
ABSTRACT
The development of a multivalent outer membrane vesicle (OMV) vaccine where each strain contributes multiple key protein antigens presents numerous analytical challenges. One major difficulty is the ability to accurately and specifically quantitate each antigen, especially during early development and process optimization when immunoreagents are limited or unavailable. To overcome this problem, quantitative mass spectrometry methods can be used. In place of traditional mass assays such as enzyme-linked immunosorbent assays (ELISAs), quantitative LC-MS/MS using multiple reaction monitoring (MRM) can be used during early-phase process development to measure key protein components in complex vaccines in the absence of specific immunoreagents. Multiplexed, label-free quantitative mass spectrometry methods using protein extraction by either detergent or 2-phase solvent were developed to quantitate levels of several meningococcal serogroup B protein antigens in an OMV vaccine candidate. Precision was demonstrated to be less than 15% RSD for the 2-phase extraction and less than 10% RSD for the detergent extraction method. Accuracy was 70 to 130% for the method using a 2-phase extraction and 90–110% for detergent extraction. The viability of MS-based protein quantification as a vaccine characterization method was demonstrated and advantages over traditional quantitative methods were evaluated. Implementation of these MS-based quantification methods can help to decrease the development time for complex vaccines and can provide orthogonal confirmation of results from existing antigen quantification techniques.
Abbreviations:
- OMV, outer membrane vesicle
- BCA, bicinchoninic acid
- ELISA, enzyme-linked immunosorbent assay
- LC-MS/MS, liquid chromatography, tandem mass spectrometry
- MRM, multiple reaction monitoring
- RSD, relative standard deviation
- MenB, Meningococcal serogroup B
- PorA, major outer membrane protein (class I outer membrane protein)
- fHbp, factor H binding protein
- NadA, Neisseria adhesin A
- HPLC –high performance liquid chromatography
- SDS-PAGE, sodium dodecyl sulfate, polyacrylamide gel electrophoresis
- MS, mass spectrometry
- QC, quality control
- OMPC, outer membrane protein complex
- WRAIR, Walter Reed Army Institute of Research
Introduction
Vaccines are used worldwide for the prevention of diseases. They have tremendous impact on public health by providing immunity against many illnesses that cause severe disease and death. While many of the first vaccines were attenuated viral vaccines, newer technologies have enabled the development of subunit vaccines including complex component vaccines. These advances have resulted in a larger vaccine development space ranging from vaccines composed of single recombinant proteins to vaccines composed of multiple protein variants and/or strains of live or attenuated whole viruses. Furthermore, the complexity of a vaccine candidate can increase through development stages due to the addition of components and/or strains or serotypes to potentially offer broader coverage to strain diversity of a pathogen. When looking at many of the current unmet vaccine targets (dengue, influenza, meningitidis), significant heterogeneity between strains/variants makes it difficult for a single component based vaccine to provide worldwide protection.Citation1 Therefore, many of the current vaccines in development contain multiple antigens (sequence variants of homologous proteins or different serotypes of capsular polysaccharides) from pathogen variants to provide broader protection. For example, pneumococcal and human papillomavirus vaccines currently marketed offer protection against multiple serotypes.Citation2-5
Meningococcal serogroup B (MenB) is an important vaccine target and continues to cause significant mortality worldwide.Citation6 Unlike other groups of Neisseria meningitidis, the native capsular polysaccharide of group B is a poor immunogen and shares a similar structure to components of human glycoprotein.Citation7 Therefore, conserved membrane protein antigens have been an important focus of many MenB vaccine designs. These antigens can be effective, but antigenic variability among MenB strains makes the development of a broadly effective protein-based vaccine a significant challenge.Citation6 Recent work has focused on development of a multivalent native OMV-based vaccine that has been shown to be broadly effective against multiple strains. Three strains of MenB were genetically modified to enhance safety, increase expression levels of certain antigens, and add antigenic variability to major outer membrane protein PorA. This vaccine candidate was shown to elicit bactericidal activity to multiple serogroup B strains as well as other serogroups.Citation8-10
One challenge to the development of this multivalent MenB OMV-based vaccine is the availability of specific, quantitative methods for each of the key protein antigens. Of the many possible surface protein antigens, 4 were identified for quantitative analysis: factor H binding protein (fHbp), Neisseria adhesin A (NadA), class 5C major outer membrane protein (OPC), and class 1 major outer membrane porin (PorA). These four antigens have been proposed to be critical for a broadly protective MenB vaccine.Citation11 Therefore, a quantitative and specific assay for each antigen variant present in the vaccine candidate was needed to support vaccine development.
Modern analytical methods to quantitate protein antigens in vaccine candidates include ELISA, quantitative Western blotting, fluorescence-based assays, high-performance liquid chromatography (HPLC) assays, or sodium dodecyl sulfate PAGE (SDS-PAGE).Citation12-15 These methods have the capability of addressing complex protein mixtures, in order to quantitate specific antigens. However, because this multivalent MenB OMV vaccine is comprised of multiple strains with each strain containing protein variants that only differ slightly in sequence, utility of many of these methods is limited. Although a range of different methods have emerged, the standard ELISA remains the most common for antigen quantification during vaccine development. The main challenge associated with the ELISA is the availability of immunoreagents which is most problematic during early development. The ELISA assay development cycle can extend to over a year with much of the timeline being dictated by generation of suitable immunoreagents. Additionally, traditional plate-based ELISAs are unable to quantitate multiple antigens simultaneously.
Mass spectrometry (MS) has played a key role in proteomics profiling and in determination of expression levels in proteomics. Two-dimensional electrophoresis coupled to MS has been explored as a tool to support vaccine development.Citation16-18 Gel electrophoresis is used to resolve and quantitate proteins while MS is used for identification of the gel spots or bands. There are significant limitations to this approach for routine product development due to assay throughput and variability. Given the availability of more powerful software for database searching along with modern instrumentation, non-gel based “shotgun” proteomics has become an important part of protein quantification.Citation19 Two main approaches to quantitative mass spectrometry have surfaced: those that rely on stable isotope labeling for quantification and label-free methods. Isotope labeling techniques have proven useful but the labeling steps can add a significant amount of time and complexity to the analysis. Label free quantification allows for faster and less complex sample preparation and does not include any limitation in number of proteins or samples that can be analyzed simultaneously.Citation19 One type of label free MS-based quantification relies on multiple reaction monitoring (MRM) where the analyte is quantified by selecting and measuring a parent mass with a unique fragment ion. Accuracy and precision over wide dynamic ranges are achieved along with sensitivity.Citation20 MRM-based quantification methods have been routinely used in small molecule and bioanalytical laboratories for many years and have become more common for peptide and protein quantification.Citation21 Recently, mass spectrometry-based approaches have been used for antigen quantification in vaccines including measurements of hemagluttinin and neuraminidase in influenza vaccines by multiple MS-based methods including MRM.Citation22,23 Additionally, OMV proteins in a Meningococcal vaccine (Bexsero®) were quantified via LC-MSe which differs from the MRM-based approach in both instrumentation and the means by which quantification is performed.Citation24 While quantitative MRM-based methods rely on standard curves consisting of the analyte to evaluate a peptide's individual response factor, LC-MSe uses a universal signal response from an unrelated protein digest.Citation25
In order to support the development and optimization of a multivalent OMV-based MenB vaccine, 2 related MRM-based LC-MS/MS methods were developed for fHbp, NadA, OPC, and PorA. Quantification is performed using a standard curve prepared from one or more of the 4 recombinant antigens (fHbp, NadA, OPC, and PorA) spiked into the appropriate matrix with a protein based internal standard. These methods were shown to be specific, reproducible, and accurate. This approach for antigen quantification allowed for rapid analysis of these antigens in early stage process development and can further increase the depth of characterization performed on these complex vaccine candidates.
Results and Discussion
LC-MS/MS was used to develop a highly specific quantitative method that could accurately determine fHbp, NadA, PorA, and OPC amounts from a vaccine candidate that consisted of OMV derived from 3 engineered strains of group B N. meningitidis.Citation8-10 Although all 4 of these antigens are membrane proteins, the way and extent to which they associate or are embedded in the membrane differ. PorA is an integral transmembrane protein with a β-barrel structure and has a high degree of surface loop sequence variability across strains. Two different variants of PorA (native PorA and recombinant PorA) are expressed in each of the 3 strains of the vaccine. fHbp is a surface exposed lipoprotein that is anchored to the membrane through an N-terminal lipid and exhibits less sequence variability across strains. OPC is a more conserved integral transmembrane protein with a β-barrel structure and exposed surface loops. NadA is a trimeric adhesin and contains an extened coiled-coil motif anchored into the membrane with transmembrane β-strands. An approach that consisted of protein extraction followed by proteolysis and quantification using MRM was employed for each of the 4 antigens. fHbp and NadA, which have significant structure outside the membrane, are analyzed using a centrifugation step followed by a detergent extraction and digestion. The method for PorA and OPC consisted of a 2-phase liquid extraction method using a fluorinated alcohol (HFIP) followed by dry down and digestion.
In order to determine which peptides would be monitored and quantified as a surrogate for protein antigen concentration, samples were trypsinized overnight at 37°C. The digested proteins were analyzed by nanoLC-MS/MS on a linear ion trap. Peptides that are unique to the protein were chosen for specificity, which is especially important in cases where multiple variants of a protein exist. For fHbp, 2 variants (v1 and v2) are expressed across the 3 vaccine strains. For PorA, each strain contains its native form and a second recombinant variant which was added to increase strain coverage.Citation9,10 Therefore, across the 3 vaccine strains, 6 different variants of PorA are expressed. Only a single variant of NadA and OPC were expressed among the 3 vaccine strains. Peptides which displayed strong signal intensities along with their dominant fragment ions were used as the transitions for quantification. The MRM transitions used for each of the proteins is shown in . Ideally, 2 peptides were chosen for each of the target antigens, but for fHbp only one unique transition was possible due to high sequence homology among the variants. shows an example chromatogram from a MRM experiment for an OMV sample analyzed with the detergent extraction method for fHbp and NadA simultaneously.
Figure 1. Example LC-MS/MS chromatogram for 4 monitored MRM transitions. Two peptides for NadA, one peptide for fHbp, and one peptide for the myoglobin internal standard are shown.
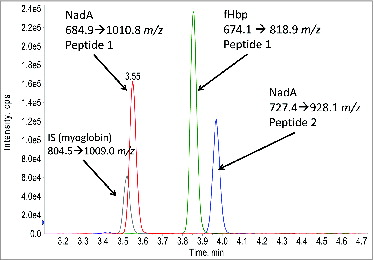
Table 1. List of MS/MS transitions used for routine MRM analysis
Recombinant versions of each of the antigens were spiked into the appropriate matrix to generate a standard curve and quality control (QC) samples. This is in contrast to a previously reported MRM-based method used to quantitate hemagglutinin and neuraminidase from a trivalent inactivated influenza vaccine which relied on standard curves consisting of synthetic peptides in a matrix of 0.1% formic acid.Citation23 In order to account for potential sample matrix effects (i.e. matrix specific signal suppression or enhancement), a well-known MS phenomenon, a blank matrix was used that consisted of a Neisseria meningitidis Outer Membrane Protein Complex (OMPC) that lacked the targeted antigens. Recombinant protein antigens were chosen for the standard curve as opposed to synthetic peptides in order to better match the effects of the sample preparation conditions of the actual OMV sample and to account for variation in digestion efficiencies that can skew accuracy when quantitating with synthetic peptides. Additionally, heavy labeled peptides are commonly used as an internal standard in protein quantification applications. These stable-labeled peptides are synthetically made with incorporated heavy isotopes. As a result, they have a distinct and heavier mass than the peptides being monitored in the assay, but have the same chromatographic and ionization behavior. This makes them well suited for use as an internal standard.
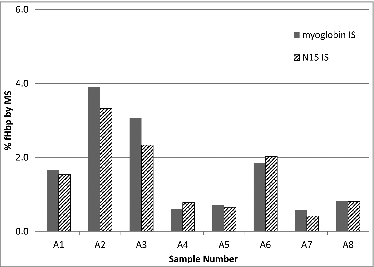
In this case however, the protein analytes are membrane proteins and not well behaved. Additionally, the sample matrix is complex and subject to change during process development and optimization. Because of these factors, alternate internal standards that take into account digestion efficiency were also evaluated. Two different types of protein internal standards were compared. Either heavy labeled full length antigens (expressed in N15 containing media) or myoglobin was used as the internal standard. The heavy labeled protein provides all the benefits of an N15 labeled peptide while correcting for any possible variation in the digestion efficiency due to slight changes in the sample matrix. Although heavy labeled proteins are an ideal choice as the internal standard, the long lead time and cost of such reagent can be prohibitive. Therefore, a simple inexpensive purified protein, myoglobin, was also considered as an internal standard. shows a comparison of myoglobin to a heavy labeled protein as the internal standard for one variant of fHbp. Both approaches gave similar results for the eight 8 samples analyzed and demonstrated that a generic purified protein such as myoglobin can be considered as an alternative to sequence specific protein internal standard. Although this alternative would need to be assessed for specific sample matrices and antigens, it can lead to faster and more inexpensive method development by eliminating the need for a stable isotope labeled recombinant protein.
Figure 2. Comparison of percent abundance of fHbp determined using myoglobin or heavy labeled (N15) recombinant fHbp as the internal standard. Percent abundance was determined by dividing the measured concentrations of fHbp by the total protein concentration as assessed by BCA assay. Each internal standard experiment was run independently (on separate days). Results generated with the myoglobin internal standard are consistent with those from the recombinant heavy labeled protein and demonstrate the feasibility of this approach.
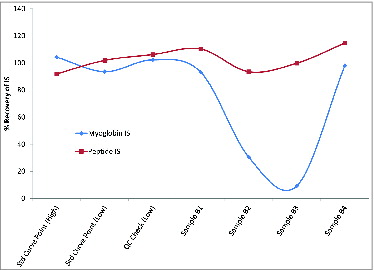
Conversely, a sequence specific N15 labeled peptide can be used as an internal standard when isotope labeled proteins are unavailable. However, when an N15 labeled peptide was used as the internal standard, no corrections could be made for samples that showed poor digestion efficiency. shows an example of data, from a set of 4 samples generated during bioprocess optimization, using a heavy labeled peptide versus myoglobin as the internal standard. Samples B2 and B3 showed poor recovery of myoglobin indicating a reduction in digestion efficiencies as a result of the sample matrix of these 2 specific samples. However, the heavy labeled peptide gave no indication that these samples were poorly digested and its use as the internal standard would result in an inaccurate measurement of the antigens in these 2 samples. Therefore, during bioprocess optimization, when digestion efficiencies can be impacted by changes in the process, a marker of digestion should be implemented if an N15 labeled peptide is chosen as an internal standard.
Figure 3. Impact of low digestion efficiencies on a peptide internal standard (IS). Recovery results are shown for either a peptide internal standard or myoglobin IS. Percent recovery was calculated using the area of the IS from the sample divided by the average of the internal standard area of all points of the standard curve. For controls, 2 calibration points (Std Curve) and one QC sample are shown along with 4 samples, 2 of which showed low digestion efficiency (samples B2 and B3) as demonstrated by the decreased recovery of the myoglobin peptide. As expected, this was not reflected in the recovery of the heavy labeled peptide IS. Because of this, the use of heavy labeled peptide IS would result in the reporting of an inaccurate antigen concentration in the cases where the sample matrix interferes with the digestion efficiency.
The analytical merits of the MRM-based methods were evaluated. Linearity of each extraction method was assessed by diluting each recombinant antigen into the negative control matrix followed by MRM analysis. Linearity of the detergent extraction method was evaluated at 8 to 250 μg/mL for NadA and 0.5 to 50 μg/mL for fHbp with R2 of 0.9994 and 0.9999 respectively. The two-phase extraction method was shown to be linear from 0.5 to 25 μg/mL for OPC and 2 to 100 μg/mL for PorA with R2 of 0.9982 and 0.9996, respectively. Accuracy of each of the methods was determined by spiking known amounts of recombinant antigen into the negative control matrix or OMV vaccine samples. For the 2-phase extraction method, spikes of recombinant PorA were performed at 15 and 40 μg/mL and OPC was spiked in at 2, 8, and 16 μg/mL. Accuracy of the detergent extraction method was assessed with spikes of recombinant fHbp at 0.5 and 1.7 μg/mL. Accuracy was calculated as a percent of the measured concentration to the theoretical concentration. The accuracy of the detergent extraction method was routinely 90 to 110% while the accuracy of the 2-phase extraction was 70 to 130%. The improved accuracy for the detergent extraction method is expected given the added variability associated with the additional steps required in the 2-phase extraction method. Intermediate precision was measured over 3 d from 4 technical replicates of a vaccine sample per day. The RSD of the detergent extraction method was determined to be less than 10% whereas the RSD of the 2-phase extraction was determined to be less than 15%.
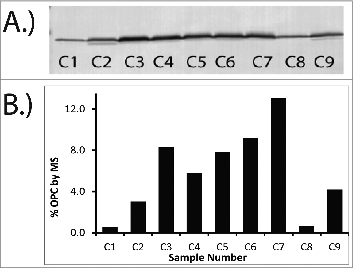
To determine if the method was accurately trending differences between samples, comparisons to orthogonal assays were made. The MS results for a series of samples generated during bioprocess optimization were compared to a Western blot against OPC and are shown in . Qualitatively, the density of the Western bands tracked well with the results from MS analysis. Samples C1 and C8 showed the lowest levels of OPC content by both MS and Western followed by samples C2 and C9. Although samples C3-C7 exhibited the highest expression of OPC by both MS and Western, MS analysis was able to discriminate between the OPC levels in samples C3-C7 whereas Western did not. This is likely due to saturation of the signal in the Western blot.
Figure 4. Comparison of Western Blot (A) and MS data (B) for OPC. Similar trends in OPC levels are observed between the Western Blot and the MS data when datasets are qualitatively evaluated. MS results (B) are reported as percentage of the total protein for a series of OMV samples. Percentage of total protein was determined by dividing the measured concentration of OPC by the total protein concentration. Sample numbers on gel (A) correspond to sample numbers in the graph (B).
A second orthogonal comparison of the MS results was made by comparing the PorA MS quantification data against data from SDS-PAGE analysis. Since PorA is one of the most abundant proteins in the OMV preparation, the total amount of PorA can be assessed by SDS-PAGE densitometry. Results for the MS method were generated from the specific peptides for each of the 2 variants (native and engineered) and summed. A summary of the comparison of the data from the 2 methods is shown in . Results are shown across a range of PorA values. At the highest level, Sample P1 contained 34.8% total PorA when measured by MS and 29.5% when measured by SDS-PAGE. At the lower end, Sample P4 had 7.5% total PorA by MS and 6.7% by SDS-PAGE. These results indicated consistency between the 2 methods. In contrast to SDS-PAGE analysis, individual levels of the 2 PorA variants can also be determined (). Although the amount of total PorA is similar between samples P2 (24.5%) and P3 (17.9%), the ratio between the 2 PorA variants are markedly different. Sample P2 had a ratio of 1:10 for the 2 variants whereas sample P3 had approximately similar concentrations of the 2 variants. These results demonstrate the sequence-based specificity achieved by MS that allows for simultaneous quantification of highly homologous protein antigens which is not routinely attainable by electrophoretic methods.
Table 2. Comparison of MS and SDS-PAGE results for PorA content for a series of OMV samples. For SDS-PAGE analysis, PorA content is expressed as a percentage of the total density. In order to compare the results from the MRM-based assay to the gels, the PorA concentration derived by MS was converted to percentage of total protein concentration. Levels of each PorA variant as well as the total PorA levels (sum of each variant) by MS are shown
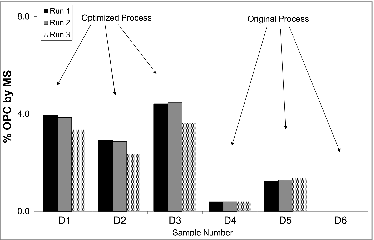
The methods described above were employed routinely to support optimization of antigen levels during the development of upstream and downstream processes for this vaccine. The results for OPC quantification, using the 2-phase extraction method, over a series of development samples are shown in . OPC content, as measured by MS, was significantly increased as a result of process improvements likely increasing the immunogenicity of this vaccine candidate. This rapid and specific approach to quantification allowed for quicker, better-informed, early-stage process decisions and rivaled the throughput of standard immunoassays due to multiplexing. Additionally, this analysis does not rely on immunoreagents so it can be employed before availability of these reagents.
Figure 5. MS results for OPC content for a series of process samples. Three samples before and after process optimization are shown. Three replicates performed on successive days with independent standard curves of each sample are shown. Results are expressed as percent of OPC.
A key difference to note is that quantification by MRM reports antigen mass and does not address antigen structure. Therefore, it cannot be used to measure key epitope stability unless the changes in stability are due to a change in the peptide sequence being monitored such as degradation or proteolysis of the peptide. Additionally, other peptide modifications in the samples such as O-glycosylation will change the mass of the peptide and skew the results because these peptides would not be detected. Because of this, monitoring more than one peptide can be beneficial in detecting disparities in measured peptides from the same protein. For instance, comparisons of measured concentrations of peptide 1 to peptide 2 for OPC in MenB vaccine samples showed that the concentrations of peptide 1 and peptide 2 were comparable, within the 95% confidence intervals, and not significantly different for all of the samples. If there had been differences detected in concentrations between peptides 1 and 2, it could have reflected potential degradation or modification of the peptide in those samples. Given the advantages of absolute quantification, ability to multiplex, throughput, specificity, and speed of development, the MRM-based approach for protein quantification in complex vaccines can significantly improve characterization knowledge and aid in many stages of product development.
Conclusions
A quantitative mass spectrometry method using traditional MRM quantification was developed for multiple variants of 4 different antigens in an OMV-based MenB vaccine. These antigens could be measured with good precision and accuracy and with throughput that rivals conventional plate-based ELISA. This approach for quantification of key protein antigens led to a quicker way to measure key antigens in early stage vaccine development. In this work, the MS results were used as a primary means for quantification of some key protein antigens. Given the results, MRM-based LC-MS/MS is an attractive option for routine protein quantification during bioprocess development due to the specificity, no requirement for immunoreagents, and ability to multiplex numerous protein antigens.
Materials and Methods
Identification of peptides and optimization of transitions used for quantification
Both recombinant proteins (NadA, fHbp, OPC, PorA) and vaccine preparations were digested with trypsin (Worthington) and analyzed on a nano-LC-MS/MS (LTQXL ion trap instrument, Thermo). Samples with total protein concentrations ranging from 0.5 to 1 mg/mL were denatured in 4 M urea and then diluted to 1 M urea and digested overnight at 37°C using a 1:10 trypsin to protein ratio (w/w). After digestion, 4 μL of sample were injected onto the nano-LC system which consisted of a PicoFrit BioBasic C18 (75 μm × 100 mm, New Objective) with a nanoAcquity trap column (180 μm × 20 mm, Symmetry C18, Waters). A 5 minute desalting step at 15 μL/min with 99% (v/v) water, 1% (v/v) acetonitrile and 0.1% (v/v) formic acid (FA, Pierce) and 0.02% (v/v) trifluoroacetic acid (TFA, Pierce) was used after samples were loaded onto the HPLC. Next, peptides were eluted from the column with a linear gradient over 85 minutes at 300 nL/min to 35% (v/v) water, 65% (v/v) acetonitrile, 0.1% (v/v) FA and 0.02% (v/v) TFA. The spray voltage was 2.5 kV. The capillary temperature was 220°C. The capillary voltage was 47 V and the tube lens was 155 V. Data dependent analysis (DDA) was performed using a single full MS scan from 300 to 2000 m/z followed by MS/MS analysis of the 5 highest ions detected in the full scan. Dynamic exclusion was applied after 2 MS/MS scans of the same ion. Peptides that were identified for quantification had to be specific for that particular protein, fall in an acceptable mass range, yield a reasonable series of fragment ions, and not contain any problematic amino acids such as methionine or N-glycosylation motifs. Once the peptides and fragments were chosen, the same digest was run on a AB Sciex 4000 Q-Trap hybrid triple quadrupole/linear ion trap instrument and the masses and collision energies were optimized for each transition. Transition optimization was performed manually by varying ionization and collision parameters (CE, CXP, DP, EP) within a single MRM run. Ideally, 2 peptides with 2 transitions each were selected for each protein. Transitions for heavy labeled peptides/proteins were adjusted according to the number of heavy isotopes incorporated.
Detergent extraction method (fHbp and NadA Antigens)
The standard curve for the detergent extraction method was prepared by spiking recombinant protein (in-house) into OMPC, a sample of similar composition that does not contain the proteins of interest. The standard curve ranged from 0.5 to 50 μg/mL for fHbp and 8 to 250 μg/mL for NadA. OMV vaccine samples were diluted with water to a common starting concentration of 200 μg/mL total protein. A total of 480 μL of sample was placed in a 1 mL ultracentrifuge tube. Ultracentrifugation was performed at 4°C for 75 minutes at 50,000 rpm. After ultracentrifugation, the supernatant (410 μL) was removed and discarded. The pelleted sample was then extracted with 0.5% (w/v) Zwittergent 3–12 (Calbiochem) by adding 7 uL of a 10X stock solution followed with vortexing. The internal standard (N15 grown recombinantly expressed NadA and fHbp, Nexomics) was added at a fixed amount (20 μL of 50 μg/mL) to each sample and standard. For the comparison of an antigen specific internal standard to a generic standard consisting of myoglobin, 20 μL of 100 μg/mL of myoglobin was added in place of the recombinant N15 fHbp. Next, samples and standards were digested with the addition of trypsin at a 1:10 (w/w) ratio overnight at 37°C. Following digestion, samples and standard were quenched by acidification with formic acid to a final concentration of 1% formic acid.
15 μL of detergent extracted digests or standards were injected on a 2.1 × 100 mm POROS R2/10 column (Applied Biosystems). The flow rate was 0.7 mL/min and the column was kept at 23°C. The mobile phase A was 0.1% (v/v) formic acid in water and mobile phase B consisted of 0.1% (v/v) formic acid in acetonitrile. A gradient was run from 2% B to 30% B in 3.5 minutes after an initial hold of 30 seconds. The column was then cleaned and re-equilibrated. MS detection for quantification was performed in positive ion mode on an AB Sciex 4000 QTrap (ABSciex) operated in MRM mode with both quads operated at low resolution. MRM acquisition parameters used were CUR-30, IS-4500, TEM-550, GS1-45, GS2-50, CAD-4, DP-70, EP-10, CXP-15. Transitions used for quantification are shown in . Results are calculated using a ratio of the target peptide area to the appropriate internal standard (heavy peptide or myoglobin peptide). Quantification was performed using the Analyst software (version 1.4.2) with a 1/x curve fit.
Two-phase extraction method (PorA and OPC Antigens)
Vaccine samples were diluted with water to 200 μg/mL total protein. Then 50 μL of OMV vaccine sample was extracted by adding 200 μL hexafluoroisopropanol (HFIP, Sigma) and 100 μL of petroleum ether. The internal standard (either 20 μL of 100 μg/mL myoglobin, Sigma or 5 uL of 2.5 pmol/μL N15 labeled peptide, Sigma) was added prior to extraction. Proteins were extracted by vortexing for one-hour at room temperature. Next, 200 μL of the bottom HFIP layer was removed and dried in a separate tube. This dried protein extract was resolubilized in 24 μL of 6 M urea with shaking for 15 minutes. Trypsin (Promega) is added (1:5 enzyme:protein ratio by mass) in 100 mM ammonium bicarbonate (80 μL) and the samples were digested overnight at room temperature.
A standard curve ranging from 0.5 to 25 μg/mL of OPC and 2 to 100 μg/mL of PorA was generated using recombinant OPC and PorA (in-house or BlueSky BioServices) with 4 M urea into OMPC, a sample of similar composition that does not contain the proteins of interest. Standards followed the same preparation as a vaccine sample. A quality control sample (QC), consisting of a blank bacterial matrix spiked with recombinant antigen, was run at each occasion to assess accuracy and ensure reproducibility of the results. QCs were performed at the low, middle and high ranges of the standard curve.
Following digestion, the reactions were quenched with 10% (v/v) formic acid to yield a final concentration of 1% (v/v) formic acid. The tryptic digest was analyzed by MRM on the a 4000 Q-Trap mass spectrometer. A Waters BEH C18 column (2.1 × 50 mm, 1.7 μm particle size) was used with 0.1% (v/v) formic acid in water as mobile phase A and 0.1% (v/v) formic acid in acetonitrile as mobile phase B. After an initial hold at 2% B for 30 seconds, a gradient of 2% B to 40% B was run in 5 minutes followed by a wash with 90% B for 1.2 minutes. The column was then returned to the starting conditions and re-equilibrated. The flow rate was 0.2 mL/minute during the gradient and increased to 0.3 mL/minute during the wash and re-equilibration steps. The column temperature was 40°C. Samples and standards were injected at 10 μL. The first 1.5 minutes of the run was diverted to waste. The 4000 QTrap was operated in MRM mode to scan for specific parent and daughter ions. MRM data was acquired in positive ion mode at low resolution in both Q1 and Q3 to maximize sensitivity. Examples of specific transitions can be found in the body of the document. Acquisition parameters used were the same as previously described except for the following: IS-5000, GS1-30, GS2-40. Transitions used for quantification are shown in . Results are calculated using a ratio of the target peptide area to the appropriate internal standard (heavy peptide or myoglobin peptide). All data were processed using Analyst 1.4.2 software. A 1/x curve fit was used.
SDS-page and western blot
MenB OMV samples were mixed with reducing sample buffer (62.5 mM Tris, 2% SDS, 9.5% glycerol, 0.002% bromophenol blue, 100 mM DTT, pH 6.8), heated at 70°C for 10 minutes and loaded at 5 μg/lane onto a Novex® 4–20% Tris-glycine 10-well mini-gel (Invitrogen). Proteins were separated under a constant voltage of 125 V for 1 hour 55 minutes. Proteins were fixed with 12% trichloroacetic acid (TCA, Fisher) for 1 hour then washed with water multiple times to remove the acid. Proteins were stained overnight with G-250 Coomassie stain (Thermo Scientific) and subsequently destained thoroughly with water for at least 6 hours. Gels were then scanned using Molecular Dynamic Personal Densitometer SI and peak quantification was performed using Image Quant software (GE Healthcare).
Western Blot was performed according to Invitrogen's instructions. In brief, after protein electrophoresis SDS-PAGE the proteins were transferred to PVDF membranes with iBlot® at 20 V for 7 minutes. The membranes were blocked with 5% non-fat milk (BioRad) in TTBS (20 mM Tris-HCl, 500 mM NaCl, 0.05% Tween 20, pH 7.5) for 2 hours, probed with the primary mouse anti-rOPC sera overnight at a dilution of 1:2,000, washed, and incubated with the secondary goat anti-mouse IgG alkaline phosphatase (AP) conjugate (Pierce) at a dilution of 1:2,000 for 1.5 hours. Membranes were thoroughly washed and developed with NBT/BCIP substrate (Pierce) for 2–3 minutes.
Samples
MenB OMV vaccine samples were provided by the process development group. A total of 3 different MenB strains were present. Each of the 3 strains contained native PorA, fHbp, NadA, and OPC. Native fHbp and PorA antigen variants were specific for the strain whereas NadA and OPC native sequences were conserved across the 3 strains. Each of the strains was engineered to overexpress either NadA or one the 2 variants of fHbp.Citation8 Additionally, a second variant of PorA was introduced recombinantly into each strain for broader immunity. Original strains were obtained from WRAIR.Citation8-10 Further details on the multivalent vaccine can be found in previous publications.Citation8-10 Protein concentrations for OMV samples were determined using a BCA assay according to the manufacturer's instructions (Pierce).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Hui Xu and Jing Lin for providing recombinant antigens and Eberhard Durr for discussions on the 2-phase extraction technique. We also would like to thank all the scientists in MRL BioProcess Research and Development who have supported this vaccine development.
References
- Koff WC, Burton DR, Johnson PR, Walker BD, King CR, Nabel GJ, Ahmed R, Bhan MK, Plotkin SA. Accelerating next-generation vaccine development for global disease prevention. Science 2013; 340:1232910-1-7; PMID:23723240; http://dx.doi.org/10.1126/science.1232910
- Jefferies JM, Macdonald E, Faust SN, Clarke SC. 13-valent pneumococcal conjugate vaccine (PCV13). Human Vaccines 2011; 7:1012-18; PMID:21941097; http://dx.doi.org/10.4161/hv.7.10.16794
- Grabenstein JD, Manoff SB. Pneumococcal polysaccharide 23-valent vaccine: Long-term persistence of circulating antibody and immunogenicity and safety after revaccination in adults. Vaccine 2012; 30:4435-44; PMID:22542818; http://dx.doi.org/10.1016/j.vaccine.2012.04.052
- Bryan JT. Developing an HPV vaccine to prevent cervical cancer and genital warts. Vaccine 2007; 25:3001-6; PMID:17289220; http://dx.doi.org/10.1016/j.vaccine.2007.01.013
- Villa LL. HPV prophylactic vaccination: The first years and what to expect from now. Cancer Lett 2011; 305:106-12; PMID:21190794; http://dx.doi.org/10.1016/j.canlet.2010.12.002
- Ceasar NM, Myers KA, Fan X. Neisseria meningitidis serogroup B vaccine development. Microb Pathog 2013; 57:33-40; PMID:23416222; http://dx.doi.org/10.1016/j.micpath.2013.02.003
- Finne J, Leinonen M, Mäkelä PH. Antigenic similarities between brain components and bacteria causing meningitis. Implications for vaccine development and pathogenesis. Lancet 1983; 2:355-7; PMID:6135869; http://dx.doi.org/10.1016/S0140-6736(83)90340-9
- Zollinger WD, Donets MA, Schmiel DH, Pinto VB, Labrie JE, Moran EE, Brandt BL, Ionin B, Marques R, Wu M, et al. Design and evaluation in mice of a broadly protective meningococcal group B native outer membrane vesicle vaccine. Vaccine 2010; 28:5057-67; PMID:20653107; http://dx.doi.org/10.1016/j.vaccine.2010.05.006
- Pinto VB, Moran EE, Cruz F, Wang XM, Fridman A, Zollinger WD, Przysiecki CT, Burden R. An experimental outer membrane vesicle vaccine from N. meningitidis serogroup B strains that induces serum bactericidal activity to multiple serogroups. Vaccine 2011; 29:7752-8; PMID:21827811; http://dx.doi.org/10.1016/j.vaccine.2011.07.124
- Moran EE, Burden R, Labrie JE, Wen Z, Wang XM, Zollinger WD, Zhang L, Pinto VB. Analysis of the bactericidal response to an experimental Neisseria meningitidis vesicle vaccine. Clin Vaccine Immunol. 2012; 19:659-65; PMID:22461527; http://dx.doi.org/10.1128/CVI.00070-12
- Feavers IM, Pizza M. Meningococcal protein antigens and vaccines. Vaccine 2009; 27 Suppl 2: B42-50 ; PMID:19481315; http://dx.doi.org/10.1016/j.vaccine.2009.05.001
- van de Waterbeemd B, Zomer G, Kaaijk P, Ruiterkamp N, Wijffels RH, van den Dobbelsteen GP, van der Pol LA. Improved production process for native outer membrane vesicle vaccine against Neisseria meningitidis. PLoS One 2013; 8:e65157; PMID:23741478; http://dx.doi.org/10.1371/journal.pone.0065157
- Rustandi RR, Loughney JW, Hamm M, Hamm C, Lancaster C, Mach A, Ha S. Qualitative and quantitative evaluation of Simon, a new CE-based automated Western blot system as applied to vaccine development. Electrophoresis 2012; 33:2790-7; PMID:22965727; http://dx.doi.org/10.1002/elps.201200095
- Lorbetskie B, Wang J, Gravel C, Allen C, Walsh M, Rinfret A, Li X, Girard M. Optimization and qualification of a quantitative reversed-phase HPLC method for hemagglutinin in influenza preparations and its comparative evaluation with biochemical assays. Vaccine 2011; 29:3377-89; PMID:21397719; http://dx.doi.org/10.1016/j.vaccine.2011.02.090
- Gutierrez-Granados S, Cervera L, Godia F, Carrillo J, Segura MM. Development and validation of a quantitation assay for fluorescently tagged HIV-1 virus-like particles. J Virol Methods 2013; 193:85-95; PMID:23707924; http://dx.doi.org/10.1016/j.jviromet.2013.05.010
- Maxwell KL, Frappier L. Viral proteomics. Microbiol Mol Biol Rev 2007; 71:398-411; PMID:17554050; http://dx.doi.org/10.1128/MMBR.00042-06
- Roepstorff P. Mass spectrometry based proteomics, background, status and future needs. Protein & Cell 2012; 3:641-7; PMID:22926765; http://dx.doi.org/10.1007/s13238-012-2079-5
- Wittmann-Liebold B, Graack H-R, Pohl T. Two-dimensional gel electrophoresis as tool for proteomics studies in combination with protein identification by mass spectrometry. Proteomics 2006; 6:4688-703; PMID:16933336; http://dx.doi.org/10.1002/pmic.200500874
- Zhu W, Smith JW, Huang CM. Mass spectrometry-based label-free quantitative proteomics. J Biomed Biotechnol 2010; 2010:840518; PMID:19911078
- Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Sys Bio 2008; 4:222; PMID:18854821
- Kitteringham NR, Jenkins, RE, Lane CS, Elliott vL, Park BK. Multiple reaction monitoring for quantitative biomarkers analysis in proteomics and metabolomics. J Chromatogr B Anlyt Techonol Biomed Life Sci 2009; 13:1229-39; PMID:19041286; http://dx.doi.org/10.1016/j.jchromb.2008.11.013
- Creskey MC, Li C, Wang J, Girard M, Lorbetskie B, Gravel C, Farnsworth A, Li X, Smith DG, Cyr TD. Simultaneous quantification of the viral antigens hemagglutinin and neuraminidase in influenza vaccines by LC-MSE. Vaccine 2012; 30:4762-70; PMID:22643214; http://dx.doi.org/10.1016/j.vaccine.2012.05.036
- Williams TL, Pirkle JL, Barr JR. Simultaneous quantification of hemagglutinin and neuraminidase of influenze virus using isotope dilution mass spectrometry. Vaccine 2012; 30:2475-82; PMID:22197963; http://dx.doi.org/10.1016/j.vaccine.2011.12.056
- Tani C, Stella M, Donnarumma D, Biagini M, Parente P, Vadi A, Magagnoli C, Costantino P, Rigat F, Norias N. Quantification by LC-MSE of outer membrane vesicle proteins of the Bexsero® vaccine. Vaccine 2014; 32:1273-9; PMID:24462403; http://dx.doi.org/10.1016/j.vaccine.2014.01.011
- Silva JC, Denny R, Dorschel C, Gorenstein MV, Li GZ, Richardson K, Wall D, Geromanos SJ. Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol Cell Proteomics 2006; 5:589-607; PMID:16399765; http://dx.doi.org/10.1074/mcp.M500321-MCP200