use in APC vaccines that will inform the utility of this strategy.Citation15,Citation68,Citation92,Citation93
In summary, vaccines in cancer immunotherapy are an innovative and attractive strategy given their safety profile and proven efficacy in other disease contexts. However, clinical benefit has remained elusive as improvements in the abilities of vaccines to induce a dual CD4+ and CD8+ mediated immune response, increased immunogenicity with addition of co-stimulatory adjuncts, and the use of multiple epitopes have produced only modest gains in antitumor efficacy. While vaccination as monotherapy may not be sufficient to treat DMG, its use as an adjuvant in combination with other forms of immunotherapy may prove efficacious.
Immune checkpoint blockade
One of the earliest forms of cancer immunotherapy, immune checkpoint inhibitors (ICIs) enhance anti-tumoral adaptive immunity by restoring cytotoxic T cell activity (). The most common ICIs currently in clinical use employ antibodies against cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), programmed cell death protein-1 (PD-1), or programmed cell death ligand protein-1 (PD-L1).Citation94 These proteins and others are involved in regulation of T cell anergy and are co-opted by malignant cells to escape T cell-mediated destruction.Citation94
The success of ICIs in other solid tumors, most notably metastatic melanoma, has generated significant interest in their application to pediatric brain tumors such as DMGs. Unfortunately, preliminary use of ICIs in DIPG has yet to prove successful; for instance, one cohort study found that anti-PD-1 treatment worsened symptomology and outcomes in patients with progressive DIPG.Citation43 Another institutional study found no significant difference in outcomes between progressive DIPG patients treated with combination PD-1 blockade and re-radiation therapy vs. re-radiation therapy alone.Citation95 This lack of response may be explained by the lack of PD-L1 expression on DIPG cells,Citation96 suggesting that immunosuppression in these tumors may not act via the PD-1/PD-L1 axis. Given the rapid development of checkpoint inhibitors and the small patient pool from which to draw clinical trial participants, further study will be required to assess the utility of classic ICIs as monotherapies in DIPG/DMG. To this end, several ongoing clinical trials are exploring the safety profiles and survival benefits of anti-PD-1, anti-PD-L1, and anti-CTLA-4 therapies either as single agents or in combination, including nivolumab, durvalumab, avelumab, pembrolizumab, and ipilimumab.Citation97
Outside of the PD-1/PD-L1 axis, other immune checkpoint pathways have emerged as potential immunotherapeutic targets in DMG. For instance, recent attention has turned to inhibiting immunosuppressive tumor-myeloid interactions, such as the CD47-signal retention protein alpha (SIRPa) signaling pathway. CD47, also known as integrin associated protein, is a known anti-phagocytic ligand that is overexpressed on a wide variety of solid and hematological tumors.Citation98 Binding between tumor cell-bound CD47 and macrophage or dendritic cell-bound SIRPa decreases the phagocytic capacity of tumor-infiltrating myeloid cells and thus promotes immune evasion by the tumor.Citation98,Citation99 CD47 overexpression has been correlated with increased tumor progression and worse patient prognoses across tumor types, making it an attractive target for recent therapeutic design.Citation100 Thus far, humanized anti-CD47 and anti-SIRPa antibodies comprise the predominant strategy of blocking the interactions of these two proteins and inducing macrophage reactivation within the tumor environment. Translational evidence has demonstrated both safety and efficacy in inhibiting CD47/SIRPa in the microenvironments of pediatric GBM and DIPG.Citation100 CD47 was found to be highly expressed in cell lines, gene expression datasets, and primary frozen tumors derived from pediatric GBM and DIPG patients.Citation100 Administration of a humanized anti-CD47 antibody, Hu5F9-G4, in mouse models of these two malignant tumor types significantly increased phagocytosis and overall survival as compared to control-treated tumors.Citation100 These promising results provided some of the preclinical rationale for a clinical trial employing the anti-CD47 antibody magrolimab to disrupt the CD47-SIRPalpha axis in recurrent or progressive malignant brain tumors in children and adults. However, this trial excludes DIPG/DMG patients from participating, meaning that additional studies will be necessary to understand the role of anti-CD47 therapy in these aggressive tumors (NCT05169944).
Another emerging immunotherapy target is indoleamine 2,3-dioxygenase (IDO), a catalyst of tryptophan catabolism that has been implicated in both normal immune tolerance mechanisms and tumor immune evasion.Citation101 IDO degrades intratumoral tryptophan into secreted kynurenine; the increased kynurenine:tryptophan ratio in the immune microenvironment contributes to regulatory T cell induction, T cell proliferation inhibition, and establishment of an overall anti-inflammatory milieu.Citation102,Citation103 IDO overexpression has been identified across solid tumor types, including melanoma, pancreatic cancer, squamous cell carcinoma, and GBM, and is associated with poor patient prognosis.Citation104 Two small molecule inhibitors have been developed to target the IDO pathway: epacadostat competitively blocks IDO without interfering with other tryptophan catabolism enzymes, while indoximod opposes the downstream effects of IDO signaling by reactivating mTOR and inhibiting regulatory T cell differentiation.Citation105,Citation106 Indoximod has been trialed as part of a combination therapy with radiation and chemotherapy in pediatric patients with newly diagnosed DIPG, the preliminary results of which were published in 2021.Citation107 The 13 patients in this phase IB trial experienced longer median overall survival compared to historical patient data, which correlated with increased circulating monocytes following administration of idoximod.Citation107 These data have motivated an ongoing phase 2 study, which will further elucidate the capacity for idoximod to augment current standards of care for newly diagnosed DIPG patients (NCT04049669).
The immune checkpoint pathways discussed above represent only a small portion of those currently being targeted by experimental or approved ICIs. However, limited or no data exists describing the role of other immunosuppressive pathways including Tim3/Gal9, CD155/TIGIT, or LAG3 in DMG. Despite the accelerated pace of immunotherapy development, assessing the utility of current and new ICIs will require similar advancements in the understanding of the composition of and interactions between the immune microenvironment and DMGs.
Combination therapy approaches
Across adult and pediatric oncology, interest is accelerating in the implementation of immunotherapies in combination with other standards of care, such as chemotherapy and radiation, and/or with other immunomodulatory agents. Motivation for this approach stems from mounting data that suggests high levels of resistance in patients treated with singular immunotherapy and a limited duration of response to immunotherapy in general. Moreover, combining different treatment modalities may augment the efficacy of individual immunotherapies through auxiliary molecular and immune mechanisms. Ongoing clinical trials in patients with newly diagnosed and recurrent or progressive DMG employ several combinatorial approaches to understand the optimal usage of immunotherapy in the treatment of these aggressive tumors.
One strategy currently being assessed in DMG patients is the combination of immunotherapy with radiotherapy. This combination has been applied to tumors across the anatomic spectrum, as radiation is an integral component of many cancer treatment algorithms.Citation108–110 In addition to inducing apoptosis of rapidly dividing cells, radiation also influences the immune microenvironment by several tumor-intrinsic and -extrinsic mechanisms. First, radiotherapy increases MHC I expression on tumor cells, which can prime cytotoxic CD8+ T cells for an antitumoral response.Citation111 Additionally, radiation may stimulate pro-inflammatory, type I interferon responses in myeloid and cytotoxic T cells via activation of the stimulator of interferon genes (STING/cGAS) pathway.Citation112,Citation113 Not only does activating this pathway promote tumor infiltration by immune cells, but it also enhances the anti-tumoral efficacy of these cells.Citation114 Radiation may also promote downregulation of CD47 on the surface of tumor cells, augmenting the effect of anti-CD47 or anti-SIRPa antibody therapies.Citation115 However, one of the strongest rationales for combining radio- and immunotherapies is the immunosuppressive sequelae that radiation produces in the tumor environment that are optimally targeted by ICIs. For example, post-radiation STING activation may also upregulate the activity of IDO, motivating the combination approach of indoximod and chemotherapy/radiotherapy in the aforementioned active clinical trial.Citation113 Radiated tumor cells may also upregulate expression of immune checkpoint targets such as PD-L1, which can be targeted by anti-PD-L1 antibodies.Citation116 Finally, radiotherapy has been posited to sensitize tumor cells to CAR T-mediated apoptosis; the combination of these two therapies has primarily been interrogated in the setting of hematological malignancies, which may motivate future studies in DMG.Citation117
Another promising future combinatorial approach for ICIs in brain tumors may include the use of OVs. As discussed above, OV therapy promotes not only direct tumor lysis, but also recruitment and proliferation of activated T lymphocytes. Recent studies in non-CNS and CNS tumors suggest that OV treatment may upregulate expression of immune checkpoint proteins on tumor cells,Citation118,Citation119 promoting resistance to virotherapy. Ongoing clinical trials are exploring combined OV/ICI therapy in a wide range of cancer types including recurrent gliomas/GBM;Citation120 early results from these trials already demonstrate the promise of this combinatorial approach.Citation121,Citation122
A third combinatorial strategy utilizes ICIs with other immunomodulatory agents to maximize the antitumor immune response. For example, combining a humanized anti-CD47 antibody with an agonistic anti-CD40 antibody increased macrophage recruitment and decreased tumor burden in mice bearing DIPG patient-derived xenografts.Citation123 CD40 agonism promotes the activation of cytotoxic T cells, macrophages, and other myeloid cells; combining anti-CD47 therapy with CD40 activation may therefore increase the phagocytic, anti-tumoral activity of the DIPG immune microenvironment.Citation124 Another ongoing clinical trial combines nivolumab, an anti-PD-1 therapy, with lirilumab, which targets the killer-cell immunoglobulin like receptor KIR2DL1/KIR2L3 (NCT02813135). KIR2DL1/KIR2L3 are expressed on the surface of NK cells and recognize MHC class I molecules on tumor cells, suppressing the NK cell antitumoral response. In theory, the application of these two separate therapies would alleviate both T cell and NK inhibition in the tumor microenvironment, enhancing innate and adaptive cytotoxicity and subsequent tumor destruction. The combination of nivolumab and lirilumab has also been studied in bladder cancer and squamous cell carcinoma of the head and neck, demonstrating excellent patient tolerance but mixed clinical results.Citation125–127 Finally, another trial is currently determining the safety and toxicity profiles of combining nivolumab and bempegaldesleukin in malignant pediatric brain tumors, including DIPG (NCT04730349). Bempegaldesleukin is a polyethylene glycol-bound IL-2 agonist that promotes cytotoxic CD8+ T cell activation over regulatory T cell activation.Citation128 Combining nivolumab and bempegaldesleukin may not only alleviate immunosuppression of cytotoxic T cells, but also prime them for antitumoral activity, producing a synergistic effect compared to ICI monotherapy. Thus far, the nivolumab/bempegaldesleukin combination has been trialed in advanced solid tumors including melanoma, renal cell carcinoma, and non-small cell lung cancer.Citation129 Although the phase III trial of nivolumab/bempegaldesleukin in metastatic melanoma failed to meet statistical significance of its primary endpoint, the ongoing trial in pediatric high-grade brain tumors will determine the efficacy of this combinatorial approach in DIPG/DMG.Citation130
Conclusion & future directions
The ever-increasing understanding of the genetic and molecular underpinnings of DMG has informed recent advances in immunotherapy trials. Adoptive cell transfer, OVs, vaccines, and ICB are the major immunotherapy approaches whose pre-clinical and clinical trials demonstrate promise in treating DIPG/DMG.
A fundamental question that remains incompletely addressed in the field of pediatric neuro-oncology is how the cellular and molecular biology of DMG, and therefore the potential responsiveness to immunotherapy, changes between initial diagnosis and tumor recurrence and/or metastasis. Retrospective clinical analyses have demonstrated some efficacy in re-radiating recurrent DIPG, an approach also under investigation in an ongoing clinical trial (NCT03126266).Citation131 However, no direct evidence yet exists to inform differential algorithms for immunotherapy application in newly diagnosed versus recurrent disease across DMG patients due to limited availability of representative animal models and access to primary tumors samples from recurrent DMG. As the use of immunotherapy continues to advance in DMG, charting the biological evolution of these tumors in response to therapeutic pressure should therefore be a central focus of future basic and translational studies.
Despite the persistent poor prognosis associated with DMG, the compelling results of recent translational and clinical trials suggest the promise of immunotherapy as a powerful new avenue for treating these aggressive tumors. Numerous ongoing clinical trials in DMG and other malignant pediatric brain tumors will elucidate not only the efficacy and safety of immunotherapeutic approaches, but also shed light on risk factors and biomarkers that will guide the design of future precision medicine endeavors.Citation97 Importantly, the success of immunotherapy in DMG will require significant investment from academic and pharmaceutical institutions, as the rarity and lethality of DMG limits the number of patients and primary samples available for study. Continued advancements in basic, translational, and clinical investigations of these treatment modalities will ultimately reveal the true benefit of immunotherapy in the management of DMG and other intractable tumors of the CNS.
Acknowledgments
The author wish to thank Dr. Saibaba Guggilapu (Bangalore Medical College and Research Institute) for his assistance with the figures presented in the manuscript.
Disclosure statement
J.D.B. has an equity position in Treovir LLC, an oHSV clinical stage company and is a member of the POCKiT Diagnostics Board of Scientific Advisors. M.G.F. is a consultant for Twentyeight-Seven, Inc., and Blueprint Medicines Corporation. The remaining authors declared that no conflict of interest exists.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, Stearns DS, Wolff JE, Wolinsky Y, Letterio JJ. Alex’s lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2015;16(Suppl 10):x1–17. doi:10.1093/neuonc/nou327.
- Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro Oncol. 2019;21(Supplement_5):v1–v100. doi:10.1093/neuonc/noz150.
- Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M, Lane A, Fuller C, Miles L, Hawkins C. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse Intrinsic pontine glioma (DIPG): a collaborative report from the international and european society for pediatric oncology DIPG registries. J Clin Oncol. 2018;36(19):1963–1972. doi:10.1200/JCO.2017.75.9308.
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231–1251. doi:10.1093/neuonc/noab106.
- Gururangan S, McLaughlin CA, Brashears J, Watral MA, Provenzale J, Coleman RE, Halperin EC, Quinn J, Reardon D, Vredenburgh J, et al. Incidence and patterns of neuraxis metastases in children with diffuse pontine glioma. J Neurooncol. 2006;77(2):207–212. doi:10.1007/s11060-005-9029-5.
- Jaradat A, Nowacki A, Fichtner J, Schlaeppi JA, Pollo C. Stereotactic biopsies of brainstem lesions: which approach? Acta Neurochir (Wien). 2021;163(7):1957–1964. doi:10.1007/s00701-021-04733-2.
- Jung IH, Chang KW, Park SH, Moon JH, Kim EH, Jung HH, Kang SG, Chang JH, Chang JW, Chang WS. Stereotactic biopsy for adult brainstem lesions: a surgical approach and its diagnostic value according to the 2016 world health organization classification. Cancer Med. 2021;10(21):7514–7524. doi:10.1002/cam4.4272.
- Puget S, Beccaria K, Blauwblomme T, Roujeau T, James S, Grill J, Zerah M, Varlet P, Sainte-Rose C. Biopsy in a series of 130 pediatric diffuse intrinsic Pontine gliomas. Childs Nerv Syst. 2015;31(10):1773–1780. doi:10.1007/s00381-015-2832-1.
- Williams JR, Young CC, Vitanza NA, McGrath M, Feroze AH, Browd SR, Hauptman JS. Progress in diffuse intrinsic pontine glioma: advocating for stereotactic biopsy in the standard of care. Neurosurg Focus. 2020;48(1):E4. doi:10.3171/2019.9.FOCUS19745.
- Gupta N, Goumnerova LC, Manley P, Chi SN, Neuberg D, Puligandla M, Fangusaro J, Goldman S, Tomita T, Alden T. Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma. Neuro Oncol. 2018;20(11):1547–1555. doi:10.1093/neuonc/noy070.
- Langmoen IA, Lundar T, Storm-Mathisen I, Lie SO, Hovind KH. Management of pediatric pontine gliomas. Childs Nerv Syst. 1991;7(1):13–15. doi:10.1007/BF00263825.
- Amsbaugh MJ, Mahajan A, Thall PF, McAleer MF, Paulino AC, Grosshans D, Khatua S, Ketonen L, Fontanilla H, McGovern SL. A phase 1/2 trial of reirradiation for diffuse intrinsic Pontine Glioma. Int J Radiat Oncol Biol Phys. 2019;104(1):144–148. doi:10.1016/j.ijrobp.2018.12.043.
- Massimino M, Biassoni V, Miceli R, Schiavello E, Warmuth-Metz M, Modena P, Casanova M, Pecori E, Giangaspero F, Antonelli M, et al. Results of nimotuzumab and vinorelbine, radiation and re-irradiation for diffuse pontine glioma in childhood. J Neurooncol. 2014;118(2):305–312. doi:10.1007/s11060-014-1428-z.
- Lu VM, Welby JP, Mahajan A, Laack NN, Daniels DJ. Reirradiation for diffuse intrinsic pontine glioma: a systematic review and meta-analysis. Childs Nerv Syst. 2019;35(5):739–746. doi:10.1007/s00381-019-04118-y . 2019.
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124(3):439–447. doi:10.1007/s00401-012-0998-0.
- Leszczynska KB, Jayaprakash C, Kaminska B, Mieczkowski J. Emerging advances in combinatorial treatments of epigenetically altered pediatric high-grade H3K27M gliomas. Front Genet. 2021;12:12.
- Johung TB, Monje M. Diffuse intrinsic pontine glioma: new pathophysiological insights and emerging therapeutic targets. Curr Neuropharmacol. 2017;15(1):88–97. doi:10.2174/1570159X14666160509123229.
- Buczkowicz P, Hawkins C. Pathology, molecular genetics, and epigenetics of diffuse intrinsic pontine glioma. Front Oncol. 2015;5:147. doi:10.3389/fonc.2015.00147.
- Rechberger JS, Lu VM, Zhang L, Power EA, Daniels DJ. Clinical trials for diffuse intrinsic pontine glioma: the current state of affairs. Child’s Nervous Sys. 2020;36(1):39–46. doi:10.1007/s00381-019-04363-1.
- Kaufmann SHE. Immunology’s coming of age. Front Immunol. 2019;10:684. doi:10.3389/fimmu.2019.00684.
- Aziz-Bose R, Monje M. Diffuse intrinsic pontine glioma: molecular landscape and emerging therapeutic targets. Curr Opin Oncol. 2019;31(6):522–530. doi:10.1097/CCO.0000000000000577.
- Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, Richards RM, Jiang L, Barsan V, Mancusi R. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603(7903):934–941. doi:10.1038/s41586-022-04489-4.
- Haumann R, Videira JC, Kaspers GJL, van Vuurden Dg, Hulleman E, van Vuurden DG. Overview of current drug delivery methods across the blood-brain barrier for the treatment of primary brain tumors. CNS Drugs. 2020;34(11):1121–1131. doi:10.1007/s40263-020-00766-w.
- van Tellingen O, Yetkin-Arik B, de Gooijer Mc, Wesseling P, Wurdinger T, de Vries HE, de Gooijer MC. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1–12. doi:10.1016/j.drup.2015.02.002.
- Lin GL, Nagaraja S, Filbin MG, Suva ML, Vogel H, Monje M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2018;6(1):51. doi:10.1186/s40478-018-0553-x.
- Ross JL, Chen Z, Herting CJ, Grabovska Y, Szulzewsky F, Puigdelloses M, Monterroza L, Switchenko J, Wadhwani NR, Cimino PJ. Platelet-derived growth factor beta is a potent inflammatory driver in paediatric high-grade glioma. Brain. 2021;144(1):53–69. doi:10.1093/brain/awaa382.
- Titov A, Petukhov A, Staliarova A, Motorin D, Bulatov E, Shuvalov O, Soond SM, Piacentini M, Melino G, Zaritskey A. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018;9(9):897. doi:10.1038/s41419-018-0918-x.
- Prasad V. Immunotherapy: tisagenlecleucel - the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2018;15(1):11–12. doi:10.1038/nrclinonc.2017.156.
- Scarfo I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer. 2017;5(1):28. doi:10.1186/s40425-017-0230-9.
- Petersen CT, Krenciute G. Next generation CAR T cells for the immunotherapy of high-grade glioma. Front Oncol. 2019;9(69). doi:10.3389/fonc.2019.00069.
- Choi BD, Curry WT, Carter BS, Maus MV. Chimeric antigen receptor T-cell immunotherapy for glioblastoma: practical insights for neurosurgeons. Neurosurg Focus. 2018;44(6):E13. doi:10.3171/2018.2.FOCUS17788.
- Berlow NE, Svalina MN, Quist MJ, Settelmeyer TP, Zherebitskiy V, Kogiso M, Qi L, Du Y, Hawkins CE, Hulleman E. IL-13 receptors as possible therapeutic targets in diffuse intrinsic pontine glioma. PloS one. 2018;13(4):e0193565. doi:10.1371/journal.pone.0193565.
- Li G, Mitra SS, Monje M, Henrich KN, Bangs CD, Nitta RT, Wong AJ. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J Neurooncol. 2012;108(3):395–402. doi:10.1007/s11060-012-0842-3.
- Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 2019;10:128. doi:10.3389/fimmu.2019.00128.
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497.
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. doi:10.1126/scitranslmed.aaa0984.
- Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA oncol. 2017;3(8):1094–1101. doi:10.1001/jamaoncol.2017.0184.
- Brown CE, Aguilar B, Starr R, Yang X, Chang W-C, Weng L, Chang B, Sarkissian A, Brito A, Sanchez JF. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther J Am Soc Gene Ther. 2018;26(1):31–44. doi:10.1016/j.ymthe.2017.10.002.
- Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, Labanieh L, Hulleman E, Woo PJ, Rietberg SP. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med. 2018;24(5):572–579. doi:10.1038/s41591-018-0006-x.
- Yu RK, Tsai Y-T, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides–an overview. J Oleo Sci. 2011;60(10):537–544. doi:10.5650/jos.60.537.
- Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. 2020;10:1000. doi:10.3389/fonc.2020.01000.
- Dhillon S. Dinutuximab: first global approval. Drugs. 2015;75(8):923–927. doi:10.1007/s40265-015-0399-5.
- Vitanza N, Wilson A, Gust J, Huang W, Perez F, Albert C, Pinto N, Gardner R, Orentas R, Berens M. Immu-11. clinical updates and correlative findings from the first patient with dipg treated with intracranial car T cells. Neuro-Oncology. 2021;23(Supplement_1):i29–i. doi:10.1093/neuonc/noab090.119.
- Mochizuki A, Ramakrishna S, Good Z, Patel S, Chinnasamy H, Yeom K, Schultz L, Richards R, Campen C, Reschke A. OMIC-11. SINGLE cell rna sequencing from the csf of subjects with H3K27M+ DIPG/DMG treated with GD2 car T-cellular therapy. Neuro-Oncology. 2021;23(Supplement_1):i39–i. doi:10.1093/neuonc/noab090.158.
- Majzner RG, Mahdi J, Ramakrishna S, Patel S, Chinnasamy H, Yeom K, Schultz L, Barsan V, Richards R, Campen C. CT001 - Major tumor regressions in H3K27M-mutated diffuse midline glioma (DMG) following sequential intravenous (IV) and intracerebroventricular (ICV) delivery of GD2-CAR T cells. Cancer Res. 2022, May 16;82(12_Supplement).
- Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. doi:10.1038/nrclinonc.2017.148.
- Bagley SJ, Desai AS, Linette GP, June CH, O’Rourke DM. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro-Oncology. 2018;20(11):1429–1438. doi:10.1093/neuonc/noy032.
- Heipertz EL, Zynda ER, Stav-Noraas TE, Hungler AD, Boucher SE, Kaur N, Vemuri MC Current perspectives on “Off-The-Shelf” ALLOGENEIC NK and CAR-NK Cell Therapies. Front Immunol. 2021;12:732135. doi:10.3389/fimmu.2021.732135.
- Lu H, Zhao X, Li Z, Hu Y, Wang H. From CAR-T CELLS to CAR-NK CELLS: a developing immunotherapy method for hematological malignancies. Front Oncol. 2022;11.
- Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. eBioMedicine. 2020;59:59.
- Lieberman NAP, DeGolier K, Kovar HM, Davis A, Hoglund V, Stevens J, Winter C, Deutsch G, Furlan SN, Vitanza NA. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol. 2019;21(1):83–94. doi:10.1093/neuonc/noy145.
- Bailey CP, Figueroa M, Gangadharan A, Yang Y, Romero MM, Kennis BA, Yadavilli S, Henry V, Collier T, Monje M, et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro Oncol. 2020;22(9):1302–1314. doi:10.1093/neuonc/noaa058.
- Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, Hughes DPM, Kleinerman ES, Lee DA. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res. 2015;32(3):779–792. doi:10.1007/s11095-013-1231-0.
- Pfefferle A, Huntington ND. You have got a fast CAR: chimeric antigen receptor NK cells in cancer therapy. Cancers. 2020;12(3):706. doi:10.3390/cancers12030706.
- Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2016;15(9):660. doi:10.1038/nrd.2016.178.
- Totsch SK, Schlappi C, Kang KD, Ishizuka AS, Lynn GM, Fox B, Beierle EA, Whitley RJ, Markert JM, Gillespie GY. Oncolytic herpes simplex virus immunotherapy for brain tumors: current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene. 2019;38(34):6159–6171. doi:10.1038/s41388-019-0870-y.
- Seymour LW, Fisher KD. Oncolytic viruses: finally delivering. Br J Cancer. 2016;114(4):357–361. doi:10.1038/bjc.2015.481.
- Ghajar-Rahimi G, Kang KD, Totsch SK, Gary S, Rocco A, Blitz S, Kachurak K, Chambers MR, Li R, Beierle EA. Clinical advances in oncolytic virotherapy for pediatric brain tumors. Pharmacol Ther. 2022;239:108193. doi:10.1016/j.pharmthera.2022.108193.
- Friedman GK, Langford CP, Coleman JM, Cassady KA, Parker JN, Markert JM, Yancey Gillespie G. Engineered herpes simplex viruses efficiently infect and kill CD133+ human glioma xenograft cells that express CD111. J Neurooncol. 2009;95(2):199–209. doi:10.1007/s11060-009-9926-0.
- Friedman GK, Bernstock JD, Chen D, Nan L, Moore BP, Kelly VM, Youngblood SL, Langford CP, Han X, Ring EK. Enhanced sensitivity of patient-derived pediatric high-grade brain tumor xenografts to oncolytic HSV-1 virotherapy correlates with nectin-1 expression. Sci Rep. 2018;8(1):13930. doi:10.1038/s41598-018-32353-x.
- Bernstock JD, Bag AK, Fiveash J, Kachurak K, Galal Elsayed GC, Gessler F, Gessler F, Valdes PA, Madan-Swain A, Whitley R. Design and rationale for first-in-human phase 1 immunovirotherapy clinical trial of oncolytic HSV G207 to treat malignant pediatric cerebellar brain tumors. Hum Gene Ther. 2020;31(19–20):1132–1139. doi:10.1089/hum.2020.101.
- Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I, Kachurak K, Nan L, Kang K-D, Totsch S. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med. 2021;384(17):1613–1622. doi:10.1056/NEJMoa2024947.
- Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore C, Tufaro F. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867–874. doi:10.1038/sj.gt.3301205.
- Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17(1):199–207. doi:10.1038/mt.2008.228.
- Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, Nabors LB, Markiewicz M, Agee BS, Coleman JM. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014;22(5):1048–1055. doi:10.1038/mt.2014.22.
- Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, Xu J, Kondo Y, Bekele N, Colman H, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99(18):1410–1414. doi:10.1093/jnci/djm102.
- Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, Shi Y-X, Levin VA, Yung WKA, Kyritsis AP. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19(1):2–12. doi:10.1038/sj.onc.1203251.
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–450.
- Allen C, Vongpunsawad S, Nakamura T, James CD, Schroeder M, Cattaneo R, Giannini C, Krempski J, Peng K-W, Goble JM. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006;66(24):11840–11850. doi:10.1158/0008-5472.CAN-06-1200.
- Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, Prabhu SS, Rao G, Fuller GN, Aldape KD. Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419–1427. doi:10.1200/JCO.2017.75.8219.
- Martínez-Vélez N, Garcia-Moure M, Marigil M, González-Huarriz M, Puigdelloses M, Gallego Pérez-Larraya J, Zalacaín M, Marrodán L, Varela-Guruceaga M, Laspidea V. The oncolytic virus delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun. 2019;10(1):2235. doi:10.1038/s41467-019-10043-0.
- Pérez-Larraya J G, Garcia-Moure M, Labiano S, Patiño-García A, Dobbs J, Gonzalez-Huarriz M, Zalacain M, Marrodan L, Martinez-Velez N, Puigdelloses M. Oncolytic DNX-2401 virus for pediatric diffuse intrinsic pontine glioma. N Engl J Med. 2022;386(26):2471–2481. doi:10.1056/NEJMoa2202028.
- Choi KJ, Zhang SN, Choi IK, Kim JS, Yun CO. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012;19(7):711–723. doi:10.1038/gt.2011.125.
- Hellums EK, Markert JM, Parker JN, He B, Perbal B, Roizman B, Whitle RJ, Langford CP, Bharara S, Gillespie GY. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-oncology. 2005;7(3):213–224. doi:10.1215/S1152851705000074.
- Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum Gene Ther Clin Dev. 2016;27(2):69–78. doi:10.1089/humc.2016.031.
- Hersh DS, Wadajkar AS, Roberts N, Perez JG, Connolly NP, Frenkel V, A. Winkles J, F. Woodworth G, J. Kim A. Evolving drug delivery strategies to overcome the blood brain barrier. Curr Pharm Des. 2016;22(9):1177–1193. doi:10.2174/1381612822666151221150733.
- Morshed RA, Gutova M, Juliano J, Barish ME, Hawkins-Daarud A, Oganesyan D, Vazgen K, Yang T, Annala A, Ahmed AU. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther. 2015;22(1):55–61. doi:10.1038/cgt.2014.72.
- Ahmed AU, Tyler MA, Thaci B, Alexiades NG, Han Y, Ulasov IV, Ulasov IV, Lesniak MS. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol Pharm. 2011;8(5):1559–1572. doi:10.1021/mp200161f.
- Chen SR, Chen MM, Ene C, Lang FF, Kan P. Perfusion-guided endovascular super-selective intra-arterial infusion for treatment of malignant brain tumors. J Neurointerv Surg. 2021;neurintsurg-2021–018190. doi:10.1136/neurintsurg-2021-018190.
- Chastkofsky MI, Pituch KC, Katagi H, Zannikou M, Ilut L, Xiao T, Han Y, Sonabend AM, Curiel DT, Bonner ER. Mesenchymal stem cells successfully deliver oncolytic virotherapy to diffuse intrinsic pontine glioma. Clin Cancer Res. 2021;27(6):1766–1777. doi:10.1158/1078-0432.CCR-20-1499.
- Carceller F, Aleu A, Casasco A, Guimaraens L, López-Pino MA, Madero L, Ramírez M. Superselective intracerebral catheterization for administration of oncolytic virotherapy in a case of diffuse intrinsic pontine glioma. J Pediatr Hematol Oncol. 2014;36(7):e430–2. doi:10.1097/MPH.0000000000000084.
- Van Gool SW, Makalowski J, Bonner ER, Feyen O, Domogalla MP, Prix L, Schirrmacher V, Nazarian J, Stuecker W. Addition of multimodal immunotherapy to combination treatment strategies for children with DIPG: a single institution experience. Med (Basel). 2020;7(5). doi:10.3390/medicines7050029.
- Guo Y, Lei K, Tang L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front Immunol. 2018;9:1499. doi:10.3389/fimmu.2018.01499.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696. doi:10.1038/nature14426.
- Shanmugam MK, Arfuso F, Arumugam S, Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS, Sethi G. Role of novel histone modifications in cancer. Oncotarget. 2017;9(13):11414–11426. doi:10.18632/oncotarget.23356.
- Ochs K, Ott M, Bunse T, Sahm F, Bunse L, Deumelandt K, Sonner JK, Keil M, von Deimling A, Wick W. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology. 2017;6(7):e1328340. doi:10.1080/2162402X.2017.1328340.
- Olsen HE, Lynn GM, Valdes PA, Cerecedo Lopez CD, Ishizuka AS, Arnaout O, Bi WL, Peruzzi PP, Chiocca EA, Friedman GK. Therapeutic cancer vaccines for pediatric malignancies: advances, challenges, and emerging technologies. Neuro-oncol adv. 2021;3(1). doi:10.1093/noajnl/vdab027.
- Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4(1):7. doi:10.1038/s41541-019-0103-y.
- Nair SK, Snyder D, Rouse BT, Gilboa E. Regression of tumors in mice vaccinated with professional antigen-presenting cells pulsed with tumor extracts. Int J Cancer. 1997;70(6):706–715. doi:10.1002/(SICI)1097-0215(19970317)70:6<706::AID-IJC13>3.0.CO;2-7.
- Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184(2):465–472. doi:10.1084/jem.184.2.465.
- Heimberger AB, Crotty LE, Archer GE, McLendon RE, Friedman A, Dranoff G, Bigner DD, Sampson JH. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J Neuroimmunol. 2000;103(1):16–25. doi:10.1016/S0165-5728(99)00172-1.
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DAK, Tönjes M. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. doi:10.1038/nature10833.
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–253.
- Sharma P, Allison JP. Dissecting the mechanisms of immune checkpoint therapy. Nat Rev Immunol. 2020;20(2):75–76. doi:10.1038/s41577-020-0275-8.
- Kline C, Liu SJ, Duriseti S, Banerjee A, Nicolaides T, Raber S, Gupta N, Haas-Kogan D, Braunstein S, Mueller S. Reirradiation and PD-1 inhibition with nivolumab for the treatment of recurrent diffuse intrinsic pontine glioma: a single-institution experience. J Neurooncol. 2018;140(3):629–638. doi:10.1007/s11060-018-2991-5.
- Walia P, D’Arcy C, Hawkins C, Warren KE. IMMU-25. Programmed cell death-ligand 1 (PD-L1) is not expressed in diffuse intrinsic pontine glioma (DIPG) tumor cells. Neuro-Oncology. 2018;20(suppl_6):vi126–vi. doi:10.1093/neuonc/noy148.528.
- Hwang EI, Sayour EJ, Flores CT, Grant G, Wechsler-Reya R, Hoang-Minh LB, Kieran MW, Salcido J, Prins RM, Figg JW. The current landscape of immunotherapy for pediatric brain tumors. Nature Cancer. 2022;3(1):11–24. doi:10.1038/s43018-021-00319-0.
- Jiang Z, Sun H, Yu J, Tian W, Song Y. Targeting CD47 for cancer immunotherapy. J Hematol Oncol. 2021;14(1):180. doi:10.1186/s13045-021-01197-w.
- Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–6667. doi:10.1073/pnas.1121623109.
- Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, Esparza R, Richard C, Ramaswamy V, Remke M. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med. 2017;9(381):eaaf2968. doi:10.1126/scitranslmed.aaf2968.
- Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, Li Y. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11(1):100. doi:10.1186/s13045-018-0644-y.
- Mellor AL, Lemos H, Huang L, Guan W-X, Du J-F. Indoleamine 2,3-dioxygenase and tolerance: where Are We Now? Front Immunol. 2017;8:8. doi:10.3389/fimmu.2017.00008.
- Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37(3):193–207. doi:10.1016/j.it.2016.01.002.
- Wang S, Wu J, Shen H, Wang J. The prognostic value of IDO expression in solid tumors: a systematic review and meta-analysis. BMC Cancer. 2020;20(1):471. doi:10.1186/s12885-020-06956-5.
- Brincks EL, Adams J, Wang L, Turner B, Marcinowicz A, Ke J, Essmann M, Mautino LM, Allen CV, Kumar S. Indoximod opposes the immunosuppressive effects mediated by IDO and TDO via modulation of AhR function and activation of mTORC1. Oncotarget. 2020;11(25):2438–2461. doi:10.18632/oncotarget.27646.
- Jochems C, Fantini M, Fernando RI, Kwilas AR, Donahue RN, Lepone LM, Grenga I, Kim Y-S, Brechbiel MW, Gulley JL. The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget. 2016;7(25):37762–37772. doi:10.18632/oncotarget.9326.
- Johnson T S, Pacholczyk R, Aguilera D, Al-Basheer A, Bajaj M, Berrong Z, Castellino RC, Eaton BR, Esiashvili N, Foreman N IMMU-04. First-in-children phase 1B study using the IDO pathway inhibitor indoximod in combination with radiation and chemotherapy for children with newly diagnosed DIPG (NCT02502708, NLG2105). Neuro-Oncology. 2021;23(Supplement_1):i27–i. doi:10.1093/neuonc/noab090.112.
- Ukleja J, Kusaka E, Miyamoto DT. Immunotherapy combined with radiation therapy for genitourinary malignancies. Front Oncol. 2021;11
- Badiyan S, Kaiser A, Eastman B, Forsthoefel M, Zeng J, Unger K, Chuong M. Immunotherapy and radiation therapy for gastrointestinal malignancies: hope or hype? Transl Gastroenterol Hepatol. 2020;5:21. doi:10.21037/tgh.2019.10.07.
- Rajani KR, Carlstrom LP, Parney IF, Johnson AJ, Warrington AE, Burns TC. Harnessing radiation biology to augment immunotherapy for glioblastoma. Front Oncol. 2019;8:656. doi:10.3389/fonc.2018.00656.
- Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, K.Wansley E, Camphausen K, Luiten RM, de Ru AH, Neijssen J. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–1271. doi:10.1084/jem.20052494.
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–852. doi:10.1016/j.immuni.2014.10.019.
- Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, Lin SH. Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front Pharmacol. 2018;9:185. doi:10.3389/fphar.2018.00185.
- Ou L, Zhang A, Cheng Y, Chen Y. The cGAS-STING pathway: a promising immunotherapy target. Front Immunol. 2021;12.
- Vermeer DW, Spanos WC, Vermeer PD, Bruns AM, Lee KM, Lee JH. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int J Cancer. 2013;133(1):120–129. doi:10.1002/ijc.28015.
- Sato H, Okonogi N, Nakano T. Rationale of combination of anti-PD-1/PD-L1 antibody therapy and radiotherapy for cancer treatment. Int J Clin Oncol. 2020;25(5):801–809. doi:10.1007/s10147-020-01666-1.
- Fang PQ, Gunther JR, Wu SY, Dabaja BS, Nastoupil LJ, Ahmed S, Neelapu SS, Pinnix CC. Radiation and CAR T-cell therapy in lymphoma: future frontiers and potential opportunities for synergy. Front Oncol. 2021;11:648655. doi:10.3389/fonc.2021.648655.
- Woller N, Gürlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, Manns MP, Wirth TC. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. 2015;23(10):1630–1640. doi:10.1038/mt.2015.115.
- Bernstock JD, Vicario N, Rong L, Valdes PA, Choi BD, Chen JA, DiToro D, Osorio DS, Kachurak K, Gessler F. A novel in situ multiplex immunofluorescence panel for the assessment of tumor immunopathology and response to virotherapy in pediatric glioblastoma reveals a role for checkpoint protein inhibition. Oncoimmunology. 2019;8(12):e1678921. doi:10.1080/2162402X.2019.1678921.
- Hwang JK, Hong J, Yun C-O. Oncolytic viruses and immune checkpoint inhibitors: preclinical developments to clinical trials. Int J Mol Sci. 2020;21(22):8627. doi:10.3390/ijms21228627.
- Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, Chastain M, Gorski KS, Anderson A, Chou J. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619–2626. doi:10.1200/JCO.2016.67.1529.
- Nakayama T, Yamashita M, Suzuki T, Shimomura M, Nakatsura T, Aoki K, Yamamoto N, Yamazaki N, Isei T, Uhara H. Immunological impact of canerpaturev (C-REV, formerly HF10), an oncolytic viral immunotherapy, with or without ipilimumab (Ipi) for advanced solid tumor patients (pts). J Clin Oncol. 2019;37(15_suppl):2610. doi:10.1200/JCO.2019.37.15_suppl.2610.
- Gholamin S, Kahn S, Esparza R, Weissman I, Mitra S, Cheshier SIMMU-18. Humanized ANTI-CD47 antibody combined with an agonist ANTI-CD40 antibody is an effective treatment for dipg xenografts with craniospinal dissemination. Neuro-Oncology. 2017;19(suppl_4):iv31–iv. doi:10.1093/neuonc/nox083.128.
- Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med. 2020;71(1):47–58. doi:10.1146/annurev-med-062518-045435.
- Hanna GJ, O’Neill AM, Jo VY, Wong K, Lizotte PH, Annino DJ, Goguen LA, Kass JI, Rettig EM, Sethi RKV. Neoadjuvant and adjuvant nivolumab and lirilumab in patients with recurrent, resectable squamous cell carcinoma of the head and neck. J Clin Oncol. 2021;39(15_suppl):6053. doi:10.1200/JCO.2021.39.15_suppl.6053.
- Grivas P, Yin J, Koshkin VS, Cole S, Jain RK, Dreicer R, Cetnar JP, Sundi D, Gartrell BA, Galsky MD. PrE0807: a phase Ib feasibility trial of neoadjuvant nivolumab (N) without or with lirilumab (L) in cisplatin-ineligible patients (pts) with muscle-invasive bladder cancer (MIBC). J Clin Oncol. 2021;39(15_suppl):4518. doi:10.1200/JCO.2021.39.15_suppl.4518.
- Grivas P, Puligandla M, Cole S, Courtney KD, Dreicer R, Gartrell BA, Cetnar JP, Dall’era M, Galsky MD, Jain RK. PrE0807 phase Ib feasibility trial of neoadjuvant nivolumab (N)/lirilumab (L) in cisplatin-ineligible muscle-invasive bladder cancer (BC). J Clin Oncol. 2019;37(15_suppl): TPS4594–TPS. doi:10.1200/JCO.2019.37.15_suppl.TPS4594.
- Charych D, Khalili S, Dixit V, Kirk P, Chang T, Langowski J, Rubas W, Doberstein SK, Eldon M, Hoch U, et al. Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PloS one. 2017;12(7):e0179431. doi:10.1371/journal.pone.0179431.
- Diab A, Tannir NM, Bentebibel SE, Hwu P, Papadimitrakopoulou V, Haymaker C, Kluger HM, Gettinger SN, Sznol M, Tykodi SS, et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 2020;10(8):1158–1173. doi:10.1158/2159-8290.CD-19-1510.
- Squibb BM 2022.
- Wolff JE, Rytting ME, Vats TS, Zage PE, Ater JL, Woo S, Kuttesch J, Ketonen L, Mahajan A. Treatment of recurrent diffuse intrinsic pontine glioma: the MD anderson cancer center experience. J Neurooncol. 2012;106(2):391–397. doi:10.1007/s11060-011-0677-3.