
Abstract
Sexual reproduction involves fusion of 2 haploid gametes to form diploid offspring with genetic contributions from both parents. Gamete formation represents a unique developmental program involving the action of numerous germline-specific proteins. In an attempt to identify novel proteins involved in reproduction and embryonic development, we have carried out a proteomic characterization of the process in Caenorhabditis elegans. To identify candidate proteins, we used 2D gel electrophoresis (2DGE) to compare protein abundance in nucleus-enriched extracts from wild-type C. elegans, and in extracts from mutant worms with greatly reduced gonads (glp-4(bn2) worms reared at 25°C); 84 proteins whose abundance correlated with germline presence were identified. To validate candidates, we used feeding RNAi to deplete candidate proteins, and looked for reduction in fertility and/or germline cytological defects. Of 20 candidates so screened for involvement in fertility, depletion of 13 (65%) caused a significant reduction in fertility, and 6 (30%) resulted in sterility (<5 % of wild-type fertility). Five of the 13 proteins with demonstrated roles in fertility have not previously been implicated in germline function. The high frequency of defects observed after RNAi depletion of candidate proteins suggests that this approach is effective at identifying germline proteins, thus contributing to our understanding of this complex organ.
Abbreviations
| 2DGE | = | 2D gel electrophoresis |
| MALDI-TOF/TOF MS | = | matrix-assisted laser desorption ionization tandem time of flight mass spectrometry |
| SAGE | = | serial analysis of gene expression |
| RNAi | = | RNA interference |
| GO | = | gene ontology |
| COG | = | clusters of orthologous groups |
Introduction
Sexual reproduction in diploid eukaryotes involves formation of haploid gametes through a specialized cell division known as meiosis. Fusion of 2 haploid gametes at fertilization recreates diploidy in the next generation. Accurate segregation of chromosomes during meiosis is critical for successful sexual reproduction; errors in chromosome segregation result in aneuploid gametes, which have an incorrect chromosome complement (For review, see ref.Citation1). Errors during meiosis are common in humans, and result in fertility issues and birth defects.Citation2 In most eukaryotes, including humans, successful formation of haploid gametes involves completion of a choreographed series of events involving pairing of and genetic exchange between homologous chromosome pairs in germ cells undergoing meiosis. This process is conserved among eukaryotes, and is fundamental for reproductive success in most eukaryotes.Citation3
The production of gametes requires major changes to cellular activity; in addition to an altered cell cycle, many germline-specific proteins are also involved.Citation4 Despite years of study, fundamental gaps still remain in our understanding of this complex process. Traditional genetic approaches have identified many proteins involved in meiosis/gametogenesis, but the infertility associated with mutations in proteins involved in these processes complicates traditional forward genetic analyses. Cytological examination of cells undergoing meiosis can directly reveal defects in chromosome behavior during meiosis; however, the labor-intensive nature of cytology makes its application in large-scale screens challenging. The advent of genome sequencing has enabled systems biology approaches such as transcriptomics and proteomics. Here, we describe a combined approach involving differential proteomic identification of germline proteins, followed by validation through RNAi-mediated depletion of candidates, using the tractable metazoan Caenorhabditis elegans.
C. elegans is an ideal model organism for the study of sexual reproduction.Citation5 Adult nematodes are roughly half gonad (). Thus, gamete production represents a substantial proportion of total cellular activity in this organism.Citation6,7 The worm gonad includes stem cells, mitotically dividing cells, and cells undergoing meiosis and gametogenesis; these cell types are arranged in a spatiotemporal gradient within the gonad, such that cell position within the gonad can be used to infer cell type / stage. The transparent nature of these animals, coupled with their large gonads, facilitates cytological examination of chromosome behavior during gamete production and meiosis. And finally, the fact that hermaphrodites are self-fertile simplifies genetic assays for detection of germline defects.
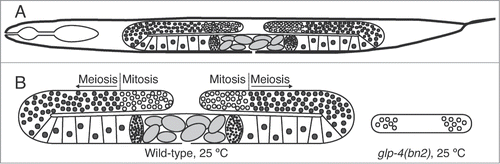
Figure 1. C. elegans gonad anatomy. (A) schematic depiction of hermaphrodite anatomy, showing the 2 gonad arms. (B) Schematic depiction of normal gonads (left) and gonads derived from glp-4(bn2) worms reared at 25°C. In a normal gonad, each arm contains roughly 1000 germ cells. In glp-4(bn2) animals, each gonad arm contains roughly 10 germ cells.
Large-scale RNA expression profiling of germline gene expression in C. elegans has identified many RNA transcripts that are enriched in the germline.Citation8-10 This list of candidate genes has been used to direct more targeted analysis of the roles of candidate gene products in meiosis and gametogenesis.Citation21-23 However, expression profiling is blind to much post-transcriptional regulation; in addition, cellular protein levels do not always directly correspond to RNA transcript levels.Citation11 Thus, there is value in direct determination of protein abundance in the germline through a proteomic approach.
The production of gametes involves extensive alterations to the cellular proteome—for example, a large multiprotein complex known as the synaptonemal complex assembles along homologous chromosomes during prophase I of meiosis.Citation12 Thus, gamete production is particularly amenable to a proteomic approach. A number of previous studies have used proteomics to identify proteins present in the C. elegans germline.Citation13-16 However, these previous studies have either been relatively limited in scope,Citation13,14 or required cellularization of gametes and thus were targeted toward the latter stages of gametogenesis.Citation15,16 The present work describes an in-depth proteomic characterization of all stages of gamete production in C. elegans.
The gene glp-4 (GermLine Proliferation) encodes a protein involved in germ cell proliferation.Citation6 In the experiments described herein, we take advantage of a specific temperature-sensitive allele of the glp-4 gene. glp-4(bn2ts) embryos raised at 25°C develop into adults with normal somatic structures but underdeveloped gonads containing roughly 10 germ nuclei (which are arrested prior to entry into meiosis; ).Citation6 In contrast, glp-4(bn2) worms raised at 16°C or wild-type worms raised at 16°C or 25°C contain roughly 2000 germ nuclei.Citation6,7 This allows large-scale preparation of worms with normal somatic structures, but with a ∼200-fold reduction in the number of germ nuclei. In an effort to identify proteins involved in gamete production, we isolated proteins from glp-4(bn2) worms reared at the restrictive temperature (25°C), representing the germline (−) sample, and wild type reared at 25°C (germline (+)). A major focus of research in our lab is meiotic chromosome behavior; thus, we carried out subcellular fractionation to enrich for nuclei (and thus chromosomes and chromosome-associated proteins). Comparison of proteins present in these samples, through 2-dimensional gel electrophoresis (2DGE) and matrix-assisted laser desorption ionization tandem time of flight mass spectrometry (MALDI-TOF/TOF MS), allows identification of candidate proteins involved in germline function, as they will be present in higher abundance in the germline (+) sample, while controlling for temperature-induced changes in protein abundance. A subset of identified proteins was then subjected to RNAi-mediated depletion, followed by phenotypic characterization. In this way, candidate proteins were directly evaluated for roles in germline function.
Through this combined approach, we expand the known C. elegans germline proteome, identifying 84 proteins whose abundance is correlated with the presence of germ cells. Functional analyses suggest that several previously uncharacterized proteins identified herein play a role in reproduction and/or embryonic development. Further characterization of these proteins is thus likely to enhance our understanding of the conserved process of gamete production.
Results and Discussion
Identification of germline-enriched proteins
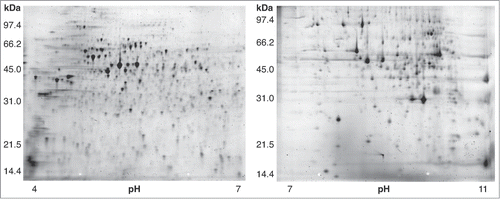
To identify proteins involved in reproduction and embryonic development, we used 2D gel electrophoresis to compare protein abundance in nucleus-enriched fractions isolated from age-matched young adult wild-type (germline(+)) and glp-4(bn2) (germline(−)) worms reared at 25°C (). Isolated proteins were first separated by pI on immobilized pH gradient strips (pH ranges used: 4–7 and 7–11); proteins were then separated by molecular weight via polyacrylamide gel electrophoresis. Each sample was analyzed in replicate; for each sample (germline (+) or (−)) and pH range (4–7 or 7–11), a minimum of 8 2D gels was run. Each gel was stained and imaged; gel images were then quantified through a software-based analysis that involved warping of gel images (to control for differences in migration) and quantification.
963 distinct protein spots could be resolved under our experimental conditions (507 in pH range 4–7; 456 in pH range 7–11) (). Of these spots, 36% (349) were significantly more abundant in the germline(+) samples (P < 0.05); 31% (294) were significantly more abundant in germline(−) samples.
Figure 2. 2D gels of nucleus-enriched protein extracts from wild type C. elegans. Left gel: isoelectric focusing done in pH range 4–7. Right gel: pH range 7–11. A total of 963 protein spots were resolved in this analysis (507 in pH range 4–7; 456 in pH range 7–11).
As our goal was to identify proteins involved in germline function, we focused our MS-based identification efforts on proteins whose abundance was positively correlated with the presence of a germline (see Experimental Section). In total, 144 protein spots (15% of total) were further analyzed (81 in pH range 4–7; 63 in pH range 7–11). From these spots, 84 unique proteins were successfully identified (50 in pH range 4–7; 36 in pH range 7–11; ; Table S1. Two proteins were identified from both pH 4–7 and pH 7-11 gels). These 84 germline-enriched proteins represent candidates for involvement in fertility.
Table 1. Identified proteins
Each of the 84 germline enriched proteins was manually assigned to a COG class, based on information present in WormBase.Citation17 Three major categories were identified: information storage and processing (23 proteins, 27% of all proteins), including proteins involved in chromosome segregation, nucleic acid metabolism, and protein synthesis; cellular processes and signaling (34 proteins, 40%), including proteins involved in membrane transport and cytoskeletal structure; and metabolism (24 proteins, 28%), including mitochondrial proteins. Five proteins (6%) are “poorly characterized.” These results underscore the fact that production of gametes requires large-scale changes in the proteome. For example, 15 of 84 proteins we identified (18%) are predicted to function in the mitochondrion. Similar results have been seen in other studies—of the 50 proteins identified as being part of the yeast meiosis proteome, 30% are mitochondrial in function.Citation18,19 This is not surprising: in adult worms, the majority of cell growth and division, which are both energy-intensive processes, takes place in the germline.Citation20-22 Thus, the abundance of mitochondrial proteins in adult worms will correlate with the presence of a germline. However, it is important to note that mitochondrial function can directly impact gamete production: for example, mutations in the mitochondrial protein SPD-3 have recently been demonstrated to affect meiotic chromosome behavior in C. elegans.Citation23
Comparison with germline transcriptome
To validate our approach, we compared our germline-enriched protein dataset (84 proteins) with the set of 1063 germline-enriched genes identified by Wang et al through Serial Analysis of Gene Expression (SAGE) analysis.Citation10 Of our 84 proteins, 25 (30%) correspond to genes identified as germline-enriched by SAGE. The observed overlap between the protein and SAGE data sets (25/84) is significantly different from that expected due to random chance (4/84; p = 2.2 × 10−5).
We also compared our germline-enriched dataset with the set of genes identified by microarray analysis as germline-enriched in adults.Citation8-10 17 proteins are present in both data sets; 9 proteins would be expected to be present in both sets based on random chance. This difference is not significant (p = 0.081). The lack of significant overlap between our dataset and that of Reinke et al likely reflects in part fundamental differences between the germline proteome and the germline transcriptome; RNA expression levels do not always correlate well with protein abundance.Citation11,24 However, the extensive and significant overlap between our data set and the germline SAGE dataset argues against this as sole explanation. Instead, we suggest that the lack of overlap between out data set and that of Reinke et al likely also reflects differences in sensitivity—our analysis was specifically targeted toward abundant proteins, while microarray analysis can evaluate expression differences for low-abundance transcripts as well (as discussed in ref.Citation10).
Gene ontology of germline proteome
As a first step in characterization, we used the DAVID classification tool to look for enriched gene ontology (GO) terms, by comparing GO terms associated with proteins in our dataset with those associated with all C. elegans proteins.Citation25 Of the 84 proteins in our data set, 41 (49%) are associated with the GO term ‘embryonic development ending in birth or hatching’ (GO:0009792); enrichment is also seen for terms related to aging/lifespan, growth, reproduction, and energy generation (; Table S2). With the exception of the aging/lifespan lists, more than 80% of proteins are shared between the smaller categories and “embryonic development.” This supports the idea that we have identified a suite of proteins expressed in the germline that are important in reproduction and early development of C. elegans.
Table 2. Gene ontology enrichment–main categories
Validation of candidate proteins through RNA interference
Our proteomic approach identified a list of germline-enriched proteins; however, demonstration that the identified proteins are involved in germline function requires phenotypic validation. To do so, we used RNAi to deplete candidate proteins, and then assayed fertility by determining the number of progeny produced. A reduction in fertility (compared to control worms) can reveal a role for the depleted protein in gamete production or embryogenesis, although depletion of somatic proteins can also result in reduced fertility. The 20 candidates selected for initial testing included proteins shown by prior analyses to contribute to fertility, as well as proteins for which roles in fertility had not previously been reported.
Of the 20 genes selected, depletion of 13 resulted in a significant reduction in fertility (). Six candidates resulted in production of less than 5% of control levels of progeny after RNAi; 7 other candidates had more moderate effects on fertility (35–75% of control).
Table 3. Phenotypic analyses of selected candidate proteins
Eleven of the candidates selected had no previously reported RNAi evidence for a role in fertility. Our experiments revealed a significant reduction in fertility for 5/11. For C31B8.8, a substantial reduction seen (to 38% of wild-type). For the other 4, a more modest reduction was seen (to 50–75% of control). These five proteins represent novel gametogenesis / embryogenesis factors in C. elegans.
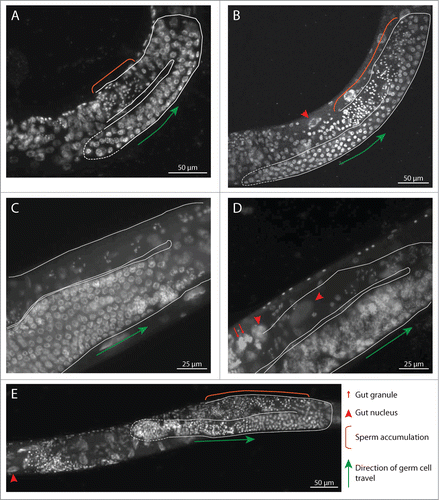
For nine of the candidate genes selected for RNAi, previous analyses had suggested roles in fertility or embryogenesis. Our RNAi experiments revealed a reduction in fertility for 8/9 (). For a subset of these, cytological examination of gonads from depleted animals recapitulated and/or extended known germline cellular defects associated with depletion of the cognate protein. For example, RNAi depletion of KLE-2 causes multiple germline defects indicative of problems with organization and segregation of chromosomes during germline cell divisions (; see also ref.Citation26). Cytological evidence for germline defects was also seen in gonads of animals depleted for B0393.1, F17C11.9, K07F5.13, R02D3.3, and T10F2.4 (; ).
Figure 3. Meiotic progression and nuclear morphology are aberrant in kle-2—depleted germlines. Gonads were dissected from wild type (top) or kle-2—depleted young adults, fixed, and DAPI stained. In each image, the distal tip of the gonad is on the left; nuclei proceed through meiosis and gametogenesis as they travel down the gonad. In the wild-type control, mitotic nuclei can be seen near the distal tip. The transition zone, characterized by asymmetrical chromatin within nuclei, marks the entry into meiosis. Adjacent to the transition zone, pachytene nuclei with paired chromosomes can be seen, with numerous diakinesis nuclei evident at the proximal end of the gonad (lower right). After kle-2 RNAi (bottom), the gonad architecture is dramatically altered: the overall number of nuclei is greatly reduced, suggesting germline proliferation defects. Most nuclei in the mitotic zone appear enlarged with punctate DNA staining, suggestive of chromosome fragmentation. Mitotic figures are absent, suggesting cell-cycle delay or arrest. Although some nuclei have a pachytene-like appearance, the transition zone is absent and the majority of nuclei in the meiotic region appear disorganized. There appear to be no diakinesis nuclei, and mature oocytes are absent. Thus, depletion of kle-2 results in dramatic defects in gamete production and fertility.
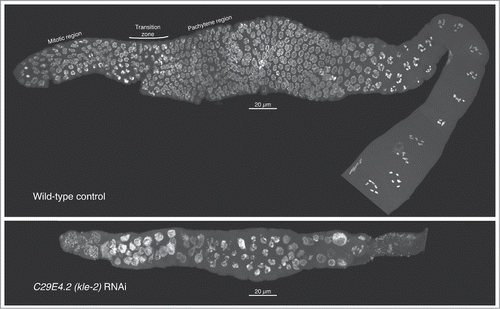
Figure 4. RNAi depletion of selected candidate proteins results in defects in gonad development. Whole hermaphrodites were ethanol fixed, DAPI stained, and imaged. In each image, the gonad is outlined in white; the distal tip is outlined with a dashed line. Orange brackets in (A, B, and D) demarcate inappropriate accumulation of sperm in the proximal gonad, suggestive of delayed development. Red arrows indicate gut granules; red arrowheads indicate gut nuclei. (A) B0393.1 RNAi, 24 hours post-L4. The gonad is smaller than WT, suggesting proliferation defects; in addition, sperm production continues (orange bracket). (B) F17C11.9 RNAi, 24 hrs post-L4. (C) WT control, 48 hrs post-L4. (D) K07F5.13 RNAi, 48 hrs post-L4. Chromatin condensation defects can be seen in early prophase nuclei (adjacent to green arrow); compare with similar region of gonad in (C) (E) T10F2.4 RNAi, hrs post-L4.
In this analysis, we saw a substantially higher proportion of phenotypes than has been seen in genome-wide RNAi screens; for example, in the genome-wide RNAi analysis carried out by Kamath et al.Citation27, 2.2% of RNAi targets resulted in a 10-fold or greater reduction in fertility. In this work, 30% of candidates tested by RNAi gave the same phenotype. Thus, our proteomic approach leads to a greater than 10-fold enrichment for this fertility phenotype over genome-wide RNAi screens. It is possible that this increased efficiency reflects our use of the C. elegans strain AZ212 as host for feeding RNAi, as this strain has been demonstrated to be more sensitive to RNAi.Citation28 However, it also likely reflects the direct advantage of targeted screening (the genes assayed were preselected for likely involvement in fertility); further, smaller-scale screens such as this also allow increased sensitivity of assays—by screening only a limited number of candidates, we can carry out assays allowing detection of more modest reductions in fertility than could be assayed in genome-wide screening.
In a related example of a targeted RNAi screen based on proteomic data, Skop et al. identified 160 candidate proteins for involvement in mammalian cytokinesis through a proteomic approach, and then used RNAi in C. elegans to validate their candidates.Citation29 33% of their candidates had been previously reported to display no RNAi phenotype (based on data from genome-wide RNAi screens), yet had demonstrable phenotypes in their targeted screen. Likewise, 25% (5/20) of candidates tested herein gave reduced fertility after RNAi, but had no previous RNAi phenotype reported from genome-wide screening. This demonstrates the advantage of targeted screening, and also underscores the fact that genome-wide RNAi screens have not detected all possible phenotypes.
In summary, these data indicate that our approach has successfully identified numerous proteins that contribute to fertility in C. elegans, including several proteins for which no previous role had been determined. As many of the identified proteins have homologs/ orthologs in mammals, it is possible that investigation of these proteins in higher organisms may lead to identification of novel mammalian fertility proteins.
Methods/Materials
Strains and culture conditions
C. elegans strains used for protein isolation were N2 (wild-type) and SS104 (glp-4(bn2)). Feeding RNAi was done in strain AZ212 (reported to have higher sensitivity to RNAi).Citation28 Standard strain propagation for proteomics took place at 16°C on nematode growth medium (NGM) plates seeded with E. coli strain OP50; liquid culture was done in S medium supplemented with OP50 at 25°C.Citation30 Feeding RNAi was carried out at 20°C.
Nematode culture for Protein Isolation
L4 hermaphrodite worms (N2 or SS104) were moved onto 100 mm NGM plates seeded with OP50 (10 L4 per plate; 8 plates per liter of liquid culture). Plates were incubated at 16°C until starved (roughly 10 days); at that point, the vast majority of worms on each plate were L1 larvae.
Worms were washed from starved plates into 250 or 500 mL baffle flasks containing S medium supplemented with OP50 bacteria; flasks were kept at 25°C, shaking at 200 rpm for 72 hours. After 72 hours, the vast majority of worms in each flask were age-matched young adults; at this point, most wild-type adults had not yet initiated embryogenesis (based on Nomarski microscopy). Worms were harvested by centrifugation, floated on 30% sucrose (to remove E. coli), washed with M9 buffer to remove sucrose, and then flash-frozen by adding dropwise to liquid N2.
Nucleus enrichment, protein isolation and two-dimensional gel electrophoresis
Subcellular fractionation and nucleus enrichment followed ref.Citation31, with some modifications. 1–2 grams of flash-frozen worm balls were ground with mortar and pestle using dry ice as grinding agent. Following grinding, samples were resuspended in buffer A+ (250 mM Sucrose, 10 mM MgCl2, 10 mM Tris, 1 mM EGTA (pH 8.0) supplemented with 0.35% 2-mercaptoethanol and Complete protease inhibitor cocktail (Roche, Indianapolis, IN, USA)). Nuclei were pelleted by centrifugation at 4000 × G (5 minutes, 4°C). Pelleted nuclei were resuspended in buffer A++ (A+ supplemented with 0.25% NP-40 and 0.1% Triton X-100), and Dounce homogenized (10 strokes with coarse pestle, 100 strokes with fine pestle).
Following Dounce homogenization, the solution was centrifuged at 50 × G (5 minutes, 4°C); this spin pellets cuticle shards and other large debris, but not nuclei. After removing the supernatant, the pellet was washed with buffer A+ (resuspend in buffer A+, pellet at 50 × G, collect supernatant) until few nuclei were seen in the supernatant after 50 × G centrifugation. The pooled, nucleus-enriched supernatant fraction was then pelleted at 4000 × G (5 minutes, 4°C), washed once with buffer A+, and pelleted again.
The resulting pellet was enriched for nuclei, based on Nomarski microscopy. However, the pellet collected also contained a significant number of mitochondria, as evidenced by the proteins with predicted mitochondrial function identified by MS (Results and Discussion).
After pelleting, the nucleus-enriched fraction was lysed in homogenization buffer (7 M urea, 2 M thiourea, 1% ASB-14 (amidosulfobetaine-14), 40 mM Tris base, 0.001% Bromophenol Blue, 0.5% immobilized pH gradient (IPG) buffer (either pH 4–7 or pH 7–11; GE Healthcare, Piscataway, NJ, USA), 40 mM DTT (dithiothreitol)) at a ratio of 1:4. Following lysis, proteins were precipitated by addition of 4 volumes 10% trichloroacetic acid in acetone, followed by overnight incubation at −20°C and centrifugation at 4°C. Pelleted proteins were washed with acetone and air-dried. To remove DNA, precipitated samples were resuspended in 1× DNase I buffer (10 mM Tris-HCl pH 7.6, 2.5 mM MgCl2, 0.5 mM CaCl2) + 1 mM PMSF; 0.01 volumes DNase I (New England Biolabs, Ipswich, MA, USA) was then added, and samples were incubated on ice 15 minutes. After DNase treatment, proteins were again TCA/acetone precipitated as above and then resuspended in rehydration buffer (7 M urea, 2 M thiourea, 2% CHAPS, 2% NP-40, 0.002% Bromophenol blue, 100 mM dithioerythritol, 0.1% IPG buffer). Protein concentration was determined using a 2D-Quant kit according to manufacturer's instructions (GE Healthcare, Uppsala, Sweden).
For isoelectric focusing, proteins were separated by isoelectric point on 11-cm IPG gel strips of the appropriate range (pH 4–7 or pH 7–11; GE Healthcare). pH 4–7 strips were first rehydrated passively in DeStreak rehydration solution + 0.5% IPG buffer (GE Healthcare) with 100 μg protein for 16 hours and subsequently focused with the following steps: hold at 100 V, 2 hours; hold at 500 V, 2 hours; gradient to 1000 V, 1 hour; gradient to 6000 V, 2.5 hours; hold at 6000 V, 2 hours (IEF done in Ettan IPGPhor 3 (GE Healthcare)). pH 7–11 strips were first rehydrated passively without protein in DeStreak rehydration solution + 0.5% IPG buffer for 16 hours. Protein samples (150 μg) were applied to strips using anodic cup loading, and subsequently focused in an Ettan IPGPhor 3 using the following steps: Hold at 100 V, 2 hours; hold at 500 V, 2 hours; gradient to 1000 V, 1 hour; gradient to 6000 V, 2.5 hours; hold at 6000 V, 2 hours.
After IEF, proteins were separated by molecular weight on 12% Tris-HCl gels in a Criterion Dodeca Cell (Bio-Rad Laboratories, Hercules, CA, USA) at 200 V for 1 hour at 10°C. Protein spots were stained with SYPRO Ruby protein gel stain (Life Technologies, Grand Island, NY, USA). For pH range 4–7, 8 gels of N2 protein (germline (+)) were run, and 8 gels of glp-4(bn2) protein were run. For pH range 7–11, 12 gels of N2 protein were run, and 8 gels of glp-4(bn2) protein were run. Gels were scanned using a Typhoon imager (GE Healthcare) and analyzed with the 2D gel image analysis software, Delta2D (version 3.6; Decodon, Greifswald, Germany). In Delta2D, gels were warped to line up corresponding spots (correcting for running differences between different gels). Proteins that were more abundant in WT (germline(+)) samples were identified with the statistical analysis package within Delta2D as has been described previously for this type of comparison.Citation32 Spot volumes were analyzed with a t-test according to a null distribution that was generated with 200 permutations to account for unequal variance and non-normal distribution of the data.
Mass spectrometry—protein identification
MS-based identification of low-abundance proteins is difficult; thus, we focused our identification efforts on relatively abundant proteins. We also focused our efforts on protein spots that were well-resolved from neighboring spots, to facilitate single-protein identification. Protein spots were excised from 2D gels with the use of a 1.5-mm manual tissue puncher (Beecher Instruments, Sun Prairie, WI, USA) or by a ProteomeWorks spot cutter system (Bio-Rad). Excised spots were placed into 96-well plates. Gel pieces were washed twice in destaining buffer (25 mM NH4HCO3/50% acetonitrile) for 30 min, washed with 100% acetonitrile, and digested with trypsin solution (1 μg/μL MS-grade porcine trypsin gold (Promega, Madison, WI) in 40 mM NH4HCO3/10% acetonitrile) overnight at 37°C. Peptides were eluted from the gel material by incubation with shaking for 30 minutes (room temperature) in analyte solution (0.1% trifluoroacetic acid (TFA)/acetonitrile 2:1); this step was repeated. Protein samples were dehydrated in a vacuum centrifuge at 45°C and immediately rehydrated with 1 μL of analyte solution. Matrix solution (0.2 mg/mL α-cyano-4-hydroycinnamic acid in acetonitrile) was added; each sample was then plated on an Anchorchip target (Bruker Daltonics, Billerica, MA, USA). Plated spots were washed with 0.1% TFA /10 mM ammonium phosphate, and then washed with recrystallization buffer (ethanol, acetone, 0.1% TFA 6:3:1). The peptides were analyzed with a MALDI-TOF/TOF mass spectrometer (Ultraflex II; Bruker Daltonics) as in refs.Citation33,34. Peptide mass fingerprints (PMFs) were acquired using with FlexControl software (Bruker Daltonics) and externally calibrated; trypsin peaks were used to calibrate spectra internally. Spectral processing was accomplished with FlexAnalysis software (Bruker Daltonics). PMFs were analyzed with MASCOT server (version 2.2; Matrix Science Inc.., Boston, MA, USA) launched from BioTools software (Bruker Daltonics) against the National Center for Biotechnology Information (NCBI) database. When searching MASCOT, carbamidomethylation and oxidation were set as fixed modifications, a maximum of one missed cleavage site was allowed, and taxonomy category was restricted to metazoa. Peptide mass fingerprint spectra were further analyzed with tandem MS (MS/MS) spectra with the use of 5 to 10 of the largest peaks per sample, excluding keratin and trypsin. Peptide mass fingerprint and MS/MS spectra were combined and queried as described for PMF spectra analysis with the use of the MS/MS spectra.
DAVID analyses
DAVID analysis was carried out as described in ref.Citation25. Briefly, gene ontology (GO) terms (related to biological processes) associated with each of our 84 proteins were compared to GO terms associated with all C. elegans genes; enrichment for a certain term occurs when that term is associated with a higher proportion of genes in our dataset than among C. elegans genes overall. To minimize errors associated with small samples, we only selected for further analysis GO terms that were associated with a minimum of 5 genes in our germline-enriched data set. The significance of the observed enrichment for a given GO term can be evaluated by a modified Fisher's exact test (P value, ; Table S2); we limited our analyses to those terms with P < 0.05. To correct for multiple comparisons, a Benjamini-Hochberg P value is also calculated (Benjamini, ; Table S2). As this multiple testing correction technique is known to be conservative, we do not apply a strict cutoff of P < 0.05 for the Benjamini-Hochberg P value (as discussed in ref.Citation25). Due to the redundancy of GO terms, enrichment was seen for multiple terms that contained similar sets of genes (for example: GO:0045927∼positive regulation of growth; GO:0040010∼positive regulation of growth rate; GO:0040009∼regulation of growth rate). To resolve this, we manually sorted similar GO terms into groups, and then identified the GO term associated with the largest protein set; these always included the vast majority of proteins associated with the related GO terms. These “lead” GO terms are reported in . All GO terms significantly enriched in our dataset, and genes associated with each term, can be found in Table S2.
Comparison of proteome and expression data
To calculate the expected overlap between the germline proteome data set and RNA expression-based datasets, we determined the proportion of all C. elegans genes present in the data set of interest; random overlap would predict the same proportion of overlap between the proteome dataset and the data set in question. SAGE: 1063 adult germline-enriched genes found (4.8% of all C. elegans genes); expected random overlap: 4 (4.8% of 84).Citation10 Microarray: 2304 adult germline-enriched genes found (10.4% of all C. elegans genes); expected random overlap: 9 (10.4% of 84).Citation8-10 The difference between observed and expected dataset overlap was evaluated using Fisher's exact test.
RNAi
Our objective was identification of novel proteins with roles on gametogenesis and/or embryogenesis. To that end, we selected candidates for RNAi depletion through the following process. First, well-characterized proteins of known function were eliminated. Among the remainder, any proteins of unknown function were subjected to RNAi. In addition, candidates with predicted functions in nucleic acid metabolism were selected. As our analysis proceeded, several candidates were characterized elsewhere. Feeding RNAi was carried out as described in ref.Citation35, with slight modification. L1 larvae of strain AZ212 were placed on NGM+100 μg/mL Ampicillin + 1.0 mM IPTG (NGM + Amp + IPTG) plates that had been seeded with bacteria carrying the appropriate dsRNA-expressing plasmid and allowed to develop to the L4 stage. L4 larvae were then placed on individual seeded NGM + Amp + IPTG plates, and moved to fresh plates each 24 hours. The number of eggs laid and progeny produced on each plate were determined; these numbers were used to determine the fertility of RNAi-treated worms (relative to worms fed the control plasmid L4440, which does not trigger RNAi). Significance was determined using a 2-tailed t-test assuming unequal variance. Differences were considered significant at P < 0.05.
Cytology
AZ212 hermaphrodites were placed on NGM + Amp + IPTG plates that had been seeded with bacteria carrying a plasmid producing dsRNA to the gene of interest (or L4440 control plates) and allowed to develop to the L4 stage. 25 L4 larvae were then moved to a fresh seeded NGM + Amp + IPTG plates and cultured until fixed for microscopy. Confocal imaging (): after 48 hours, hermaphrodites were dissected on microscope slides. Following dissection, samples were fixed in 3.7% formaldehyde and then freeze-cracked into ice-cold methanol. For staining, slides in methanol were brought to room temperature, drained, and stained with 12.5 μg/mL DAPI in 1 × PBST for 15 minutes. After staining, samples were washed 3 × 20 minutes with 1× PBST and mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA). Images were collected as stacks of optical sections acquired at 0.2 μm intervals using an Olympus FV-1000 confocal microscope. For , partial projections encompassing roughly half of the gonad were generated. Whole-animal imaging (): after 24 or 48 hours, hermaphrodites were ethanol fixed and DAPI stained as in ref.Citation36, and imaged on an Olympus BX53 microscope.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
The manuscript was written by B. E. T. and K. J. H., based on contributions from all authors. Experiments were conceived and designed by B. E. T. and K. J. H., and carried out by all authors.
1008903_Supplementary_Materials.zip
Download Zip (33.6 KB)Acknowledgments
The authors thank Stephanie Skidanenko, Marcus Zuzow, Lauren Hitt, and Lars Tomanek for technical assistance; Anne Villeneuve for strains, advice, and comments on the manuscript; Lori Robins for comments on the manuscript; Dave Hansen for data and comments on the manuscript; Ed Himelblau for assistance with figure preparation; WormBase; and the C. elegans Genetic Center (CGC) for strains.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by National Institutes of Health grant R15HD059093 to K.J.H; additional support was provided by the Cal Poly College-Based Fee program and Extramural Funds Initiative.
References
- Petronczki M, Siomos MF, Nasmyth K. Un ménage à quatre: the molecular biology of chromosome segregation in meiosis. Cell 2003; 112:423-40; PMID:12600308; http://dx.doi.org/10.1016/S0092-8674(03)00083-7
- Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2001; 2:280-91; PMID:11283700; http://dx.doi.org/10.1038/35066065
- Villeneuve AM, Hillers KJ. Whence meiosis? Cell 2001; 106:647-50; PMID:11572770; http://dx.doi.org/10.1016/S0092-8674(01)00500-1
- Roeder GS. Meiotic chromosomes: it takes two to tango. Genes Dev 1997; 11:2600-21; PMID:9334324; http://dx.doi.org/10.1101/gad.11.20.2600
- Hubbard EJA, Greenstein D. Introduction to the germ line. WormBook 2005; 1:1-4; PMID:18050415
- Beanan MJ, Strome S. Characterization of a germ-line proliferation mutation in C. elegans. Development 1992; 116:755-66; PMID:1289064
- Crittenden SL, Leonhard KA, Byrd DT, Kimble J. Cellular analyses of the mitotic region in the Caenorhabditis elegans adult germ line. Mol Biol Cell 2006; 17:3051-61; PMID:16672375; http://dx.doi.org/10.1091/mbc.E06-03-0170
- Reinke V, Smith HE, Nance J, Wang J, Van Doren C, Begley R, Jones SJ, Davis EB, Scherer S, Ward S, et al. A global profile of germline gene expression in C. elegans. Mol Cell 2000; 6:605-16; PMID:11030340
- Reinke V, Gil IS, Ward S, Kazmer K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 2004; 131:311-23; PMID:14668411; http://dx.doi.org/10.1242/dev.00914
- Wang X, Zhao Y, Wong K, Ehlers P, Kohara Y, Jones SJ, Marra MA, Holt RA, Moerman DG, Hansen D. Identification of genes expressed in the hermaphrodite germ line of C. elegans using SAGE. BMC Genomics 2009; 10:213; PMID:19426519; http://dx.doi.org/10.1186/1471-2164-10-213
- De Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol Biosyst 2009; 5:1512-26; PMID:20023718
- Page SL, Hawley RS. The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol 2004; 20:525-58; PMID:15473851; http://dx.doi.org/10.1146/annurev.cellbio.19.111301.155141
- Krijgsveld J, Ketting RF, Mahmoudi T, Johansen J, Artal-Sanz M, Verrijzer CP, Plasterk RHA, Heck AJR. Metabolic labeling of C. elegans and D. melanogaster for quantitative proteomics. Nat Biotechnol 2003; 21:927-31; PMID:12858183; http://dx.doi.org/10.1038/nbt848
- Bantscheff M, Ringel B, Madi A, Schnabel R, Glocker MO, Thiesen H-J. Differential proteome analysis and mass spectrometric characterization of germ line development-related proteins of Caenorhabditis elegans. Proteomics 2004; 4:2283-95; PMID:15274122; http://dx.doi.org/10.1002/pmic.200400807
- Chu DS, Liu H, Nix P, Wu TF, Ralston EJ, Yates JR, Meyer BJ. Sperm chromatin proteomics identifies evolutionarily conserved fertility factors. Nature 2006; 443:101-5; PMID:16943775; http://dx.doi.org/10.1038/nature05050
- Chik JK, Schriemer DC, Childs SJ, McGhee JD. Proteome of the Caenorhabditis elegans oocyte. J Proteome Res 2011; 10:2300-5; PMID:21452892; http://dx.doi.org/10.1021/pr101124f
- Harris TW, Baran J, Bieri T, Cabunoc A, Chan J, Chen WJ, Davis P, Done J, Grove C, Howe K, et al. WormBase 2014: new views of curated biology. Nucleic Acids Res 2014; 42:D789-93; PMID:24194605; http://dx.doi.org/10.1093/nar/gkt1063
- Grassl J, Scaife C, Polden J, Daly CN, Iacovella MG, Dunn MJ, Clyne RK. Analysis of the budding yeast pH 4-7 proteome in meiosis. Proteomics 2010; 10:506-19; PMID:20029842; http://dx.doi.org/10.1002/pmic.200900561
- Scaife C, Mowlds P, Grassl J, Polden J, Daly CN, Wynne K, Dunn MJ, Clyne RK. 2-D DIGE analysis of the budding yeast pH 6-11 proteome in meiosis. Proteomics 2010; 10:4401-14; PMID:21136594; http://dx.doi.org/10.1002/pmic.201000376
- Killian DJ, Hubbard EJA. Caenorhabditis elegans germline patterning requires coordinated development of the somatic gonadal sheath and the germ line. Dev Biol 2005; 279:322-35; PMID:15733661; http://dx.doi.org/10.1016/j.ydbio.2004.12.021
- McCarter J, Bartlett B, Dang T, Schedl T. On the control of oocyte meiotic maturation and ovulation in Caenorhabditis elegans. Dev Biol 1999; 205:111-28; PMID:9882501; http://dx.doi.org/10.1006/dbio.1998.9109
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 1977; 56:110-56; PMID:838129; http://dx.doi.org/10.1016/0012-1606(77)90158-0
- Labrador L, Barroso C, Lightfoot J, Müller-Reichert T, Flibotte S, Taylor J, Moerman DG, Villeneuve AM, Martinez-Perez E. Chromosome movements promoted by the mitochondrial protein SPD-3 are required for homology search during Caenorhabditis elegans meiosis. PLoS Genet 2013; 9:e1003497; PMID:23671424; http://dx.doi.org/10.1371/journal.pgen.1003497
- Schrimpf SP, Weiss M, Reiter L, Ahrens CH, Jovanovic M, Malmström J, Brunner E, Mohanty S, Lercher MJ, Hunziker PE, et al. Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biol 2009; 7:e48; PMID:19260763; http://dx.doi.org/10.1371/journal.pbio.1000048
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/10.1038/nprot.2008.211
- Csankovszki G, Collette K, Spahl K, Carey J, Snyder M, Petty E, Patel U, Tabuchi T, Liu H, McLeod I, et al. Three distinct condensin complexes control C. elegans chromosome dynamics. Curr Biol 2009; 19:9-19; PMID:19119011; http://dx.doi.org/10.1016/j.cub.2008.12.006
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003; 421:231-7; PMID:12529635; http://dx.doi.org/10.1038/nature01278
- Hayashi M, Mlynarczyk-Evans S, Villeneuve AM. The synaptonemal complex shapes the crossover landscape through cooperative assembly, crossover promotion and crossover inhibition during Caenorhabditis elegans meiosis. Genetics 2010; 186:45-58; PMID:20592266; http://dx.doi.org/10.1534/genetics.110.115501
- Skop AR, Liu H, Yates J, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science 2004; 305:61-6; PMID:15166316; http://dx.doi.org/10.1126/science.1097931
- Stiernagle T. Maintenance of C. elegans. WormBook 2006; 11:1-11; PMID:18050451
- Dixon DK, Jones D, Candido EP. The differentially expressed 16-kD heat shock genes of Caenorhabditis elegans exhibit differential changes in chromatin structure during heat shock. DNA Cell Biol 1990; 9:177-91; PMID:2160246; http://dx.doi.org/10.1089/dna.1990.9.177
- Berth M, Moser FM, Kolbe M, Bernhardt J. The state of the art in the analysis of two-dimensional gel electrophoresis images. Appl Microbiol Biotechnol 2007; 76:1223-43; PMID:17713763; http://dx.doi.org/10.1007/s00253-007-1128-0
- Tomanek L, Zuzow MJ, Hitt L, Serafini L, Valenzuela JJ. Proteomics of hyposaline stress in blue mussel congeners (genus Mytilus): implications for biogeographic range limits in response to climate change. J Exp Biol 2012; 215:3905-16; PMID:22899524; http://dx.doi.org/10.1242/jeb.076448
- Johnson TL, Tomanek L, Peterson DG. A proteomic analysis of the effect of growth hormone on mammary alveolar cell-T (MAC-T) cells in the presence of lactogenic hormones. Domest Anim Endocrinol 2013; 44:26-35; PMID:23017303; http://dx.doi.org/10.1016/j.domaniend.2012.08.001
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 2003; 30:313-21; PMID:12828945; http://dx.doi.org/10.1016/S1046-2023(03)00050-1
- Hillers KJ, Villeneuve AM. Analysis of meiotic recombination in Caenorhabditis elegans. Methods Mol Biol 2009; 557:77-97; PMID:19799178; http://dx.doi.org/10.1007/978-1-59745-527-5_7
- Green RA, Kao H-L, Audhya A, Arur S, Mayers JR, Fridolfsson HN, Schulman M, Schloissnig S, Niessen S, Laband K, et al. A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell 2011; 145:470-82; PMID:21529718; http://dx.doi.org/10.1016/j.cell.2011.03.037
- Norman KR, Fazzio RT, Mellem JE, Espelt M V, Strange K, Beckerle MC, Maricq A V. The Rho/Rac-family guanine nucleotide exchange factor VAV-1 regulates rhythmic behaviors in C. elegans. Cell 2005; 123:119-32; PMID:16213217; http://dx.doi.org/10.1016/j.cell.2005.08.001
- Simmer F, Moorman C, van der Linden AM, Kuijk E, van den Berghe PVE, Kamath RS, Fraser AG, Ahringer J, Plasterk RH. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol 2003; 1:E12; PMID:14551910; http://dx.doi.org/10.1371/journal.pbio.0000012
- Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, Hengartner MO, Ravichandran KS. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol 2008; 10:556-66; PMID:18425118; http://dx.doi.org/10.1038/ncb1718
- Cui M, Kim EB, Han M. Diverse chromatin remodeling genes antagonize the Rb-involved SynMuv pathways in C. elegans. PLoS Genet 2006; 2:e74; PMID:16710447; http://dx.doi.org/10.1371/journal.pgen.0020074
- Robert VJP, Sijen T, van Wolfswinkel J, Plasterk RH. Chromatin and RNAi factors protect the C. elegans germline against repetitive sequences. Genes Dev 2005; 19:782-7; PMID:15774721; http://dx.doi.org/10.1101/gad.332305
- Waters K, Yang AZ, Reinke V. Genome-wide analysis of germ cell proliferation in C.elegans identifies VRK-1 as a key regulator of CEP-1/p53. Dev Biol 2010; 344:1011-25; PMID:20599896; http://dx.doi.org/10.1016/j.ydbio.2010.06.022
- Colaiácovo MP, Stanfield GM, Reddy KC, Reinke V, Kim SK, Villeneuve AM. A targeted RNAi screen for genes involved in chromosome morphogenesis and nuclear organization in the Caenorhabditis elegans germline. Genetics 2002; 162:113-28.