
ABSTRACT
Hepatocellular carcinoma (HCC) is the fourth leading cause of cancer-related deaths worldwide due to high recurrence rates after curative treatment and being frequently diagnosed at an advanced stage. Immune-checkpoint inhibition (ICPI) has yielded impressive clinical successes in a variety of solid cancers, however results in treatment of HCC have been modest. Vaccination could be a promising treatment to synergize with ICPI and enhance response rates. Cancer testis antigens (CTAs) were recently discovered to be widely expressed in HCC and expression in macroscopically tumor-free tissues correlated with recurrence, implying the presence of micro-satellites. To determine whether CTAs are immunogenic in HCC patients, we analyzed systemic T-cell and humoral responses against seven CTAs in 38 HCC patients using a multitude of techniques; flowcytometry, ELISA and whole antigen and peptide stimulation assays. CTA-specific T-cells were detected in all (25/25) analyzed patients, of which most had a memory phenotype but did not exhibit unequivocal signs of chronic stimulation or recent antigen encounter. Proliferative CD4+ and CD8+ T-cell responses against these CTAs were found in 14/16 analyzed HCC patients. CTA-peptide stimulation-induced granzyme B, IL2, and TNFa in 8/8 analyzed patients, including two MAGEA1 peptides included based on in silico prediction. Finally, IgG responses were observed in 13/32 patients, albeit with low titers. The presence of CD4+ and CD8+ T-cells and IgG responses shows the immunogenicity of these CTAs in HCC-patients. We hypothesize that vaccines based on these tumor-specific antigens may boost preexisting CTA-specific immunity and could enhance therapeutic efficacy of ICPI in advanced HCC.
Introduction
Hepatocellular carcinoma (HCC) is the most common occurring liver cancer and the fourth leading cause of cancer-related death worldwide.Citation1 As a result of an increased prevalence of several risk factors such as obesity and diabetes, the incidence of HCC in Western countries is rising.Citation2 Early HCC can be treated with curative intent by either radiofrequency ablation (RFA) or surgical resection. However, up to 70% of these patients experience cancer recurrence, and currently there is no therapy to prevent this.Citation3 In addition, most patients are diagnosed at a late stage and can only be offered palliative treatments such as transarterial chemoembolization (TACE) or multikinase inhibitors, which have limited survival benefits.Citation4 Recently, combination therapy of the ICPI atezolizumab (a programmed death-ligand 1 [PD-L1] inhibitor) and bevacizumab (blocking vascular endothelial growth factor [VEGF]) showed modest survival benefit compared to treatment with the multikinase inhibitor sorafenib in advanced HCC.Citation5
ICPI therapy is generally most effective in patients with T-cell inflamed “hot” tumors,Citation6,Citation7 and recent data indicate this is also true for HCC.Citation8 Attempts to transform immunologically “cold” tumors into “hot” tumors to sensitize them to ICPI therapy have yielded promising clinical results by combining ICPI therapy with immunogenic cell death-inducing chemotherapeutics.Citation6,Citation7,Citation9 Similarly, recent studies suggest synergistic effects of combining ICPI therapy with therapeutic cancer vaccination to enhance anti-tumor immunity and T-cell infiltration.Citation10–14 Such combinatorial studies have not been performed yet in HCC, but therapeutic vaccination with tumor-associated antigen (TAA)-derived peptides can elicit systemic TAA-directed T-cell immunity and tumor T-cell infiltration in both early and advanced HCC patients.Citation15,Citation16 Clinical responses, however, were thus far limited and call for a strategy targeting multiple different strongly immunogenic TAA at the same time. Hence, there is a need to discover additional TAA targets for example by mass spectrometry-based HLA-peptidomics.Citation17–19
Target antigens for cancer vaccines need to be selected carefully to avoid on-target/off-tumor toxicities. A subset of cancer-testis antigens (CTAs) is not expressed in adult healthy tissues, except in immune-privileged germ cells, but can reach high levels in cancer cells due to epigenetic aberrations.Citation20 CTAs have therefore been targeted with therapeutic cancer vaccines. A major advantage of CTAs over neo-antigens is the shared expression of specific CTAs in tumors of the same type from different patients, enabling their targeting with off-the-shelf vaccines for groups of patients. However, careful selection of CTAs that are highly prevalent in a specific cancer but absent in healthy tissue (except immune-privileged germ cells) is required for inclusion in a therapeutic cancer vaccine.Citation20 Until recently, clinical results of CTA-based vaccines have been largely disappointing,Citation21 but new powerful vaccination strategies in combination with ICPI therapy have lately demonstrated the potential of CTAs as vaccine targets in cancer.Citation11,Citation12
Recently, we identified a panel of 12 tumor-restricted CTAs prevalent in HCC, but absent from healthy tissue.Citation22 This panel of CTAs may therefore hold attractive targets for therapeutic vaccination. Moreover, while absent from healthy liver tissue, we detected low-level expression of one or more of these CTAs in histologically tumor-free liver tissues of 28% and 45% of HCC patients, respectively, in two independent cohorts undergoing primary tumor resection. In both cohorts, CTA expression in tumor-surrounding liver tissue was independently associated with cancer recurrence and poor survival after resection.Citation22 These data suggest the presence of CTA-expressing occult micro-metastases or (pre-)malignant cells in tumor-surrounding liver tissue that give rise to new tumors after resection. Therefore, we postulated that our panel of HCC-restricted CTAs may be targets for adjuvant vaccination strategy to prevent cancer recurrence after primary tumor resection or to sensitize advanced HCC patients to ICPI therapy.
An important prerequisite for clinical efficacy of vaccines is antigen immunogenicity in the target patient population. Yet, immunogenicity is largely unknown for our panel of CTAs in HCC patients. To determine whether these CTAs are immunogenic in HCC patients, we analyzed systemic CD4+ and CD8+ T-cell responses as well as IgG-responses against six CTAs of our panel (melanoma-associated antigen 1 [MAGEA1], melanoma-associated antigen 9 [MAGEA9], melanoma-associated antigen B2 [MAGEB2], melanoma-associated antigen C1 [MAGEC1], melanoma associated antigen C2 [MAGEC2], P antigen family member 1 [PAGE1]) in HCC patients. We also included synovial sarcoma, X breakpoint 2 (SSX2), a thoroughly studied tumor-restricted and immunogenic CTA with known expression in HCC.Citation23–25 Because systemic T-cell responses against TAAs may be transiently enhanced after local tumor ablation in HCC patients,Citation26,Citation27 we analyzed these immunological responses before and at several time points after RFA or TACE.
Materials and methods
HCC patients
Thirty-eight patients diagnosed with HCC and referred for RFA or TACE were enrolled in the Erasmus Medical Center, Rotterdam, the Netherlands, between January 2016 and May 2019. Diagnosis was based on pathognomonic radiological findings, and all lesions were classified as LI-RADS 5.Citation28 Completeness of treatment of the lesion was assessed by computed tomography (CT) and alfa feto protein (AFP) level in blood. If necessary treatment was repeated. Multiple patients experienced rapid recurrence after initial treatment, and one retreated patient (ITV-016) was included twice. HCC recurrence was evaluated by CT scan routinely after approximately 6 weeks (range 5–11 weeks), and subsequently every 3 months.
Blood was collected before treatment and after 3 (3–5), 7 (5–11), and 25 (13–47) weeks (median (range)) of treatment. Variation in blood collection time was due to differences in individual treatment plans and out-patient clinic visits. Six patients were excluded from the analysis because only blood before treatment was collected (Supplementary Table S1a). Medical records were reviewed for clinicopathological variables, dates of first cancer recurrence, HCC-specific death, liver transplantation (LTx; after which follow-up stopped) or last follow-up until April 1,st 2020 (). All patients signed informed consent before participation. The study adhered to the Helsinki declaration and was approved by the local medical ethical committee (MEC-2015-563).
Table 1. Patient characteristics.
PBMC isolation, HBsAg, and anti-HBsAg determination and flowcytometric HLA-A*02 typing
Blood was collected in Sodium Heparin tubes (BD), which were centrifuged at 120xg for 20 min to collect plasma. Consecutively, the plasma was centrifuged at 3220xg for 15 min to discard platelets. Plasma samples were stored at −80°C until use. Quantitative measurements of Hepatitis B surface antigen (HBsAg) and anti-HBsAg were performed in thawed plasma using tests from the DiaSorin LIAISON® XL HEPATITIS A and B LINE by the Department of Virology, Erasmus MC, Rotterdam, the Netherlands. Peripheral blood mononuclear cells (PBMC) were isolated from the plasma-depleted fraction by Ficoll density gradient centrifugation. To select human leukocyte antigen (HLA)-A*02-positive patients for the analysis of CTA-specific CD8+ T-cells using HLA-A*02:01 dextramers, a sample of PBMC was labeled with a fluorochrome-conjugated anti-HLA-A*02 antibody (Supplementary Table S2) and analyzed by flow cytometry (FACS Canto II [BD Biosciences]). Remaining PBMC were cryopreserved in 49% RPMI-1640 medium (Gibco), 0.5% penicillin/streptomycin (Gibco), 0.5% ultraglutamin (Lonza), 40% fetal calf serum (FCS, Sigma) and 10% DMSO (Sigma), and stored in liquid nitrogen until use.
Ex vivo CTA-specific T-cell proliferation assay
Autologous B-cell blasts were generated from 10 × 106 PBMC by culturing with 1 ug/ml soluble human trimeric CD40ligand (CD40L; kindly provided by National Institutes of Health (NIH), Bethesda, MD, USA), 40 IU/ml recombinant interleukin 4 (rIL4; Strathman) and 1 ug/ml Cyclosporin A (Novartis) for 3 weeks, as previously described.Citation29 Vitality, purity, and maturation status were checked by flow cytometry (FACS Canto II [BD Biosciences]) after antibody staining (see Supplementary Table S2 and 7-Aminoactinomycin D (7AAD; Invitrogen), shown in Supplementary Figure S1. B-cell blasts were electroporated with 10 μg of messenger RNA (mRNA) encoding MAGEA1, MAGEA9, MAGEB2, MAGEC1934-1142, MAGEC2, PAGE, SSX2, or luciferase, (Supplementary File) by electroporation using the square-wave protocol with a single pulse of 800 V and a length of 0.6 ms in a 4 mm cuvette, as previously described.Citation29 After electroporation the B cells were rested for at least two hours at 37°C. Messenger RNAs were synthesized by BioNTech SE by in vitro transcription from linearized plasmids in which the full-length (or in case of MAGEC1 amino acids 934–1142, because of its large size and the presence of known immunogenic epitopes in this partCitation30) coding sequences of the respective genes were flanked with an HLA class I signal peptide fragment (sec) at the 5’- and an HLA class I trafficking signal at the 3’-end to facilitate loading in HLA class II.Citation31 For comparison, B-cell blasts were electroporated with 10 μg glypican 3 (GPC-3) or enhanced green fluorescent protein (eGFP) encoding mRNAs, purchased from eTheRNA (Belgium), which we used in previous studies.Citation29,Citation32 In these constructs, antigens were fused to the transmembrane and luminal regions of DCLamp for endo-lysosomal targeting. mRNA encoding New York esophageal squamous cell carcinoma-1 (NY-ESO-1) was not available.
HCC patient PBMC were thawed, stained with CellTrace CFSE (Invitrogen), and co-cultured with transfected autologous B-cell blasts at a 1:10 B-cell:PBMC ratio in RPMI1640 supplemented with ultraglutamine (Lonza), 10% normal human AB serum (Sigma), 1% penicillin/streptomycin (Gibco), 10 mM Hepes buffer (Lonza), 1 mM Sodium Pyruvate (Invitrogen) and 1% MEM Non-Essential Amino Acids Solution 100x (MEM-NEAA, Invitrogen) for 6 days at 37°C in a humidified atmosphere supplemented with 5% CO2. Triplicate cultures were set up for each CTA as well as for luciferase, GPC-3, and eGFP. As a positive control, PBMC were stimulated with 25 ng/ml Staphylococcal Endotoxin B (SEB; Sigma). Total numbers of PBMC varied between 1.1 and 2.2 × 105 cells per well, depending on the number of available PBMC. After 6 days cells were harvested, technical triplicate cultures were pooled, and CFSE-dilution in CD4+ and CD8+ T-cells was analyzed by flow cytometry (FACS Canto II [BD Biosciences]), after surface labeling for 20 min at 4°C with antibodies (anti-CD3 PE-Cy7, anti-CD4 APC-Cy7, anti-CD8 PerCP-Cy5.5, and anti-CD56 PE; see Supplementary Table S2), followed by 7AAD (Invitrogen).Citation29 Percentages of proliferated (CFSE-low) CD4+ and CD8+ T-cells were determined. Antigen-specific T-cell responses were considered positive when percentages of CFSE-low T-cells in cultures with CTA or GPC3 mRNA transfected B-cell blasts were at least twofold higher compared to percentages of CFSE-low T-cells in co-cultures of B-cell blasts transfected with luciferase or eGFP mRNA, respectively. Because >50% of T-cells were CFSE-low in all SEB-stimulated PBMC, all cultures could be accepted for analysis. A minimum of 50 (range 50–13845) CFSE− cells, 520 (range 520–32012) CD8+ T cells and 3091 (range 3091–107528) CD4+ T cells in the CTA-stimulated conditions were acquired.
Flow cytometric PBMC phenotyping
For phenotypic analysis without HLA-A*02:01 dextramer labeling, PBMC were thawed, stained with a CCR7-specific mAb for 1 h at room temperature (RT), followed by a fixable live/dead stain (eBioscience) for 20 min at 4°C, antibody labeling of other surface antigens for 20 min at 4°C, fixation with the FoxP3 staining buffer kit (eBioscience) for 20 min at 4°C and staining of intracellular antigens for 20 min at 4°C. For phenotypic analysis including peptide-loaded HLA-A*02:01 dextramer labeling, PBMC of HLA-A*02+ patients and HLA-A*02+ healthy blood bank donors (with written informed consent from the donors provided by Sanquin Blood bank, Amsterdam) were thawed and stained with a CCR7-specific mAb for 1 hr at RT, followed by a fixable live/dead stain (eBioscience) for 20 min at 4°C. Consecutively, before staining with CTA peptide-loaded HLA-A*02:01 dextramers, PBMC were incubated with 50 nM dasatinib (Sigma) for 30 min at 37°C to enhance T-cell receptor expression,Citation33 followed by incubation with a pool of APC-labeled HLA-A*02:01 dextramers loaded with previously described immunogenic epitopes derived from 7 different CTAs (Immudex. Supplementary Table S3), or single CTA peptide-loaded dextramers for 10 min at RT. Cytomegalovirus (CMV)pp65 peptide loaded dextramer was included as a reference. Thereafter, cells were labeled for surface antigens for 20 min at 4°C, permeabilized and fixed with the FoxP3 staining buffer kit (eBioscience) for 20 min at 4°C and stained for intracellular antigens for 20 min at 4°C. As no immunogenic epitope of PAGE1 is known, we could not include a PAGE1 dextramer labeling. For the PE-labeled CMVpp65-peptide loaded HLA-A*02:01 dextramer, pre-incubation with dasatinib was not required and therefore omitted. A threshold of a minimum of 10 events was set.
A detailed list of fluorochrome-conjugated antibodies used for phenotyping is listed in Supplementary Table S2. All antibody stains were carried out in the presence of 0,2% Fc-block (BD Biosciences). Flow cytometric phenotyping was performed using a FACS Symphony (BD Biosciences), and analysis was performed using FlowJo version 10.4 software (FlowJo).
High resolution HLA-I typing and peptide stimulation
For high-resolution HLA-typing of HCC patients selected for peptide stimulation assays, DNA was isolated from PBMC using the QIAamp DNA Mini kit (Qiagen) according to manufacturer’s instructions. High-resolution HLA-I typing was performed by the Department of Internal Medicine, Erasmus MC, Rotterdam, the Netherlands using the Illumina GSA beadchip GSA MD v2. HLA types were imputed from the obtained SNP data (after translation of Illumina IDs to Affymetrix IDs) using the Axiom HLA analysis tool (Affymetrix). Next, in silico prediction was performed (NetMHCpan3.0) to expand the CTA-peptide pool that could be tested in donors that were negative for HLA-A*02:01 (ITV-006; ITV-007 and ITV-021). The peptides that were predicted to bind (rank ≤ 2.0) at least one HLA-type of a patient were included in peptide stimulation assays (underlined in Supplementary Table S4). Cryopreserved PBMC of 8 HCC patients were subsequently thawed and stimulated with CTA and hepatitis B virus (HBV) peptides (Peptide 2.0 Inc) at 37°C in a humidified atmosphere supplemented with 5% CO2 as described previously.Citation34 Briefly, PBMC (1x106 cells/ml) were stimulated with pools of a maximum of 5 peptides of interest (Supplementary Table S4) based on HLA class I types of the patients (Supplementary Table S5) at a concentration of 5 ug/ml of each peptide in Iscove’s modified Dulbecco’s medium (IMDM; Lonza) supplemented with 2% normal human AB serum (Sanquin), 1% ultraglutamine (Lonza), 1% penicillin/streptomycin (Gibco) and 50 IU/ml hIL-2 (Miltenyi). After 14 days, cultured PBMC were re-stimulated for 48 hr at 37°C with 5 ug/ml of each peptide separately or solvent-containing medium as a negative control. Depending on cell availability, 80.000–200.000 cultured PBMC were re-stimulated in duplicate. Cytokine production was assessed in pooled duplicate supernatants by the LEGENDplex™ Human CD8/NK Panel (BioLegend) according to the manufacturer’s instructions. Plates were read using the MACSQuant Analyzer 10 (Miltenyi) and analyzed using the LEGENDplex™ data analysis software (BioLegend). Peptides for which cytokine concentrations were ≥2-fold higher as compared to the corresponding negative control cultures were determined to have evoked a peptide-specific response.
CTA IgG ELISA
Plasma samples of 32 HCC patients and 15 healthy controls (Sanquin blood bank, informed consent supplied) were diluted 1:1 in glycerol (Sigma) for storage at −20°C and used for determination of Immunoglobulin G (IgG) against recombinant full-length CTA proteins (Supplementary Table S6). Plasma IgG against CTAs was determined by indirect ELISA according to Gnjatic, et al.,Citation35 with minor adjustments made in preliminary testing of sera from melanoma patients which contained MAGEA1 or NY-ESO-1 specific IgG, kindly provided by Prof. Dr. S.B. Eichmüller (German Cancer Research Center, Heidelberg, Germany).Citation36 For screening of IgG immune reactivity against CTAs in patient plasma samples, four random plasma samples of healthy subjects were included as negative controls in ELISAs for each CTA, and all measurements were performed in duplicate. Maxisorp-plates™ (ThermoFisher) were coated with 1 µg/ml full length recombinant protein or 1 µg/ml bovine serum albumin (BSA, Sigma; negative control protein) in coating buffer (0.2 M sodium carbonate, pH 9.5) overnight at 4°C. Plates were then washed eight times with phosphate buffered saline (PBS), and blocked with blocking buffer (5% nonfat dry milk [Bio-Rad] and 10% fetal calf serum in PBS) for 2 hours at RT. After washing the plates eight times with PBS, they were incubated with plasma samples diluted 1:20 in blocking buffer for 2 hours at RT. Consecutively, plates were washed four times with 0.1% Tween 20 (Sigma) in PBS, four times with PBS, and incubated with a goat anti-human IgG-horseradish peroxidase ([HRP]; 1:6000, BD Biosciences) detection antibody for 1 h at RT. Plates were washed four times with 0.1%Tween 20 in PBS and four times with PBS, after which TMB substrate (Invitrogen) was added for 15 minutes. The reaction was stopped with 1 N H2SO3 and absorbance was measured at 450 nm using an Infinite® 200 PRO microplate reader (Tecan).
The delta optical density (ΔOD) of each plasma sample was calculated by subtracting the OD from wells coated with BSA from those coated with the specific CTA. Patient plasma samples were considered positive if the ΔOD value exceeded the mean ΔOD plus ten times the standard deviation of the four healthy control plasma samples.Citation35
The positive plasma samples (at the screening dilution of 1:20) were then selected for determination of the CTA-specific IgG titer by testing plasma dilutions of 1:20, 1:40, 1:80, 1:160, and 1:320 using the same protocol.
Statistical analysis
Statistical analyses were performed using Graphpad (Version 8.2.1 for Windows, San Diego, CA) and R Statistical software (Version 3.6.1 for Windows, Foundation for Statistical Computing, Vienna, Austria). Heat maps were created with RStudio and the ‘gplots’ and ‘pheatmap’ packages. Used statistical tests are indicated in the figure legends. P-values < 0.05 were considered significant.
Results
Patients and healthy controls
The clinicopathological characteristics of the 32 included HCC patients and 15 age and gender-matched healthy blood bank donors are shown in . The most prevalent etiology of HCC was alcoholic liver disease (n = 13, 40.6%), second was nonalcoholic steatohepatitis (NASH; n = 6, 18.8%). Most patients were classified as Barcelona clinic liver cancer (BCLC) stage A (n = 27, 84.4%), 5 patients were classified as BCLC stage B (15.6%). Most patients were treated with RFA (n = 25, 78.1%) and five were treated with TACE (15.6%), whereas two patients underwent both treatments subsequently (6.3%, both of these patients had BCLC stage B disease). During the follow-up, 23 patients (71.9%) developed HCC recurrence, 7 (21.9%) died of HCC and 8 (25%) underwent liver transplantation. Treatments, blood collection, tumor recurrence, LTx and HCC-specific death are summarized in .
Figure 1. Patient treatment timeline. Schematic overview of events (blood sample collection, RFA/TACE, LTx, HCC-specific death) in HCC patients included in the study during the follow-up period. The colors indicate the presence or absence of detectable HCC tumor. Patient ITV-016 was retreated with RFA 13 weeks after the first treatment due to rapid recurrence. Of this patient, blood samples were collected before and after the first RFA and before and after the second RFA and were included in the study separately.
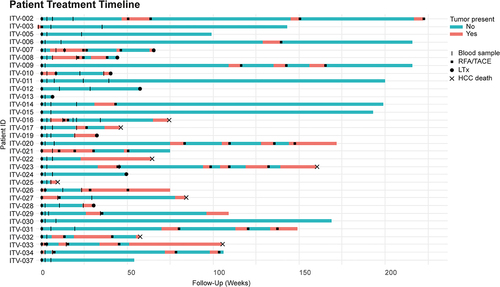
HCC patients have enhanced frequencies of circulating CTA-specific CD8+ T-cells compared to healthy subjects
It has been described that local ablative therapies can enhance frequencies of circulating tumor-specific T-cells in HCC-patients.Citation26,Citation27,Citation37 Therefore, to maximize the chance of detecting CTA-specific T-cells in HCC patients, we analyzed PBMC of 26 HCC patients by flow cytometry before treatment and at 3, 7, and 25 weeks after local ablative therapies (Supplementary Table S1A). No effect of ablative therapy on any of the major leukocyte (sub)populations was observed ( and Supplementary Figure S2a,b). Also, the main T-cell subpopulations did not change in response to ablative therapy (Supplementary Figure S2c). Next, we determined the frequencies of CTA peptide-specific CD8+ T-cells in HLA-A*02:01+ patients (n = 17, Supplementary Table S1A) and compared these with frequencies in 9 healthy HLA-A*02:01+ subjects, by staining with a pool of HLA-A*02:01 dextramers loaded with published immunogenic CTA-peptides (MAGEA1278-286, MAGEA9223-231, MAGEB2231-240, MAGEC11087-1095, MAGEC2191-200, NY-ESO-1157-165, and SSX241-49; Supplementary Figure S3a, Supplementary Table S3). A CMVpp65 dextramer was used as a non-tumor antigen reference. CTA peptide-specific CD8+ T-cells in HCC patients ranged between 0.07–1.99%, 0.12–1.45%, 0.09–1.3%, and 0.08–2.4% at t = 0, 3, 7 and 25 weeks after treatment, respectively (). A maximum of 0.06% of CD8+ T-cells recognized these pooled CTA-peptides in healthy controls (). Using > 0.06% as a cutoff, these data demonstrate that all 17 HCC patients who were included in this part of the study had enhanced frequencies of circulating CTA peptide-specific CD8+ T-cells compared to healthy subjects. However, there were no marked changes in CTA-specific CD8+ T-cell frequencies over time upon local ablative therapies ().
Figure 2. HCC patients show enhanced frequencies of circulating CTA-specific CD8+ T-cells compared to healthy subjects. PBMC from HCC patients before (t = 0), and approximately 3, 7, and 25 weeks (see for the exact time points) after RFA/TACE treatment were phenotyped by flow cytometry (n = 26). CTA-specific CD8+ T-cells were determined in HLA-A*02:01-positive patients (n = 17) using CTA peptide-loaded HLA-A*02:01 dextramers. Patient ITV-016 was included twice, before and after a first RFA and before and after a second RFA treatment. A. CTA-specific CD8+ T-cells were detected using a pool of 7 CTA peptide-loaded HLA-A*02:01 dextramers (Supplementary Table S3). Frequencies of CMVpp65495-503-specific CD8+ T-cells were measured for comparison. B. Differentiation status of CMVpp65495-503-dex+, CTA-dex+ and CTA-dex− CD8+ T-cells was determined based on CD45RA and CCR7 expression (Naïve; CD45RA+CCR7+, central memory [CM]; CD45RA−CCR7+, effector memory [EM]; CD45RA−CCR7−, and TEMRA; CD45RA+CCR7−). C. CMV-specific CD8+ T-cells were detected in healthy blood bank donors, but only a maximum of 0.06% of CD8+ T-cells recognized the pooled CTA-peptides. D. Surface expression of activation marker CD137, intracellular expression of proliferation marker Ki67, cytotoxic effector molecule granzyme B and checkpoint inhibitor PD1 were determined in CTA-dex+, CMV-dex+ and CTA-dex− CD8+ T-cells. Wilcoxon signed-rank test, * p < .05, **p < .01, ***p < .001.
![Figure 2. HCC patients show enhanced frequencies of circulating CTA-specific CD8+ T-cells compared to healthy subjects. PBMC from HCC patients before (t = 0), and approximately 3, 7, and 25 weeks (see Figure 1 for the exact time points) after RFA/TACE treatment were phenotyped by flow cytometry (n = 26). CTA-specific CD8+ T-cells were determined in HLA-A*02:01-positive patients (n = 17) using CTA peptide-loaded HLA-A*02:01 dextramers. Patient ITV-016 was included twice, before and after a first RFA and before and after a second RFA treatment. A. CTA-specific CD8+ T-cells were detected using a pool of 7 CTA peptide-loaded HLA-A*02:01 dextramers (Supplementary Table S3). Frequencies of CMVpp65495-503-specific CD8+ T-cells were measured for comparison. B. Differentiation status of CMVpp65495-503-dex+, CTA-dex+ and CTA-dex− CD8+ T-cells was determined based on CD45RA and CCR7 expression (Naïve; CD45RA+CCR7+, central memory [CM]; CD45RA−CCR7+, effector memory [EM]; CD45RA−CCR7−, and TEMRA; CD45RA+CCR7−). C. CMV-specific CD8+ T-cells were detected in healthy blood bank donors, but only a maximum of 0.06% of CD8+ T-cells recognized the pooled CTA-peptides. D. Surface expression of activation marker CD137, intracellular expression of proliferation marker Ki67, cytotoxic effector molecule granzyme B and checkpoint inhibitor PD1 were determined in CTA-dex+, CMV-dex+ and CTA-dex− CD8+ T-cells. Wilcoxon signed-rank test, * p < .05, **p < .01, ***p < .001.](/cms/asset/1097b588-a1f2-44b0-a22b-f7244a22e1e6/koni_a_2131096_f0002_oc.jpg)
Using CD45RA and CCR7 expression, we subsequently analyzed the differentiation status of circulating CD8+ T-cells (Supplementary Figure S4). In agreement with previous reports,Citation38 most CMV-specific CD8+ T-cells showed a terminally differentiated phenotype (). Most CTA peptide-specific CD8+ T-cells also showed a memory phenotype but contained more effector memory (EM) and central memory (CM) T-cells than CMV-specific CD8+ T-cells (). To investigate whether CTA-specific CD8+ T-cells in HCC patients showed signs of recent activation, we determined their expression of activation marker CD137 and proliferation marker Ki67 (). Although we observed tendencies to slightly enhanced expression of CD137 and Ki67 in CTA-specific CD8+ T-cells compared to CMV-specific CD8+ T-cells and non-CTA recognizing CD8+ T-cells, most of these differences did not reach statistical significance ().
Compared to non-CTA recognizing CD8+ T-cells, CTA-specific CD8+ T-cells showed reduced Granzyme B expression before local tumor ablation (). This was not accompanied by further signs of chronic stimulation and exhaustion as expression of the inhibitory receptors PD1, LAG3 and TIM3 was not elevated on CTA-specific CD8+ T-cells compared to their CMV-specific or non-CTA recognizing counterparts ( and Supplementary Figure S3b). Moreover, LAG3 expression on CTA-specific CD8+ T-cells was reduced compared to CMV-specific CD8+ T-cells (Supplementary Figure S3b). Finally, we measured the binding of the individual CTA peptide-loaded HLA-A*02:01 dextramers to CD8+ T-cells. For this purpose, we pre-selected 5 HCC patients in which >0.5% of CD8+ T-cells bound the pool of CTA peptide-loaded HLA-A*02:01 dextramers at a given time point (Figure S5). We detected CD8+ T-cells specific for each individual CTA in these patients. Frequencies were low, but comparable to those of CD8+ T-cells that recognized the established immunogenic NY-ESO-1157-166 epitope. However, no consistent effect of local tumor ablation was observed, which is consistent with the pooled CTA-peptide dextramer results.
Taken together, we detected higher frequencies of CTA peptide-specific circulating CD8+ T-cells in all 17 HCC patients compared to healthy subjects. Most of these cells had a memory phenotype, suggesting in vivo priming, but they did not exhibit unambiguous signs of chronic stimulation (co-inhibitory receptor expression), or recent antigen encounter (CD137 or Ki-67 expression). In addition, neither frequency nor differentiation or activation status of CTA peptide-specific CD8+ T-cells was influenced by local tumor ablation.
CD4+ and CD8+T-cells of HCC patients show in vitro proliferative responses to CTA-specific stimulation
To study whether circulating CTA-specific T-cells in HCC patients are functionally competent, we first quantified in vitro proliferative responses of peripheral T-cells from 16 HCC-patients (Supplementary Tables S1A and S1B) against CTAs. One patient (ITV-016) was included twice in the study, using blood samples collected before and after an initial RFA and a second RFA performed 13 weeks after the first due to rapid cancer recurrence. CFSE-labeled PBMC were stimulated for 6 days with expanded autologous B-cell blasts electroporated with CTA-encoding mRNA or, as a negative control, with luciferase mRNA (see Supplementary Figure S1 for phenotyping results of generated B-cell blasts). Proliferative CD4+ and CD8+ T-cell responses were subsequently measured by CFSE-dilution and observed against all of the seven tested CTAs (MAGEA1, MAGEA9, MAGEB2, MAGEC1, MAGEC2, PAGE1 and SSX2). Overall, strong proliferative CD4+ and/or CD8+ T-cell responses (≥ 4-fold over response to luciferase) against each of the CTAs were observed in at least one patient. All but two HCC patients (ITV-011 and ITV-031) showed a proliferative T-cell response to at least 1 and maximally 6 CTAs (). MAGEC2-, PAGE1 and SSX2-specific T-cell responses were most frequently observed (in 11/16, 9/16 and 9/16 patients, respectively), while T-cell responses against each of the other 4 CTAs were detected in 7 HCC patients. GPC3, a well-known immunogenic HCC-associated antigenCitation16,Citation39 was included as a reference antigen and induced a maximum fold-change in proliferation of 2.7-fold in CD4+ T-cells and 3.9-fold in CD8+ T-cells, which is lower than the maximum responses observed against each of the tested CTAs. We did not observe an uniform relation between tumor ablation and proliferative CTA-specific T-cell responses. In addition, we did not find a clear association between CTA-specific proliferative T-cell responses and etiology, AFP levels (), or other clinicopathological factors (Supplementary Figure S6).
Figure 3. CTA-specific stimulation induces proliferative responses in CD4+ and CD8+ T-cells of HCC patients. CTA-specific CD4+ and CD8+ T-cell proliferation was measured in 16 HCC patients by 6-day stimulation of CFSE-labeled PBMC with CTA mRNA or luciferase mRNA (negative control)-electroporated autologous B-cell blasts. CTA-specific proliferation was then calculated by dividing the percentage proliferated (CFSE-low) T-cells upon CTA-stimulation by the percentage proliferated T-cells upon luciferase-stimulation. A. Gating example of proliferation of CD4+ and CD8+ T-cells collected before (t = 0) and t = 3 and t = 45 weeks after RFA from patient ITV-023 upon 6 days of stimulation with MAGEA9 or luciferase mRNA electroporated B-cell blasts. B. Heat map indicates fold-changes in proliferation of peripheral CD4+ and CD8+ T-cells upon stimulation with individual CTAs compared to stimulation with luciferase (colored legend). CTA-specific T-cell responses were considered positive with a ≥ two-fold increase compared to lucifase-stimulated T-cell proliferation was observed. For comparison, proliferative responses against the onco-fetal tumor-associated antigen GPC3 were measured in 11 patients and depicted as fold changes compared to stimulation with eGFP. Patient ITV-016 was included after initial RFA and after a second RFA 13 weeks after the first treatment due to rapid cancer recurrence. Annotation above the heat map indicates the etiology of HCC, AFP levels before treatment, and the treatment (RFA or TACE) which the patients received. Annotation below the heat map indicates the actual weeks after treatment at which blood samples of the individual patients were collected (t = 0: before local tumor ablation).
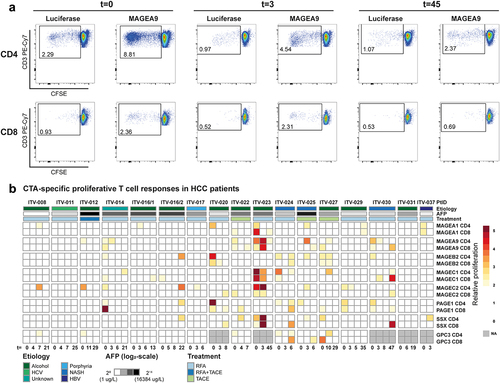
In conclusion, proliferative CD4+ T-cell and CD8+ T-cell responses were found against one or more of the tested CTAs in 14 out of 16 analyzed HCC patients, but were not enhanced by local ablative therapies.
CTA-peptide-specific stimulation induces pro-inflammatory cytokine and granzyme secretion by PBMC of HCC patients
To investigate whether our selection of CTAs could induce effector responses in T-cells, we tested the production of cytokines and cytotoxic molecules secreted upon stimulation of PBMC. For this purpose, we included 8 HCC patients (Supplementary Tables S1A and S1B) and tested 8 different HLA class I epitopes derived from five CTAs (MAGEA1, MAGEA9. MAGEB2, MAGEC1, SSX2). The selected epitopes were matched with the HLA class I types of the included HCC patients (Supplementary Tables S4-S5). Five (out of the eight) epitopes had also been included in HLA-A:02:01 dextramer binding analysis of CD8+ T-cells (Supplementary Table S3). PBMC were pre-stimulated with pools of a maximum of five peptides for 14 days and re-stimulated with single peptides for 2 days after which secreted molecules were quantified. Due to limited numbers of PBMC, we could not test each epitope in all patients. Nevertheless, we observed that all CTA-epitopes stimulated production of IL-2, Granzyme B and/or TNFα in one or more patients (). Interestingly, combined production of 2 or 3 of these pro-inflammatory factors against CTA epitopes was identified 13 and 5 times across patients and time points, respectively (). Production levels were similar to those invoked by a known immunogenic peptide of GPC3 (). It is noteworthy that cytokine production was also observed in response to two MAGEA1 peptides (MAGEA1161-169 and MAGEA1289-298) that are thus far only described as immunogenic in context of HLA-types not expressed in these 8 HCC patients. These peptides were included in our study based on in silico prediction of HLA-binding properties (indicated with an asterisk in Supplementary Table S4 and underlined in ).
Figure 4. CTA-specific peptide stimulation induces cytokine and granzyme B production in PBMC of HCC patients. Peptide-specific induction of cytokine and granzyme B (GrB) production was measured after re-stimulation of expanded PBMC of 8 HCC patients prior to local ablative treatment (t = 0) and at the indicated weeks after intervention. A. Heatmaps depicting the fold changes for production of the indicated cytokines and GrB in the peptide stimulated condition compared to the solvent-containing medium condition. Peptides with a ≥ 2 fold change were determined to have evoked a peptide-specific response and are depicted as such (color-coded legend). B. Heatmap displaying concomitant production of GrB, IL-2 and/or TNF-α in the combinations indicated by the color-coded legend. Annotation above the heat map indicates the etiology of HCC, AFP levels before treatment, the treatment (RFA or TACE) patients received, and HBV status. Underlined peptides were included in our study based on in silico prediction of HLA-binding properties.

Since 5 of the HCC-patients included in the peptide stimulation experiments had a (clinical history of) HBV infection, we additionally investigated functional responses against well-known HBV-derived HLA class I epitopes (Supplementary Figures S7-9). A total of 23 HBV-derived epitopes were tested, representing all HBV proteins and several common HLA-types. The production of IL-2, Granzyme B and/or TNFα was observed in response to 21 out of 23 epitopes, with numerous responses both in patients with an ongoing chronic (ITV-006 and ITV-007) or resolved (ITV-010 and ITV-021) HBV infection. The HBV parameters HBsAg and anti-HBsAg did not seem to relate to local ablative intervention, although numbers of patients are too few to draw definitive conclusions (Supplementary Figure S10).
HCC patients have IgG-responses against CTAs
To gain further insight into the immunogenic potential of the selected CTAs, we investigated humoral IgG responses by ELISA. We screened 105 plasma samples, obtained from 32 HCC patients, and 15 healthy subjects (negative controls) for IgG antibodies against MAGEA1, MAGEA9, MAGEB2, MAGEC2, PAGE1, and SSX2 recombinant full-length CTA proteins. We additionally investigated IgG titers against the established immunogenic tumor antigen NY-ESO-1 (Supplementary Table S6) as reference. We could not include MAGEC1 in antibody screening, because neither full-length nor MAGEC1934-1142 protein are commercially available. Compared to healthy subjects, we detected enhanced levels of specific IgG against every CTA in at least one HCC patient (). In 13 out of 32 HCC patients we found IgG specific for at least one CTA, and in 11 patients the anti-CTA antibodies were already present before tumor ablation. These preexisting humoral responses were generally stable over time, with titers ranging between 1:20 and 1:640 (). In comparison, two positive control sera from melanoma patients showed NY-ESO-1-specific IgG titers of 1:1000, and a positive control serum from a melanoma patient showed a MAGEA1-specific IgG titer of 1:200 (Supplementary Figure S11). De novo induction of CTA-specific IgG after local tumor ablation was observed in 4 patients.
Figure 5. HCC-patients mount CTA-specific IgG responses. CTA-specific IgG responses were measured in 32 HCC patients by indirect ELISA. ELISA plates were coated with human recombinant full length CTA protein, and after incubation with HCC patients’ plasma, CTA-specific IgG was detected by anti-human IgG-HRP. After screening of 1:20 plasma dilutions, the positive samples were titrated. A. Heat map displaying the CTA-specific IgG responses in individual before and at different time points (weeks) after local tumor ablation. Columns show IgG titers against the 7 different CTAs. Colors indicate the detected titers (legend). B, C. Examples of serial dilutions of plasma samples of 3 HCC-patients in MAGEA1 ELISA (b) and of 2 patients in MAGEC2 ELISA (c). The delta optical density (ΔOD) of each plasma sample was calculated by subtracting the sample’s OD in wells coated with BSA from the OD in wells coated with the specific CTA protein. In each ELISA, plasma samples of 4 healthy individuals (indicated by E numbers) were included as negative controls to determine the cutoff OD-value that was used to determine positivity of patient samples. The dotted line indicates the cutoff for each dilution, which was determined by the average plus 10 times the standard deviation of the OD-values of 4 healthy control samples.
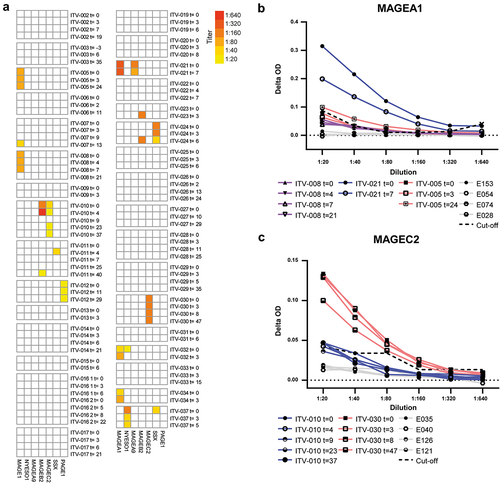
Taken together, IgG responses against all 7 CTAs were observed in our HCC patient cohort. In general, local tumor ablation may induce de novo anti-CTA IgG responses or enhance CTA-specific IgG titers; however, numbers are too small to draw any conclusions.
Discussion/conclusion
Recently, we have identified a panel of 12 CTAs prevalent in HCC, but absent from healthy tissue except immune-privileged germ cells. Moreover, we demonstrated that expression of these CTAs in non-tumor liver tissues was associated with early cancer recurrence after primary tumor resection and may indicate presence of occult micro-metastases or pre-malignant transformation of liver tissue. Therefore, we postulated that vaccination strategies targeting these CTAs in early-stage HCC may have clinical utility as adjuvant therapy to prevent cancer recurrence after primary tumor resection.Citation22 In advanced HCC such vaccination approach may synergize with ICPI therapy.
Here, we comprehensively studied the immunogenicity of six of these pre-defined CTAs and of SSX-2 in HCC patients before and after local tumor ablation. We demonstrate systemic IgG responses against one or more CTAs in 40% of HCC patients and establish the presence of CD4+ and CD8+ T-cells which mount proliferative responses against one or more CTAs in the circulation of most HCC patients. In addition, we found higher frequencies of circulating CTA peptide-specific CD8+ T-cells in HCC patients compared to healthy subjects. Moreover, we demonstrate that HLA class I epitopes derived from five of the CTAs induce cytokine and granzyme B production in PBMC, indicating that CTA-specific T-cells, most likely CD8+, in HCC patients are functionally competent.
Whereas MAGEA1-, MAGEC1, MAGEC2-, and SSX2-specific CD8+ T-cells have been detected in HCC patients in previous studies,Citation23,Citation29,Citation37,Citation40–43 the current study is the first to demonstrate MAGEA9, MAGEB2, and PAGE1-specific CD8+ T-cell responses in HCC patients. Using CTA peptide-loaded HLA class I multimers, we observed that already prior to ablative therapy more than 50% of CTA-specific CD8+ T-cells displayed a phenotype that suggested they were antigen experienced. It should be noted that for these multimer experiments CTA peptides were pooled with a peptide from NY-ESO and hence part of the responses may have been directed against this CTA that was not focus of our study. Staining for single multimers, however, indicated T cells against the other CTA contributed as well. We did not find clear signs of recent activation, chronic activation or exhaustion on CTA peptide-specific CD8+ T-cells. The observed proliferative CD8+ T-cell responses against the CTAs together with the observed cytokine and granzyme B production upon stimulation of PBMC with HLA class I CTA epitopes, indicate that CTA-specific CD8+ T-cells in HCC patients are functionally competent. Although viral epitopes, due to their foreign nature,Citation44 are considered to be more immunogenic than CTA epitopes, the strength of cytokine and granzyme B production upon stimulation of PBMC with either HBV or CTA peptides was comparable. Moreover, the maximum proliferative CD8+ T-cell responses against all CTAs tested were stronger than those against the well-known immunogenic HCC-associated antigen GPC3.Citation16,Citation39 Together, these data may suggest that tumor-restricted CTAs are sufficiently immunogenic to stimulate functional CD8+ T-cell responses in HCC patients. This is in accordance with recent data generated by us and others that show abundant presence of peptides derived from CTA such as MAGEA1, MAGEB2, and MAGEC2 in HLA class I restricted epitopes of HCC tumors.Citation45
CD4+ T-cells are needed for optimal expansion of CD8+ T-cells and for CD8+ T-cell memory and are therefore indispensable for adequate and sustained anti-tumor immunity.Citation46 Recently, also the direct anti-tumor potency of CD4+ T-cells was demonstrated, as well as their role in directing a sustained anti-tumor response.Citation47 Nevertheless, CD4+ T-cell responses against CTAs have been rarely studied in HCC. To our knowledge, we are the first to demonstrate CD4+ T-cell responses against MAGEA1-, MAGEA9, MAGEB2, MAGEC1, MAGEC2-, PAGE1, and SSX2 in HCC patients, while confirming the presence of MAGEC2-specific CD4+ T-cells in HCC.Citation29,Citation48 Similar to CD8+ T-cell responses, the maximum proliferative CD4+ T-cell responses against CTAs were stronger than those against GPC3.
In contrast to T-cell immunogenicity, the role of tumor-specific antibodies in anti-tumor immunity is not yet clear and a frequently debated topic. However, antibody presence is at least an additional indication for antigen immunogenicity, and could be a sign of the presence of CD4+ T helper cells recognizing the antigen.Citation47 Moreover, tumor-specific antibodies can form immune complexes with TAAs and thereby facilitate cross-presentation by dendritic cells.Citation49 Even antibodies against intracellular TAAs may contribute via this mechanism to anti-tumor immunity upon release of the TAA from tumor cells.Citation50 Importantly, several studies demonstrated positive associations between humoral responses against TAAs and therapeutic outcome after ICPI therapy or vaccination with TAA-containing cancer vaccines.Citation51–53 We detected IgG responses against every tested CTA in our cohort of 32 HCC patients. However, the prevalence of IgG responses against most individual CTAs was rather low. For instance, serum IgG against the established immunogenic tumor antigen NY-ESO-1 was detected in only 2 patients, but this is in line with its low prevalence in HCC.Citation48,Citation54,Citation55 The most frequently detected humoral response was directed against MAGEA1 (in 6 patients). Antibodies against these CTAs have been detected in other types of cancers,Citation36,Citation56–60 but in HCC patients humoral responses against these CTAs, excluding anti-NY-ESO-1, have not been previously assessed. The titers of CTA-specific IgGs were comparable to previously reported anti-NY-ESO-1 IgG titers in HCC patientsCitation48,Citation54,Citation55 and IgG titers against other CTAs in patients with other types of cancer.Citation58 The presence of both IgG- and T-cell responses provides further evidence for their immunogenicity of studied CTA in HCC patients.
Whereas previous studies have reported transient increases in systemic T-cell responses against TAAs between 2 and 4 weeks after local tumor ablation in HCC patients,Citation26,Citation27,Citation37 we did not find any increase in CTA-specific T-cell responses at 3 weeks or later time points after RFA and/or TACE. This discrepancy may be caused by several factors. Most importantly, we analyzed T-cell responses against tumor-restricted CTAs, whereas Zerbini et alCitation26 studied T-cell responses against autologous tumor lysate and Mizukoshi et alCitation27,Citation37 quantified T-cell responses against a panel of TAAs which are also expressed, although at lower levels, in healthy tissues.Citation61–64 Furthermore, the patient cohorts differ quite significantly, as only a few patients in our cohort had HCC caused by chronic viral hepatitis, whereas this was the major etiology in these previous studies. Finally, whereas TAA-specific T-cells were detected by IFN-γ ELISPOT assays in the previous studies, we used three other techniques. A recent study reported transiently enhanced T-cell responses against MAGEA1, MAGEC1 and a few other tumor-restricted CTAs, at 1 week after microwave ablation in 30% of HCC patients.Citation43 Because the earliest time point at which we analyzed T-cell responses was at 3 weeks after local tumor ablation, we might have missed such early T-cell responses. Changes in circulating T-cell and NK cell subsets after tumor ablation in HCC patients were recently shown to be detectable only at 1 day after RFA,Citation65 which explains that we did not observe such effects.
The present study provides a comprehensive overview of systemic adaptive immune responses against tumor-restricted CTAs in HCC published to date. Despite demonstrating CD4+, CD8+ and IgG responses against a large set of CTAs in HCC patients for the first time, we acknowledge that our study has some limitations. Not all included patients were treatment naïve before inclusion in the study, and therefore it is unclear whether the detected CTA-specific immune responses can be considered spontaneous. Second, due to unavailability of reagents such as in vitro transcribed mRNA for several CTAs, we could not study the immunogenicity of 6 additional tumor-restricted CTAs we previously described in HCC. Importantly, we were able to include the most prevalent CTAs in HCC; MAGEA1 (found in 59% of patients), MAGEC1 (48%), and MAGEC2 (56%).Citation22 Third, due to limitations in numbers of PBMC, we could not apply all three techniques for analysis of T-cell responses in all individual patients. This, combined with the usage of different techniques, makes direct comparison of the different results difficult, yet we observed that overall, some patients displayed broader responses than others. Fourth, due to unavailability of tumor biopsies, we could not analyze whether presence and/or intensity of CTA-specific immune responses in individual patients was related to CTA expression in the tumors and could not correlate CTA expression to outcome after local ablative therapies. Last but not least, to establish the therapeutic potential of each of these TAAs, further (pre-)clinical studies are needed.
In conclusion, the presence of CD4+ and CD8+ T-cell and IgG responses against seven tumor-restricted CTAs shows the immunogenicity of these CTAs in HCC-patients. Further research should elucidate whether vaccines based on these tumor-specific antigens can boost preexisting CTA-specific immunity sufficiently to combat small malignant foci remaining after primary tumor resection in early HCC and/or to enhance therapeutic efficacy of ICPI blockade in advanced HCC.
Consent to participate statement
Written informed consent was obtained from participants.
Study approval statement
This study protocol was reviewed and approved by the local medical ethical committee of the Erasmus Medical Center Rotterdam, approval number MEC-2015-563.”
Author contributions
K, DS and SB conceived the idea and designed the study. LN, MdB and SM performed most of the experiments. PB, VdR and RL provided assistance with experiments. LN and MdB analyzed the data and wrote the manuscript. LN conducted sample analysis and provided clinicopathological data. JIJ provided patient samples and obtained consent from patients. JK, DS, MB and SB supervised the study. IV and US supported experimental design. LN, MdB, SB and JK revised the manuscript with input from all authors.
Supplemental Material
Download Zip (2.5 MB)Acknowledgments
We would like to thank Dr. Hanneke van Vuuren (Department of Gastroenterology and Hepatology, Erasmus MC, for providing us with frozen plasma samples of healthy blood bank donors. Finally, we would like to thank Prof. Dr. Stefan Eichmüller from the department of GMP & T-cell therapy of the Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany, for providing us with positive control sera for the CTA-specific IgG ELISAs.
Disclosure statement
The following authors have securities from BioNTech: Isabel Vogler, Ugur Sahin. The remaining authors disclose no conflicts.
Data availability statement
All data generated or analyzed during this study are included in this article or its supplementary material files. Further enquiries can be directed to the corresponding author.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2131096
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–14. doi:10.3322/caac.21492.
- Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3.
- Brown ZJ, Greten TF, Heinrich B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy. Hepatology. 2019;70(4):1437–1442. doi:10.1002/hep.30633.
- Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30010-2.
- Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, Kudo M, Breder V, Merle P, Kaseb AO, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745.
- Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6(9):990–1000. doi:10.1158/2326-6066.CIR-18-0277.
- Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. doi:10.1038/s41573-018-0007-y.
- Sangro B, Melero I, Wadhawan S, Finn RS, Abou-Alfa GK, Cheng A-L, Yau T, Furuse J, Park J-W, Boyd Z, et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J Hepatol. 2020;73(6):1460–1469. doi:10.1016/j.jhep.2020.07.026.
- Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, Nederlof I, Kluin RJC, Warren S, Ong S, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–928. doi:10.1038/s41591-019-0432-4.
- Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67–73. doi:10.1001/jamaoncol.2018.4051.
- Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, Maurus D, Schwarck-Kokarakis D, Kuhn AN, Omokoko T, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585(7823):107–112. doi:10.1038/s41586-020-2537-9.
- De Keersmaecker B, Claerhout S, Carrasco J, Bar I, Corthals J, Wilgenhof S, Neyns B, Thielemans K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: link between T-cell activation and clinical responses in advanced melanoma. J Immunother Cancer. 2020;8(1):1. doi:10.1136/jitc-2019-000329.
- Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi:10.1038/nature23003.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi:10.1038/nature22991.
- Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, Mizuno S, Ishii H, Nakachi K, Konishi M, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18(13):3686–3696. doi:10.1158/1078-0432.CCR-11-3044.
- Tsuchiya N, Yoshikawa T, Fujinami N, Saito K, Mizuno S, Sawada Y, Endo I, Nakatsura T. Immunological efficacy of glypican-3 peptide vaccine in patients with advanced hepatocellular carcinoma. Oncoimmunology. 2017;6(10):e1346764. doi:10.1080/2162402X.2017.1346764.
- Charneau J, Suzuki T, Shimomura M, Fujinami N, Nakatsura T. Peptide-based vaccines for hepatocellular carcinoma: a review of recent advances. J Hepatocell Carcinoma. 2021;8:1035–1054. doi:10.2147/JHC.S291558.
- de Beijer MTA, Bezstarosti K, Luijten R, Doff WAS, Boor PPC, Pieterman RFA, Bouzid R, Biesta PJ, Ijzermans JNM, Doukas M, et al. Immunopeptidome of hepatocytes isolated from patients with HBV infection and hepatocellular carcinoma. JHEP Reports.2022;4(11):100576. doi:10.1016/j.jhepr.2022.100576.
- Loffler MW, Gori S, Izzo F, Mayer-Mokler A, Ascierto PA, Königsrainer A, Ma YT, Sangro B, Francque S, Vonghia L, et al. Phase I/II multicenter trial of a novel therapeutic cancer vaccine, hepaVac-101, for hepatocellular carcinoma. Clin Cancer Res. 2022;28(12):2555–2566. doi:10.1158/1078-0432.CCR-21-4424.
- Hofmann O, Caballero OL, Stevenson BJ, Chen Y-T, Cohen T, Chua R, Maher CA, Panji S, Schaefer U, Kruger A, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A. 2008;105(51):20422–20427. doi:10.1073/pnas.0810777105.
- Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21(6):360–378. doi:10.1038/s41568-021-00346-0.
- Noordam L, Ge Z, Ozturk H, Doukas M, Mancham S, Boor PPC, Campos Carrascosa L, Zhou G, van den Bosch TPP, Pan Q, et al. Expression of cancer testis antigens in tumor-adjacent normal liver is associated with post-resection recurrence of hepatocellular carcinoma. Cancers (Basel). 2021;13(10):10. doi:10.3390/cancers13102499.
- Bricard G, Bouzourene H, Martinet O, Rimoldi D, Halkic N, Gillet M, Chaubert P, MacDonald HR, Romero P, Cerottini J-C, et al. Naturally acquired MAGE-A10- and SSX-2-Specific CD8+T cell responses in patients with hepatocellular carcinoma. J Immunol. 2005;174(3):1709–1716. doi:10.4049/jimmunol.174.3.1709.
- Smith HA, McNeel DG. The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin Dev Immunol. 2010;2010:150591. doi:10.1155/2010/150591.
- Wang C, Gu Y, Zhang K, Xie K, Zhu M, Dai N, Jiang Y, Guo X, Liu M, Dai J, et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat Commun. 2016;7(1):10499. doi:10.1038/ncomms10499.
- Zerbini A, Pilli M, Penna A, Pelosi G, Schianchi C, Molinari A, Schivazappa S, Zibera C, Fagnoni FF, Ferrari C, et al. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res. 2006;66(2):1139–1146. doi:10.1158/0008-5472.CAN-05-2244.
- Mizukoshi E, Yamashita T, Arai K, Sunagozaka H, Ueda T, Arihara F, Kagaya T, Yamashita T, Fushimi K, Kaneko S, et al. Enhancement of tumor-associated antigen-specific T cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology. 2013;57(4):1448–1457. doi:10.1002/hep.26153.
- Tang A, Bashir MR, Corwin MT, Cruite I, Dietrich CF, Do RKG, Ehman EC, Fowler KJ, Hussain HK, Jha RC, et al. Evidence supporting LI-RADS major features for CT- and MR imaging-based diagnosis of hepatocellular carcinoma: a systematic review. Radiology. 2018;286(1):29–48. doi:10.1148/radiol.2017170554.
- Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, Pedroza-Gonzalez A, Polak WG, de Jonge J, Gaspersz M, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology. 2017;153(4):1107–19 e10. doi:10.1053/j.gastro.2017.06.017.
- Anderson LD Jr., Cook DR, Yamamoto TN, Berger C, Maloney DG, Riddell SR. Identification of MAGE-C1 (CT-7) epitopes for T-cell therapy of multiple myeloma. Cancer Immunol Immunother. 2011;60(7):985–997. doi:10.1007/s00262-011-1009-3.
- Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, Huber C, Türeci Ö, Sahin U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2008;180(1):309–318. doi:10.4049/jimmunol.180.1.309.
- van Beek AA, Zhou G, Doukas M, Boor PPC, Noordam L, Mancham S, Campos Carrascosa L, Heide‐Mulder M, Polak WG, Ijzermans JNM, et al. GITR ligation enhances functionality of tumor-infiltrating T cells in hepatocellular carcinoma. Int J Cancer. 2019;145(4):1111–1124. doi:10.1002/ijc.32181.
- Dolton G, Zervoudi E, Rius C, Wall A, Thomas HL, Fuller A, Yeo L, Legut M, Wheeler S, Attaf M, et al. Optimized peptide-MHC multimer protocols for detection and isolation of autoimmune T-cells. Front Immunol. 2018;9:1378. doi:10.3389/fimmu.2018.01378.
- de Beijer MTA, Jansen D, Dou Y, van Esch WJE, Mok JY, Maas MJP, Brasser G, de Man RA, Woltman AM, Buschow SI. Discovery and Selection of hepatitis B virus-derived T cell epitopes for global immunotherapy based on viral indispensability, conservation, and HLA-binding strength. J Virol. 2020;94(7662):7. doi:10.1128/JVI.01663-19.
- Gnjatic S, Old LJ, Chen YT. Autoantibodies against cancer antigens. Methods Mol Biol. 2009;520:11–19.
- Zornig I, Halama N, Lorenzo Bermejo J, Ziegelmeier C, Dickes E, Migdoll A, Kaiser I, Waterboer T, Pawlita M, Grabe N, et al. Prognostic significance of spontaneous antibody responses against tumor-associated antigens in malignant melanoma patients. Int J Cancer. 2015;136(1):138–151. doi:10.1002/ijc.28980.
- Mizukoshi E, Nakamoto Y, Arai K, Yamashita T, Sakai A, Sakai Y, Kagaya T, Yamashita T, Honda M, Kaneko S, et al. Comparative analysis of various tumor-associated antigen-specific t-cell responses in patients with hepatocellular carcinoma. Hepatology. 2011;53(4):1206–1216. doi:10.1002/hep.24149.
- Solana R, Tarazona R, Aiello AE, Akbar AN, Appay V, Beswick M, Bosch JA, Campos C, Cantisán S, Cicin-Sain L, et al. CMV and Immunosenescence: from basics to clinics. Immun Ageing. 2012;9(1):23. doi:10.1186/1742-4933-9-23.
- Wu Y, Liu H, Ding H. GPC-3 in hepatocellular carcinoma: current perspectives. J Hepatocell Carcinoma. 2016;3:63–67. doi:10.2147/JHC.S116513.
- Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, Schemmer P, Bruns H, Eiermann T, Price DA, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59(4):1415–1426.
- Gehring AJ, Ho ZZ, Tan AT, Aung MO, Lee KH, Tan KC, Lim SG, Bertoletti A. Profile of tumor antigen-specific CD8 T cells in patients with hepatitis B virus-related hepatocellular carcinoma. Gastroenterology. 2009;137(2):682–690.
- Tauber C, Schultheiss M, Luca R, Buettner N, Llewellyn-Lacey S, Emmerich F, Zehe S, Price DA, Neumann-Haefelin C, Schmitt-Graeff A, et al. Inefficient induction of circulating TAA-specific CD8+ T-cell responses in hepatocellular carcinoma. Oncotarget. 2019;10(50):5194–5206. doi:10.18632/oncotarget.27146.
- Leuchte K, Staib E, Thelen M, Godel P, Lechner A, Zentis P, Garcia-Marquez M, Waldschmidt D, Datta RR, Wahba R, et al. Microwave ablation enhances tumor-specific immune response in patients with hepatocellular carcinoma. Cancer Immunol Immunother. 2021;70(4):893–907. doi:10.1007/s00262-020-02734-1.
- Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, de Silva AD, Sette A, Kesmir C, Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9(10):e1003266. doi:10.1371/journal.pcbi.1003266.
- Dong LQ, Peng LH, Ma LJ, Liu DB, Zhang S, Luo SZ, Rao JH, Zhu HW, Yang SX, Xi SJ, et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J Hepatol. 2020;72(5):896–908.
- Borst J, Ahrends T, Babala N, Melief CJM, Kastenmüller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–647. doi:10.1038/s41577-018-0044-0.
- Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2020;28(1–2):5–17. doi:10.1038/s41417-020-0183-x.
- Korangy F, Ormandy LA, Bleck JS, Klempnauer J, Wilkens L, Manns MP, Greten TF. Spontaneous tumor-specific humoral and cellular immune responses to NY-ESO-1 in hepatocellular carcinoma. Clin Cancer Res. 2004;10(13):4332–4341. doi:10.1158/1078-0432.CCR-04-0181.
- Luetkens T, Kobold S, Cao Y, Ristic M, Schilling G, Tams S, Bartels BM, Templin J, Bartels K, Hildebrandt Y, et al. Functional autoantibodies against SSX-2 and NY-ESO-1 in multiple myeloma patients after allogeneic stem cell transplantation. Cancer Immunol Immunother. 2014;63(11):1151–1162. doi:10.1007/s00262-014-1588-x.
- Noguchi T, Kato T, Wang L, Maeda Y, Ikeda H, Sato E, Knuth A, Gnjatic S, Ritter G, Sakaguchi S, et al. Intracellular tumor-associated antigens represent effective targets for passive immunotherapy. Cancer Res. 2012;72(7):1672–1682. doi:10.1158/0008-5472.CAN-11-3072.
- Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, Xu Y, Pogoriler E, Terzulli SL, Kuk D, et al. Integrated NY-ESO-1 antibody and CD8+T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A. 2011;108(40):16723–16728. doi:10.1073/pnas.1110814108.
- Takayama K, Sugawara S, Saijo Y, Maemondo M, Sato A, Takamori S, Harada T, Sasada T, Kakuma T, Kishimoto J, et al. Randomized Phase II study of docetaxel plus personalized peptide vaccination versus docetaxel plus placebo for patients with previously treated advanced wild type EGFR non-small-cell lung cancer. J Immunol Res. 2016;2016:1745108. doi:10.1155/2016/1745108.
- Somaiah N, Block MS, Kim JW, Shapiro GI, Do KT, Hwu P, Eder JP, Jones RL, Lu H, ter Meulen JH, et al. First-in-Class, First-in-human study evaluating LV305, a dendritic-cell tropic lentiviral vector, in sarcoma and other solid tumors expressing NY-ESO-1. Clin Cancer Res. 2019;25(19):5808–5817. doi:10.1158/1078-0432.CCR-19-1025.
- Oshima Y, Shimada H, Yajima S, Nanami T, Matsushita K, Nomura F, Kainuma O, Takiguchi N, Soda H, Ueda T, et al. NY-ESO-1 autoantibody as a tumor-specific biomarker for esophageal cancer: screening in 1969 patients with various cancers. J Gastroenterol. 2016;51(1):30–34. doi:10.1007/s00535-015-1078-8.
- Nakamura S, Nouso K, Noguchi Y, Higashi T, Ono T, Jungbluth A, Chen Y-T, Old LJ, Nakayama E, Shiratori Y, et al. Expression and immunogenicity of NY-ESO-1 in hepatocellular carcinoma. J Gastroenterol Hepatol. 2006;21(8):1281–1285. doi:10.1111/j.1440-1746.2006.04271.x.
- Michels J, Becker N, Suciu S, Kaiser I, Benner A, Kosaloglu-Yalcin Z, Agoussi S, Halama N, Pawlita M, Waterboer T, et al. Multiplex bead-based measurement of humoral immune responses against tumor-associated antigens in stage II melanoma patients of the EORTC18961 trial. Oncoimmunology. 2018;7(6):e1428157. doi:10.1080/2162402X.2018.1428157.
- Djureinovic D, Dodig-Crnkovic T, Hellstrom C, Holgersson G, Bergqvist M, Mattsson JSM, Pontén F, Ståhle E, Schwenk JM, Micke P, et al. Detection of autoantibodies against cancer-testis antigens in non-small cell lung cancer. Lung Cancer. 2018;125:157–163. doi:10.1016/j.lungcan.2018.09.012.
- Daudi S, Eng KH, Mhawech-Fauceglia P, Morrison C, Miliotto A, Beck A, Matsuzaki J, Tsuji T, Groman A, Gnjatic S, et al. Expression and immune responses to MAGE antigens predict survival in epithelial ovarian cancer. PLoS One. 2014;9(8):e104099. doi:10.1371/journal.pone.0104099.
- Stockert E, Jager E, Chen YT, Scanlan MJ, Gout I, Karbach J, Arand M, Knuth A, Old LJ. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med. 1998;187(8):1349–1354. doi:10.1084/jem.187.8.1349.
- Mizukami M, Hanagiri T, Baba T, Fukuyama T, Nagata Y, So T, Ichiki Y, Sugaya M, Yasuda M, Takenoyama M, et al. Identification of tumor associated antigens recognized by IgG from tumor-infiltrating B cells of lung cancer: correlation between Ab titer of the patient’s sera and the clinical course. Cancer Sci. 2005;96(12):882–888. doi:10.1111/j.1349-7006.2005.00119.x.
- Nakao M, Shichijo S, Imaizumi T, Inoue Y, Matsunaga K, Yamada A, Kikuchi M, Tsuda N, Ohta K, Takamori S, et al. Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J Immunol. 2000;164(5):2565–2574. doi:10.4049/jimmunol.164.5.2565.
- Kuhlmann WD, Peschke P. Hepatic progenitor cells, stem cells, and AFP expression in models of liver injury. Int J Exp Pathol. 2006;87(5):343–359. doi:10.1111/j.1365-2613.2006.00485.x.
- Chai J, He Y, Cai SY, Jiang Z, Wang H, Li Q, Chen L, Peng Z, He X, Wu X, et al. Elevated hepatic multidrug resistance-associated protein 3/ATP-binding cassette subfamily C 3 expression in human obstructive cholestasis is mediated through tumor necrosis factor alpha and c-Jun NH2-terminal kinase/stress-activated protein kinase-signaling pathway. Hepatology. 2012;55(5):1485–1494. doi:10.1002/hep.24801.
- Leao R, Apolonio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: clinical impacts in cancer. J Biomed Sci. 2018;25(1):22. doi:10.1186/s12929-018-0422-8.
- Rochigneux P, Nault JC, Mallet F, Chretien A-S, Barget N, Garcia AJ, Del Pozo L, Bourcier V, Blaise L, Grando-Lemaire V, et al. Dynamic of systemic immunity and its impact on tumor recurrence after radiofrequency ablation of hepatocellular carcinoma. Oncoimmunology. 2019;8(8):1615818. doi:10.1080/2162402X.2019.1615818.