
Abstract
Cross-presentation is the process by which professional APCs load peptides from an extracellularly derived protein onto class I MHC molecules to trigger a CD8+ T cell response. The ability to enhance this process is therefore relevant for the development of antitumor and antiviral vaccines. We investigated a new TLR2-based adjuvant, Small Molecule Immune Potentiator (SMIP) 2.1, for its ability to stimulate cross-presentation. Using OVA as model antigen, we demonstrated that a SMIP2.1-adjuvanted vaccine formulation induced a greater CD8+ T cell response, in terms of proliferation, cytokine production and cytolytic activity, than a non-adjuvanted vaccine. Moreover, using an OVA-expressing tumor model, we showed that the CTLs induced by the SMIP2.1 formulated vaccine inhibits tumor growth in vivo. Using a BCR transgenic mouse model we found that B cells could cross-present the OVA antigen when stimulated with SMIP2.1. We also used a flow cytometry assay to detect activation of human CD8+ T cells isolated from human PBMCs of cytomegalovirus-seropositive donors. Stimulation with SMIP2.1 increased the capacity of human APCs, pulsed in vitro with the pp65 CMV protein, to activate CMV-specific CD8+ T cells. Therefore, vaccination with an exogenous antigen formulated with SMIP2.1 is a successful strategy for the induction of a cytotoxic T cell response along with antibody production.
Abbreviations
| SMIP | = | Small Molecule Immune Potentiator |
| OVA | = | avalbumin |
| CTL | = | cytotoxic t lymphocyte |
| BCR | = | B cell receptor |
| APC | = | antigen presenting cell |
| CMV | = | cytomegalovirus |
| MHC | = | major histocompatibility complex |
| DC | = | dendritic cell |
| TLR | = | toll like receptor |
| HCMV | = | human CMV |
| PBMC | = | peripheral blood mononuclear cell |
| KO | = | knock out |
| LN | = | lymph node |
Introduction
Although vaccination is one of the most successful public health interventions, there are still no efficacious vaccines for the prevention of many old as well as emerging infectious diseases. The increased life-expectancy of the human population worldwide adds further urgency to the development of new vaccines together with the improvement of existing vaccines.Citation1
CD8+ T cells play a critical role in host defense against viral infection and certain cancers,Citation2 and thus the development of vaccines that elicit a robust CD8+ T cell response is an area of great interest. Subunit vaccines do not usually induce CD8+ T cell immunity. Protein components in vaccine formulations are captured by APCs and presented to CD4+ T cells by class II MHC molecules, thus eliciting an adequate humoral response but a poor cytotoxic response. Activation of CD8+ T cells by extracellularly delivered (exogenous) antigens requires the induction of cross-presentation, a mechanism by which peptides derived from extracellularly derived antigens are presented by APCs on Class I MHC molecules, leading to activation of CD8+ T cells.Citation3,4
Different approaches have been pursued to successfully introduce proteins into the Class I MHC processing pathway, including conjugation of the antigen to antibodies that target dendritic cells (DCs), and by ex vivo manipulation of DCs.Citation5,6 These approaches suffer from difficulties in manufacturing, as well as the high costs.
A promising strategy is the use of adjuvants, molecules that are added to vaccine formualtions in order to modulate the immune response and ultimately increase protection. Although many experimental adjuvants have been evaluated in animal models, until 10 y ago only squalene-based oil in water emulsions and aluminum-based salt adjuvants had been licensed for inclusion in human vaccines.Citation1 These adjuvants are effective at eliciting humoral responses, but fail to stimulate CD8+ T cell immunity.
Alternative vaccine adjuvants aimed at eliciting both antibody and cellular responses are based on the activation of receptors of the innate immune system, such as TLRs. Engagement of TLRs with either natural or synthetic agonists, results in a robust activation of innate immune cells and leads to the production of proinflammatory cytokines.Citation7,8 Many pre-clinical studies support the safety of TLRs agonists in vaccine formulations as well as their ability to increase adaptive immune responses.Citation9,10 TLR agonists have also been shown to enhance therapeutic vaccination against cancer and chronic viral infections.Citation8,11,14 Indeed, vaccines containing the adjuvant AS04, made by the alum-absorbed TLR4 agonist monophosphoryl lipid A (MPL), have been approved for human use in 2005.Citation1,15
Here we explored the ability of SMIP2.1, a novel synthetic lipopeptide-based TLR2 agonist, to induce cross-presentation by both mouse and human APCs. Using in vitro and in vivo experiments we showed that SMIP2.1 can activate the innate immune system via a TLR2-dependent mechanism, induce the maturation of APCs, and elicit a strong antibody response against influenza and tetanus toxoid antigens.
In mice, TLR2 agonists can induce an antigen-specific CD8+ T cell response, especially when linked to the antigen.Citation16-18 Here, we show that SMIP2.1 is also a good inducer of a CTL response when mixed with the antigen as aqueous suspension using either mice or human cells. Mice that received OVA-specific OT-I TCR transgenic cells by adoptive transfer showed increased CD8+ T cell proliferation, cytokine production, and cytotoxic activity upon inclusion of SMIP2.1 in the OVA vaccine formulation. We investigated which APCs populations could be the target for SMIP2.1-induced cross-presentation and showed that both CD8α+ and CD8α− DCs could cross-present. While it is already known that DCs can cross-present exogenous antigens, the role of B cells in this process is less clear.Citation19-21 Using transnuclear B cells that express a BCR specific for OVA, we demonstrated for the first time that B cells can cross-present OVA upon TLR2 stimulation. Likewise, upon stimulation with SMIP2.1, human PBMCs were able to cross-present the CMV pp65 protein to human CMV (HCMV)-primed CD8+ T cells.
This study shows that SMIP2.1 could assist in the generation of antigen specific CTL along with the robust activation of CD4+ T cells, and thus could be exploited in the design of effective adjuvants for antitumor and antiviral vaccines.
Results
Identification of a new TLR2 agonist
A series of high-throughput screens on a chemical library of 1.8 million compounds were performed. Briefly, the TLR2 expressing human B cell line RI-I and monocytic cell line THP-1 were screened in arrayed, 1536 well format in single point (10 μM in DMSO) using TNFα as a readout (data not shown). Compounds able to stimulate these leukocyte cell lines were counter-screened using mouse lymphocytes as well as HEK293 clones stably transfected with the luciferase gene under control of transcription factor NF-kB and different human TLRs (data not shown). This strategy resulted in the identification of a group of triacetylated lipopeptides active only on both human and mouse TLR2 which differed in the amino acid component and in the length of the acyl chain. This class of lipopeptide bears a triacylated cysteine glycerol core, similar to the known TLR2 agonist Pam3CSK4, but differs in the serine and lysine amino acid residues.Citation22 A representative of this class of lipopeptides is shown in as SMIP2.1. The dipeptide portion of SMIP2.1 is composed of α-aminobutyric acid and glutamic acid. Alpha-aminobutyric acid can be substituted with alanine with no apparent loss of activity. Glutamic acid, which is at the C-terminus of the lipopeptide, can tolerate even a wider range of chemical modifications. SMIP2.1, as expected, is able to stimulate HEK293 NFκB-luciferase reporter cell lines stably transfected with human or mouse TLR2 (). SMIP2.1 also stimulated TNFα secretion by human MoDCs whereas the addition of an anti-TLR2 blocking antibody inhibited this effect (). To further confirm the TLR2 dependency of activation in mouse cells, BM-DCs derived from TLR2 KO, MyD88 KO, and WT C57Bl/6 mice were stimulated with SMIP2.1. No IL-6 release was detected in the supernatants of TLR2 KO and MyD88 KO BM-DCs (). Together, these results suggest that SMIP2.1 is a TLR2 agonist, active on both human and mouse receptors. Since SMIP2.1 differs from Pam3CSK4 only at the solvent exposed region of the ligand in the TLR1/2 co-crystal structure while conserves the hydrophobic acyl chains critical for TLR1/2 heterodimerization,Citation23 it is likely that these 2 molcules share a similar binding conformation.
Figure 1. Activity of a new synthetic compound on human and mouse TLR2. (A), Structure of SMIP2.1 (IUPAC Name=(4R)-4-[(2S)-2-[(2R)-3-{[(2R)-2,3bis(dodecanoyloxy)propyl]sulfanyl}-2-hexadecanamidopropanamido]butanamido]-4-carbamoylbutanoic acid). (B) Luciferase expression in HEK293-cells stable transfected with FLAG-tagged human TLR2 (dotted lines) or with HA-tagged mouse TLR2 (solid lines) and a NF-κB-luciferase reporter gene after stimulation with different doses of SMIP2.1. Pam3CSK4 was used as positive control for activation of TLR2 HEK293 transfectants. (C) Human MoDCs were stimulated for 24 hours with different concentration of SMIP2.1 in the absence or in the presence of a blocking anti-TLR2 antibody. Secretion of TNFα in the supernatants was measured as indicator of the lipopeptide activity. (D) BM-DCs from the indicated mice were stimulated for 24 hours with different concentrations of SMIP2.1. Secretion of IL-6 in the supernatants was measured as indicator of the lipopeptide activity. Group of 6 BALB/c mice were immunized intramuscularly at 0, 21 and 35 d with vaccines containing 1 μg of TT antigen (E) or with 0.2 μg of H1N1 Solomon Flu subunit antigen (F) with the addition of the indicated concentration of SMIP2.1. 2 weeks after the third immunization sera were pooled and antigen specific antibody titers were measured by ELISA.
![Figure 1. Activity of a new synthetic compound on human and mouse TLR2. (A), Structure of SMIP2.1 (IUPAC Name=(4R)-4-[(2S)-2-[(2R)-3-{[(2R)-2,3bis(dodecanoyloxy)propyl]sulfanyl}-2-hexadecanamidopropanamido]butanamido]-4-carbamoylbutanoic acid). (B) Luciferase expression in HEK293-cells stable transfected with FLAG-tagged human TLR2 (dotted lines) or with HA-tagged mouse TLR2 (solid lines) and a NF-κB-luciferase reporter gene after stimulation with different doses of SMIP2.1. Pam3CSK4 was used as positive control for activation of TLR2 HEK293 transfectants. (C) Human MoDCs were stimulated for 24 hours with different concentration of SMIP2.1 in the absence or in the presence of a blocking anti-TLR2 antibody. Secretion of TNFα in the supernatants was measured as indicator of the lipopeptide activity. (D) BM-DCs from the indicated mice were stimulated for 24 hours with different concentrations of SMIP2.1. Secretion of IL-6 in the supernatants was measured as indicator of the lipopeptide activity. Group of 6 BALB/c mice were immunized intramuscularly at 0, 21 and 35 d with vaccines containing 1 μg of TT antigen (E) or with 0.2 μg of H1N1 Solomon Flu subunit antigen (F) with the addition of the indicated concentration of SMIP2.1. 2 weeks after the third immunization sera were pooled and antigen specific antibody titers were measured by ELISA.](/cms/asset/d04f8a66-e498-4139-a995-1340fba496ac/khvi_a_1027467_f0001_b.gif)
We then examined whether SMIP2.1 could function as an adjuvant in vivo using 2 vaccines against tetanus and influenza. In animals immunized with either of these 2 vaccines, inclusion of SMIP2.1 in the vaccine formulation significantly increased the relevant antigen specific antibody titer ().
SMIP2.1 induces cross-presentation in vivo
In order to test the potential of SMIP2.1 to induce cross-presentation of soluble antigens, we assessed its effects on antigen-induced expansion of CD8+ T cells in vivo. We used TCR transgenic OT-I CD8+ T cells, which recognize an OVA-derived peptide and have been extensively used in many experimental settings to investigate cross-presentation.Citation24
We adoptively transferred CFSE-labeled OT-I CD8+ T cells into Ly5-congenic mice and 24 hours later we immunized the recipients with PBS, OVA alone or OVA formulated with SMIP2.1. After 48 h proliferation of OT-I in draining LNs was evaluated by flow cytometry. We used the extent of OT-I CD8+ T cell expansion as a measure of OVA cross-presentation and observed increased proliferation of OT-I cells in mice immunized with OVA in the presence of SMIP2.1 (on average 48.9% dividing OT-I cells) compared to OVA alone (24.5% dividing OT-I cells) (). When we immunized recipient mice with the OVA257–264 peptide, that is able to bind directly MHC class I molecules on DCs without intracellular antigen processing, no increased proliferation of OT-I cells induced by SMIP2.1 was observed (data not shown). This suggests that the increased T cell proliferation in mice immunized with OVA and SMIP2.1 is not due to the effect of the adjuvant on the up-regulation of co-stimulatory molecules or the maturation of DCs.
Figure 2. SMIP2.1 induces cross-presentation in vivo. (A), Congenic Ly5 CD45.1+ mice, injected in the tail vein with CFSE-labeled OT-I CD8+ T cells, were immunized with PBS or OVA (10 μg/mouse) alone or adjuvanted with SMIP2.1 (10 μg/mouse). Proliferation of adoptively transferred cells was assessed after 3 d in draining LNs by flow cytometry, gating on viable CD3+, CD8+, CD45.2+, CFSElow CD8+ T cells. Upper panels show flow cytometry analysis for one mouse in each group. The percentage of proliferating CD8+ T cells (mean ± SD) in each group is reported in the lower graph. Data are representative of 3 independent experiments (B-D), C57Bl/6 mice were immunized 3 times with PBS or OVA (25 μg/mouse) alone or adjuvanted with SMIP2.1 (10 μg/mouse). (B) 7 d after the first immunization OVA specific frequency of CD8+ T cells was measured in the blood by Kb/OVA257–264 tetramer staining. Upper panels show flow cytometry analysis for one mouse in each group. The percentage of tetramer+ CD8+ T cells (mean ± SD) in each group is reported in the lower graph. Data shown are representative of 2 independent experiments. (C) 7 d after the third immunization splenocytes were stimulated for 6 h with OVASIINFEKL peptide (3 μg/ml) or PBS, as negative control, then fixed and stained for intracellular IFNγ and TNFα. (D) Total anti-OVA IgG serum titers were measured by ELISA 2 weeks after the third immunization. **P < 0.01; *** P < 0.001.
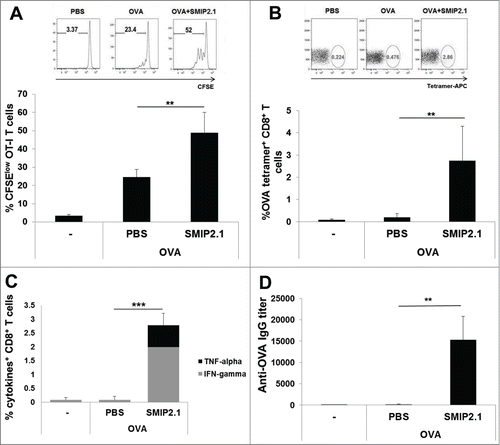
We then tested the ability of SMIP2.1 to enhance cross-presentation in normal C57Bl/6 mice immunized 3 times (at days 1, 21, 35) with PBS or OVA in the presence or absence of SMIP2.1. Priming of CTLs induced by cross-presentation of OVA protein to naive CD8+ T cells was evaluated by monitoring the percentage of Kb-OVA257–264 tetramer positive peripheral blood CD8+ T cells 7 d after the 1st (data not shown) and the 2nd immunization. While no significant expansion of antigen-specific CD8+ T cells was seen in mice immunized with OVA alone, the addition of TLR2 agonist SMIP2.1 increased the frequency of OVA-primed CD8+ T cells ().
Mice were sacrificed 7 d after the 3rd immunization, and splenocytes were re-stimulated ex vivo with PBS alone or with the OVA257–264 peptide. We compared the ability of splenic CD8+ T cells to respond to antigenic stimuli by measuring intracellular IFNγ and TNFα production by flow cytometry (). Splenic CD8+ T cells of mice immunized with OVA adjuvanted with SMIP2.1 showed strong cytokine production when re-stimulated in vitro.
Taken together, these data demonstrate that inclusion of SMIP2.1 in the vaccine formulation led to expansion of the number of functionally active antigen-specific CD8+ T cells. Moreover, as previously shown for tetanus and influenza vaccine antigens, we demonstrated that SMIP2.1 could induce high titers of OVA-specific total IgG 2 weeks after the 3rd immunization ().
SMIP2.1 induces antigen-specific CTL activity in vivo
We next evaluated whether antigen-specific CD8+ T cells, induced by immunization with OVA plus SMIP2.1, were cytolytic. We used an in vivo cytotoxicity assay in C57Bl/6 mice immunized with either PBS or OVA in the presence or absence of SMIP2.1. Seven days after immunization, mice were injected i.v. with two CFSE-labeled splenocytes populations, loaded with either the OVA257–264 peptide or an irrelevant control peptide, and mixed in a ratio of 1:1 prior to injection. The SIINFEKL-loaded splenocytes were labeled with 1 μM CFSE (CFSEhigh target population), while the control splenocytes, loaded with an irrelevant peptide, were labeled with 0.1 μM CFSE (CFSElow cell population). Twenty-four hours after target cells administration, survival of CFSE-labeled cells in the spleen of injected animals was assayed by flow cytometry and OVA-specific killing was calculated as described in Materials and Methods.
Cytolytic activity toward SIINFEKL-coated target cells was increased when mice were immunized with OVA and SMIP2.1 compared to when immunized with OVA alone (). OVA-specific CTL activity correlated with the expansion of the OVA-specific CD8+ T lymphocyte population, as demonstrated by the increase in the percentage of peripheral blood Kb-OVA257–264 tetramer positive CD8+ T cells ().
Figure 3. SMIP2.1 induces antigen-specific CTL activity in vivo. (A and B), C57Bl/6 mice were immunized with PBS or OVA (25 μg/mouse) alone or adjuvanted with SMIP2.1 (10 μg/mouse). (A), After 7 days, syngeneic splenocytes loaded with 2 different concentrations of CFSE and pulsed with either OVASIINFEKL peptide (CFSEhigh) or an irrelevant control peptide (CFSElow) were injected i.v. into recipient mice at a ratio of 1:1. Twenty-four hours later, the CTL response was assessed in draining LNs by measuring the presence of CFSEhigh target cells using flow cytometry. Upper panels show flow cytometry analysis while the graph at the bottom shows the percentage of specific lysis of fluorescent target cells in the different groups calculated as described in the Material and Methods section. (B), A peripheral blood sample was obtained from mice prior to infusion of cells. Cells were stained with Kb/OVA257–264 tetramer to measure the frequency of OVA specific CD8+ T cells. The percentage of Kb/OVA257–264 tetramer+ CD8+ T cells in mice immunized with PBS was subtracted from the other groups. (C), C57Bl/6 mice were immunized twice with PBS or OVA (25 μg/mouse) alone or adjuvanted with SMIP2.1 at the indicated concentration. Seven days after the second immunization, mice were implanted s.c. with OVA-expressing E.G7 tumor cells and mice were monitored for tumor growth. The graph shows the percentage of tumor-free mice along 47 d after tumor cells implantation. Mice were euthanized when moribund. Representative data of 3 independent experiments are shown. Statistical analysis was performed vs OVA immunized group: *P ≤ 0.05; *** P < 0.001.
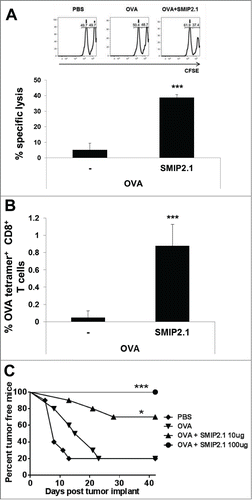
We next assessed whether CD8+ T cells elicited by immunization with OVA and SMIP2.1 could recognize and kill OVA-expressing tumor cells. We therefore evaluated the ability of mice immunized with OVA and SMIP2.1 to reject E.G7-OVA tumor cells. Mice were immunized twice with PBS, OVA protein alone or formulated with SMIP2.1 at 10 or 100 μg/mouse. Seven days after the second immunization, mice were inoculated subcutaneously with OVA-expressing E.G7 tumor cells and tumor growth was monitored. Forty-seven days after inoculation with E.G7-OVA tumor cells, 80% of non-vaccinated and OVA-immunized mice died (20% survival). Mice immunized with OVA in combination with 10 μg of SMIP2.1 showed increased survival as only 3 out of 10 animals developed tumors (70% of survival) (). The group of mice immunized with a higher dose (100 μg) of SMIP2.1 showed complete protection against tumor growth (100% of survival). The same amount of SMIP2.1 without antigen did not induce any protection (data not shown). We conclude that in a prophylactic setting immunization with OVA plus SMIP2.1 inhibits tumor growth.
SMIP2.1 induces antigen deposition
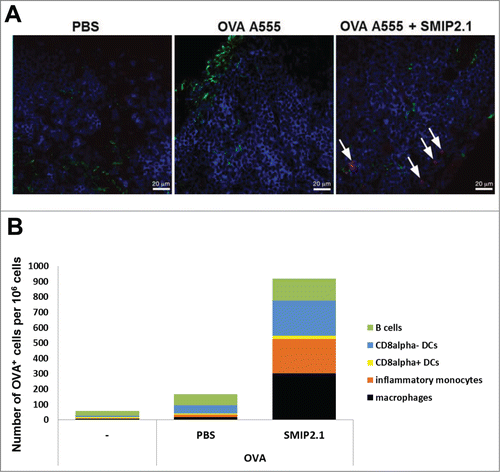
The mechanisms by which adjuvants enhance immunogenicity of the co-administered antigens have been explored previously. In some cases adjuvanticity has been linked to an increased antigen deposition in the LNs, thus prolonging antigen availability and presentation in vivo.Citation25 To explore the mechanisms that underlie the action of TLR2 agonists as vaccine adjuvants, we considered the possibility that SMIP2.1 increases antigen deposition in the draining LNs. Mice were immunized with fluorescent-labeled OVA in the presence or absence of SMIP2.1. After 24 hours draining LNs were collected to assess antigen deposition. We analyzed various tissues by confocal microscopy and found that mice immunized with OVA and SMIP2.1 showed increased deposition of OVA compared to mice immunized with OVA alone (). We then evaluated the effect of SMIP2.1 on antigen uptake by specific cell types in the draining LNs, analyzing by FACS the presence of fluorescent-OVA in CD8α− DCs (CD11bhigh, CD11c+, CD8a−, MHCII+), CD8α+ DCs (CD11bhigh, CD11c+, CD8a+, MHCII+), macrophages (Cd11b+, F4/80high), inflammatory monocytes (CD11bhigh, CD11c−, Ly6Chigh) and B cells (CD11b−, CD11c+, MHCII+). In agreement with the confocal microscopy results, SMIP2.1 induced an increase in the total number of OVA-positive cells for all cellular subtypes analyzed ().
Figure 4. SMIP2.1 increases antigen deposition in the draining LN. (A), C57Bl/6 mice were immunized with PBS or OVA A555 (25 μg/mouse) alone or adjuvanted with SMIP2.1 (100 μg/mouse) and draining LNs were collected 24 h later. 8 μm thick cryosections of draining LNs were stained for CD169 (green) and CD45R (blue) and observed under a confocal microscope. The picture (63x magnification) shows the OVA antigen deposition (red) only in mice immunized with OVA and SMIP2.1. Bar represents 20 μm. (B), Groups of 3 mice were immunized with PBS or OVA A647 (25 μg/mouse) alone or adjuvanted with SMIP2.1 (100 μg/mouse). Draining LNs were collected 24 h later and analyzed in pool by FACS to identify specific cell types and antigen-content. The graph shows the number of OVA A647+ cells per 1 × 106 total cells. The data shown are representative of 2 independent experiments.
SMIP2.1 increases cross-presentation in both CD8α+ and CD8α- DCs
Based on the expression of the CD8α marker, 2 major populations of DCs with different functions can be identified in mice. CD8α+ CD11c+ DCs have been suggested to possess the unique ability to cross-present antigensCitation26-28 whereas among human cells CD141+ (BDCA3+) DCs were described as the most efficient at cross-presenting.Citation29,30 However, evidence exists that both murine CD8α+ and CD8α− CD11c+ DCs subsets are able to present exogenous antigen to CD8+ T lymphocytes with equivalent efficacy when appropriately stimulated.Citation31,32 Since immunization with TLR2 agonist increases the antigen uptake by both CD8α+ CD11c+ and CD8α− CD11c+ DCs, we assessed the potential of SMIP2.1 to enhance the cross-presentation ability of both populations.
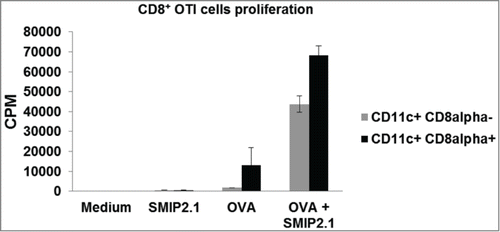
CD8α+ CD11c+ and CD8α− CD11c+ DCs were purified from C57Bl/6 splenocytes and loaded with soluble endotoxin-free OVA protein in the presence or absence of SMIP2.1 for 4 hours, prior to co-culture with OT-I CD8+ T cells in order to evaluate their capacity to induce proliferation of OVA specific CD8+ T cells. Although CD8α+ CD11c+ DCs were better at cross presentation in the absence of TLR2 agonist, both DCs populations pulsed with OVA in the presence of SMIP2.1 induced a greater OT-I proliferation compared to DCs pulsed with OVA alone ().
Figure 5. SMIP2.1 increases cross-priming of OT-(I)cells in vitro. CD8α+ CD11c+ and CD8α− CD11c+ DCs were purified by cell sorting from the spleen of C57Bl/6 mice and incubated with medium or OVA (10 μg/ml) alone or in the presence of SMIP2.1 (10 μM) for 4 hours. After washing, DCs were co-cultured with purified OT-I CD8+ T cells for 60 hours and proliferation of CD8+ OTI T cells was measured by 3H thymidine incorporation. Data in the graph indicate counts per min (CPM) and are expressed as mean ± SD of triplicate wells. Values of CPM from each cell population incubated with medium alone were subtracted from values of CPM in all the other conditions.
SMIP2.1 stimulates cross-presentation in B cells
Among APCs, DCs have the unique ability to cross-present,Citation28 while the role of B cells in CD8+ T cell cross-presentation has been more controversial. B cells are generally regarded as not being as effective as DCs for T cells priming. However, the ability of B cells to efficiently processCitation33,34 and present antigens to CD4+ T cellsCitation35,36 suggest that B cells might also possess the ability to cross-present antigens.
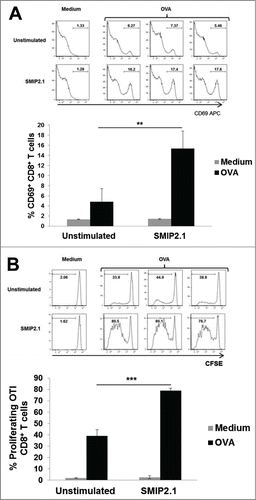
A complication in testing whether B cells can cross present antigens to CD8+ T cell arises from the low frequency of naïve B cells specific for a given antigen, making antigen recognition and receptor-mediated uptake a rare event for polyclonal B cells. To assess the potential of SMIP2.1 to enhance cross-presentation of B cells, we used OBI RAG−/− mice, generated by somatic cell nuclear transfer. These mice have a single population of B cells with a BCR that specifically recognizes an OVA epitope.Citation37 Purified CD19+ B cells from the OB1 mouse were cultured with medium alone or with SMIP2.1 for 20 hours. The B cells were then washed and loaded with soluble OVA protein for 4 hours before being added to CFSE-labeled CD8+ OT-I T. Upregulation of the activation marker CD69 and proliferation of the OT-I cells was assessed as an indication of CD8+ T cell activation. While unstimulated OBI B cells pulsed with OVA induced a modest up-regulation of CD69, stimulation of OBI B cells with SMIP2.1 prior to OVA loading rendered B cells able to induce a significant upregulation of CD69 on CD8+ OT-I T cells (). In addition, TLR2-activated OBI B cells greatly enhanced proliferation of CD8+ OTI T cells (). Therefore we conclude that B cells can present exogenous processed antigens to CD8+ T cells when activated through TLR2.
Figure 6. SMIP2.1 increases cross-presentation by OBI B cells in vitro. Isolated OBI B cells were incubated for 20 h with medium (Unstimulated) or SMIP2.1 (10 μg/ml). After washing, B cells were loaded with OVA (100 μg/ml) for 4 h and then co-cultured with purified CFSE labeled CD8+ OT-I T cells. (A), Surface expression of CD69 by OT-I cells was analyzed by flow cytometry after 20 hours. Histograms of flow cytometry data are shown in the upper panel while mean values for the percentage of CD69 positive cells from 3 samples in each condition are reported in the lower panel. (B), After 52 hours, proliferation of OT-I cells was assessed by flow cytometry detection of CFSE. Histograms of flow cytometry data are shown in the upper panel while mean values for the percentage of CFSElow cells from 3 samples in each condition are reported in the lower panel. Error bars indicate SD. **P < 0.01; *** P < 0.001.
SMIP2.1 stimulates cross-presentation in human cells in vitro
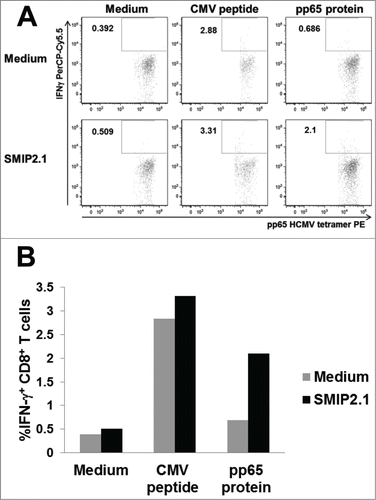
To test if SMIP2.1 could induce cross-presentation also in human cells, we set up a cell-based in vitro model. In this model the cross-priming of human PBMCs from HCMV seropositive donors by HCMV-derived recombinant pp65, was evaluated using flow cytometry to detect intracellular cytokine production by CD8+ T cells. HCMV is a highly prevalent herpes virus: CD8+ T cell responses to HCMV antigens are detectable in most HCMV-seropositive donors.Citation38 PBMCs from HCMV seropositive donors were therefore stimulated with a library of pp65 peptides to expand the HCMV-specific memory CD8+ T cell population in the presence of exogenous IL-2. The population of HCMV-specific CD8+ T cells was expanded approximately 2-fold (from 0.8% to 2.1% of the total CD8+ T cell population, data not shown) and was then restimulated with autologous PBMCs loaded with HCMV pp65 with or without SMIP2.1. After 4 hours of stimulation, cells were fixed, permeabilized and stained with CD3, CD8, IFNγ antibodies and HCMV-tetramers to detect IFNγ production by HCMV-specific CD8+ T cells.
APCs pulsed with the pp65 protein and stimulated with SMIP2.1 cross-presented pp65 more efficiently than APCs pulsed with pp65 alone (), as demonstrated by their ability to induce a larger fraction of IFNγ producing HCMV-specific CD8+ T cells.
Figure 7. SMIP2.1 induces cross-presentation in human cells in vitro. PBMCs from a HCMV seropositive donor were pulsed in vitro as indicated for 2 hours, washed and co-cultured for 4 hours with an expanded HCMV specific CD8+ T cell population from the same donor. Production of IFNγ by CD8+ T cells was quantified by intracellular staining and flow cytometry. Dot plot analysis (A) and the percent of CD3+, CD8+, pp65 HCMV tetramer+ IFNγ+ T cells (B) are shown.
Discussion
Failure to stimulate a CTL response is a major limitation of subunit vaccines. Exogenous proteins in vaccine formulations are poor inducers of CD8+ T cell immunity, as they are captured by APCs and presented primarily to CD4+ T helper cells, leading to a good humoral immune response but poor cellular CD8+ T cell immunity. Therefore, identification of vaccine adjuvants that can enhance Class I MHC presentation of purified soluble antigens to CD8+ T cells would be valuable in the development of vaccines against viral targets or cancer, for which the induction of a CTL response is critical.
Here we demonstrated that a soluble protein can be directed into the Class I MHC antigen-restricted processing and presentation pathway by the use of SMIP2.1, a TLR2-based adjuvant. Indeed, combination of OVA with SMIP2.1, in addition to enhancing antibody production, was able to increase priming of CD8+ T cells. This resulted in greater numbers of OVA-responsive CD8+ T cells, as demonstrated by both an increase of IFNγ producing cells upon ex vivo re-stimulation and the lysis of target cells in vivo. Moreover, using an OVA expressing tumor model, we show that the CTL response induced by the SMIP2.1-adjuvanted vaccine inhibits tumor growth. Therefore, SMIP2.1 could be a very useful adjuvant for subunit vaccines, especially whenever a CTL response is required.
SMIP2.1 was selected from an HTS screening in which we identified a group of triacetylated lipopeptides active on human and mouse TLR2. However, we did not analyze the effect on cross-presentation neither of those analogs, nor of other available TLR2 agonists. Future work comparing the effect of TLR2 agonists with different chemical structures on cross-presentation will definitely contribute to a better understanding of the mechanism of action of this class of adjuvants.
The mechanisms by which adjuvants can enhance the immunogenicity of vaccine antigens have not been completely elucidated. Formation of antigen depots may be one of the biological processes associated with adjuvanticity. Adjuvants would thus enhance an immune response through prolonging the persistence of antigens in vivo. Indeed, we found that in mice immunized with OVA, SMIP2.1 increases the number of APCs (CD8α− DCs, CD8α+ DCs, macrophages, inflammatory monocytes and B cells) that take up antigen and migrate to LNs. We can then speculate that this increased uptake results in enhanced cross-presentation by both CD8α− and CD8α+ DCs populations as observed in the in vitro assays and it may also account for the increased immune response against the different antigens we tested in vivo.
It has been described that only CD8α+ CD11c+ DCs can cross-present in miceCitation39,40 and most of the adjuvants that can stimulate cross-presentation act exclusively on this population,Citation41,16 which represents only ˜20% of the conventional DCs in mouse spleen.Citation27 The ability of SMIP2.1 to activate cross-presentation also in the CD8α− subset offers the advantage of acting on a larger cell population.
The role of B cells in priming CD8+ T cells in vivo has been difficult to evaluate because of the dominant role of DCs in cross-presentation and the lack of appropriate animal models. We took advantage of OB1 BCR transnuclear mice that specifically recognize an epitope of the OVA protein, in order to assess the function of B cells in T cell cross-priming. When crossed with a RAG-deficient background, OB1 mice have a single population of B cells, all of which are specific for the OVA antigen. Since B cells express several TLRs on their surface, including TLR2,Citation42 we analyzed the ability of SMIP2.1 stimulated OBI B cells to cross-present the OVA antigen captured via BCR. We clearly observed that TLR2 engagement results in induction of the ability of B cells to powerfully cross-present OVA in vitro to OVA specific T cells. Although TLR2 engagement can potentially induce up-regulation of co-stimulatory molecules in B cell, we can exclude that this effect could be responsible of the increase in T cell proliferation. In fact, presentation of OVA257–264 peptide by OB1 cells results in maximal proliferation of OT-I cells (data not shown), excluding that co-stimulation is a limiting factor in the activation of these cells. To the best of our knowledge this is the first time that engagement of TLR2 in B cells is shown to activate antigen cross presentation in these cells. Although our results will need to be confirmed in an in vivo model, this finding expands our knowledge of the adjuvant properties of SMIP2.1. Indeed, in addition to its ability to enhance antibody production, SMIP2.1 can also directly target B cells to help primary CD8+ T cell expansion. We think that by using a rational chemical approach to engineer TLR2 agonists the function of these molecules can be modified so to obtain the desired effect on the immune response.
Cross-presentation has been well-characterized in different animal models, while few reports address cross-presentation by human cells. Sub-populations of human DCs have been targeted through C-type lectin receptors to increase their ability to cross-present.Citation43-45 Moreover, it has been shown that human CD141+ DCs represent the human counterpart of the murine CD8α+ DCs, which are the cell type most effective at antigen cross-presentation.Citation29,30,46 Our data showed that SMIP2.1 adjuvant increases cross-presentation in total human PBMCs, although we could not establish which subpopulation is specifically targeted by SMIP2.1.
TLR ligands improve vaccine efficacy and a better immune response is generated when a TLR agonist and antigen are linked covalentlyCitation16,47,48 or electrostatically.Citation17 However, these 2 approaches may complicate large-scale production of vaccines. For example, scale-up of chemical conjugation of a soluble antigen with a lipopeptide can be challenging because of the different solubility characteristics of these 2 components. SMIP2.1 is a triacetylated lipopeptide, and induces a good immune response without the need to be coupled covalently to the antigen. Using HEK293 cells stably transfected with TLR2, we screened a series of TLR2 agonist compound and selected SMIP2.1 for its favorable physical-chemical characteristics and suitability of the synthetic path for a scale-up process. However, this molecule could be further refined to improve some specific activities including boosting of humoral response and further enhancement of cross-presentation.
In conclusion, our work shows that a SMIP2.1 adjuvanted vaccine induces a strong CTL response after a single immunization in mice and allows antigen access to the cross-presentation pathways in different APCs, including B cells. SMIP2.1 is therefore a promising adjuvant for the development of vaccines that aim to enhance a CD8+ T cell response.
Materials and Methods
Stimuli
The palmitoyl3Cys-Ser-Lys4 lipopeptide (Pam3CSK4) was purchased from Alexis. For the in vivo and in vitro studies reported in this manuscript, SMIP2.1 was formulated and dosed as an aqueous suspension. Aqueous suspension formulation was prepared by probe sonication of the dry powder in PBS. Size of the particle suspension was monitored by granulometric analysis (particles size analyzer Beckman Coulter LS13320) to ensure an average particles size <20 micron.
Cell Culture
All cultured cells were grown at 37°C in a humidified environment containing 5% CO2.
Human Embryonic Kidney HEK293 cells expressing FLAG-tagged humanTLR2 were cultured in complete medium (DMEM containing 4500 mg/l glucose, 10% heat-inactivated FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine) supplemented with puromycin (5 μg/ml) and Hygromycin (250 μg/ml).
HEK293 cells expressing HA-tagged mouse-TLR2 were cultured in complete medium supplemented with puromycin (5 μg/ml) and blasticidin (10 μg/ml).
Mouse cells from lymph nodes (LNs) and spleens were cultured in RPMI 1640 (GIBCO) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2mM glutamine (GIBCO), 2 × 10−5 M 2-mercaptoethanol (Sigma) and 5% heat-inactivated fetal calf serum (Hyclone).
Human primary cell isolation
Human PBMCs were isolated from buffy coats of healthy donors using Ficoll gradient (Amersham) and cultured in RPMI 1640 (GIBCO) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, 1% of non-essential amino acids (GIBCO), and 5% heat-inactivated AB human serum (Euroclone). Buffy coats from healthy donors were obtained from the Blood Transfusion Section, Empoli Hospital. Informed consent was obtained before all blood donations. The study protocol was approved by the Novartis Research Center ethical committee and conforms to the ethical guidelines of the 1975 Declaration of Helsinki.
Highly purified monocytes (>98%) were obtained from PBMCs by a one-step positive selection using MACS anti-CD14-coated magnetic microbeads (Miltenyi Biotec) according to the manufacturer's instructions. Monocyte-derived DCs (MoDCs) were differentiated from fresh isolated monocytes culturing them with GM-CSF (50 ng/ml) and human recombinant IL-4 (5 ng/ml) (both from Gentaur) for 5–7 d. MoDC differentiation was verified by staining with anti-CD1a-FITC and anti-CD14-APC (BD Biosciences) while their phenotypic maturation was evaluated by surface staining with anti-CD80-FITC and anti-CD86-APC (BD Biosciences). MoDCs were seeded in 96-well round bottom plates (1 × 105 cells/well) and stimulated over night with different concentrations of SMIP2.1. In blocking experiments, cells were pre-incubated for 20 minutes with 25 μg/ml of anti-TLR2 antibody (BD Biosciences) and then stimulated with SMIP2.1 overnight.
Production of human TLR2 expressing cells (HEK293-FLAG-hTLR2-NF-κB-Luc) and mouse TLR2 expressing cells (HEK293-mTLR2HA-NF-κB-Luc)
HEK293-NF-κB-Luc cells (clone LP58), a cell line stably transfected with a reporter vector in which the luciferase gene is under the control of an NF-κB dependent promoter, were previously produced by Cell and Molecular Technologies Inc. (CMT) for Chiron Corporation under a service contract. These cells were transfected using lipofectamine 2000 (Invitrogen) with pcDNA3.1-Hygro-FLAG-hTLR2 plasmid encoding for human TLR2 containing a FLAG epitope at the NH2 terminus and a hygromycin resistance gene for selection or pUNO-mTLR2HA (Invivogen) plasmid encoding for mouse TLR2 containing a HA epitope at the C- terminus and a blasticidin resistance gene for selection. Transfected cells were cultured in the presence of respective selection antibiotics and individual resistant clones were picked, expanded and tested for expression of luciferase upon stimulation with the TLR2 agonist Pam3CSK4.
In vitro activity on human and mouse TLR2 stable cells
For the luciferase reporter gene assay, HEK293-FLAG-hTLR2-NF-κB-Luc cells or and HEK293-mTLR2HA-NF-κB-Luc cells (25 × 103 cells/well) were seeded into 96-well flat bottom plates in 90 μl of complete medium in the absence of selection antibiotics. After overnight incubation, cells were stimulated in duplicates with different concentrations of stimuli (10 μl/well) for 6 hours.
Subsequently, medium was discarded and cells were lysed with 20 μl of Passive Lysis Buffer (Promega) for 20 min at room temperature and Luciferase levels were measured by addition of 100 μl/well Luciferase Assay Substrate (Promega) using the LMax II384 microplate reader (Molecular Devices). Relative luminescence units (RLU) from each sample (average of 2) were divided by the RLU of the control sample (PBS or DMSO) and expressed as fold induction (FI).
Preparation of Bone-marrow derived dendritic cells (BM-DCs)
BM-DCs were obtained by culturing bone marrow cells from C57/BL6, TLR2-knockout (KO) and Myd88 KO mice (Oriental Bioservice), with mouse recombinant GM-CSF (5 ng/ml) and IL-4 (10ng/ml) (both from Gentaur) for 7 d. Differentiation of BM-DCs was confirmed by anti-CD11c staining. BM-DCs were seeded into 96-well round bottom plates (2 × 105 cells/well) and stimulated over night with escalating doses of SMIP2.1.
Measurements of cytokine production
All cytokine measurements in the supernatants of cultured cells were performed using Mesoscale Assay 7-spot (MSD Technology) to detect human or mouse cytokines following manufacturer's instructions.
Mice and immunizations
Females 8 weeks old BALB/c, C57Bl/6, OT-I and B6-Ly5.2 (Charles River) mice were used for immunogenicity studies approved by the institutional animal welfare committee. Animals were immunized intra-muscularly in both quadriceps muscle with Dulbecco's PBS (DPBS 1x, GIBCO) alone, 10 or 25 μg of OVA protein (EndoGradeTM Ovalbumin, Hyglos), 0.2 μg of H1N1 Solomon Flu subunit antigen or 1 μg of Tetanus Toxoid (TT), dissolved in DPBS or in the presence of SMIP2.1. A total volume of 50 μl was injected per leg. Six to ten mice per group were used in different experiments. Mice were immunized at days 0, 21 and 35. Blood samples for antibody titer analysis were collected 2 weeks after the second and the third immunization, and serum prepared by centrifugation. For Kb/OVA257–264 tetramer analysis, blood was collected 1 week after each immunization in a tube with heparin.
In vivo proliferation of OVA-specific T cells
CD8+ T cells were separated from spleen and total LNs of OT-I mice by a positive selection using a CD8a+ T cell isolation kit (Miltenyi Biotec) accordingly to manufacturer instructions. The efficiency of enrichment was routinely 85–95%, as determined by flow cytometry. Cells were incubated with CFSE at 0.5 μM for 15 min at room temperature (RT) in the dark and CFSE staining was quenched by the addition of 5 ml of FBS. Cells were then washed extensively with PBS and injected intravenously into B6-Ly5.2mice (1 × 106 in 100 ul/mouse). After 1 day, mice were immunized as indicated. Proliferation of adoptively transferred OT-I cells in the inguinal LNs was quantified 48 h after the immunization by determining the number of CFSElow OT-I cells by flow cytometry after staining with anti-CD3 PE, anti CD8-V500, anti-CD44 APC (BD PharMingen) and anti-CD45.2 Alexa 700 (eBioscience).
Tetramer analysis of OVA-specific CD8+ T cells
Peripheral blood samples, collected from the saphenous vein at the indicated time points, were stained with APC-conjugated Kb/OVA257–264 tetramers (Beckman Coulter), anti-CD8 PE Texas Red, anti-CD3 PerCP Cy5.5, anti-CD4 V500, anti-CD44 V450 (BD PharMingen) for 30 min at RT. Samples were lysed and fixed with iTAG MHC tetramer lyse and fixative (Beckman Coulter). Cells were analyzed by flow cytometry collecting a minimum of 2.5 × 105 events.
In Vivo Killing Assay
A single-cell suspension of splenocytes from naïve B6-Ly5.2 mice was prepared, and red cells were lysed. Cells were split into 2 aliquots and labeled with either high (1 μM) or low (0.1 μM) concentration of CFSE. Cells were then suspended at a concentration of 1 × 107 cells/ml in complete RPMI containing 10 μg/ml OVA257–264 peptide (CFSEhigh) or an irrelevant control peptide derived from HCMV pp65protein (CFSElow) and incubated for 30 min at 37°C. After three washes in PBS the 2 labeled cells were mixed at a 1:1 ratio in PBS (1 × 107 cells/ml). 100 μl of cell suspension was delivered intravenously into the tail vein of C57Bl/6 mice that had been immunized 7 d before as indicated. Twenty-four hours after target cell implant, mice were euthanized and splenocytes were stained with CD45.2 Alexa 700. The percentage of CFSEhigh and CFSElow cells in the CD45.2+ population was measured by flow cytometry and the CFSEhigh/CFSElow ratio was calculated. Specific lysis in each group was assessed according to the formula: [1 − (CFSEhigh/CFSElow ratio of mice immunized with adjuvant/mean of CFSEhigh/CFSElow ratio of mice immunized with PBS)] × 100, as described by Ingulli.Citation49
Determination of antigen-specific antibody titer by ELISA
Serum anti-Ovalbumin specific total IgG were measured in individual mice by ELISA using a standard reference serum. Anti H1N1 Solomon Flu or anti-TT specific total IgG titers were measured by ELISA performed on pooled sera from each experimental group and using a standard reference serum. For the OVA IgG ELISA, Maxisorp plates (Nunc) were coated with 50 μg/ml of OVA in carbonate buffer over-night at 4°C. For H1N1 Solomon Flu or TT IgG, plates were coated with 2 μg/ml of subunit H1N1 Solomon Flu or 1 μg/ml of TT antigens in PBS overnight at 27–30°C. Plates were blocked for 1 h at 37°C with 100 μl of PBS 1% BSA. Serum samples and serum standard were serially diluted in PBS, 1% BSA, 0.05% Tween-20 and transferred into antigen coated-blocked plates. Antigen-specific IgGs were detected with alkaline phosphatase-conjugated goat anti-mouse IgG (Southern Biotech) and the substrate 4-Nitrophenyl phosphate disodium salt hexahydrate (SIGMA). Antibody titer was calculated using a 4-parameter interpolation fit as the reciprocal of the dilution of each serum sample.
E.G7 tumor challenge experiments
C57Bl/6 mice were immunized twice as indicated in the . Seven days after the second immunization, mice were subcutaneously inoculated in the right flank with 2.5 × 105 E.G7-OVA cells (ATCC) in 100 μl of PBS. Mice were monitored every 2–3 d for tumors growth. Moribund tumor bearing-mice were euthanized in accordance with Animal Care guidelines.
Confocal analysis of LNs cryosections
OCT-embedded LN was sectioned transversely at 8 μm, mounted onto Superfrost glass slides, and quickly air dried. Slides were fixed in a 4% formaldehyde solution on ice and blocked with PBS, 3% BSA and 1% saponin. Slides were then stained with anti-CD169 FITC (Serotec) and anti-CD45R Alexa700, mounted with ProLong gold antifade reagent (Invitrogen), and imaged by a LSM 710 microscope (Zeiss).
Preparation of LN single cell suspension and flow cytometry
Draining LNs were harvested, cut into small fragments and digested with collagenase D (2 mg/ml, Roche) and DNase I (0.5 mg/ml, Sigma) in RPMI 1640 (Gibco) medium for 30 min at 37°C. Digested fragments were filtered through a 70 μm nylon mesh (Becton Dickinson), before staining with fluorescently labeled antibodies and FACS analysis. Cells were stained with combinations of the following antibodies: anti-Ly6C-FITC, anti-CD11b-PE-Cy7, anti-CD11c-APC, F4/80 PacificBlue, anti-CD11c-APC-AlexaFluor750 (all from eBioscience). Flow cytometry was performed on FacsCanto or FACS LSRII instruments using DIVA software (Becton Dickinson) and data were then analyzed using Flowjo software (Treestar Inc.).
OT-I T cell proliferation in vitro by [3H] Thymidine incorporation
CD11c DCs were isolated from spleens of C57Bl/6 mice by positive cell sorting using FACS Aria sorter. Sorted DCs were plated at 2.5 × 104 cells/well in a 96 well plate with medium or OVA (10 μg/ml) alone or in the presence of SMIP2.1 (10 μM) for 4 hours. DCs were then washed twice with PBS and mixed at a ratio of 1:5 (DC:T cells) with isolated OT-I transgenic CD8α+ T cells from spleen and LNs (Miltenyi Biotec). Cells were then incubated for 54 hours, pulsed with [3H] Thymidine (Amersham Biosciences) at 0.5 μCi/well for additional 18 hours, and harvested onto filter plates (Packard Instruments). [3H]Thymidine incorporation was determined using a Top Count NXT β counter (Packard Instruments).
OT-I T cell proliferation in vitro by CFSE proliferation assay
B cells were purified by negative selection from the spleen of an OBI mouse using anti-mouse CD43 Dynabeads (Untouched B Cells kit from Invitrogen). B cells were resuspended at 2 × 106 cells/ml and incubated with medium alone or SMIP2.1 at 10 μg/ml for 24 hours. Cells were then washed with PBS, plated in a 96 well plate at 5 × 104 cells/well in the presence or absence of OVA (100 μg/ml) for 4 hours. After two washes with PBS, B cells were incubated in a ratio 1:5 (B:T) with CD8+ OT-I T cells labeled with 0.5 μM CFSE (Molecular Probes), as previously described. After 72 hours, cells were stained with Live/dead Yellow dead cells kit (Invitrogen), anti-CD3 PerCP-Cy5.5 and anti-CD8 APC. OT-I proliferation was assessed by flow cytometry by determining the number of CFSElow cells.
Experiments with human PBMCs
PBMCs were assayed for the presence of HCMV specific CD8+ T cells using a panel of HCMV-specific tetramers (Bekman Coulter). Positive samples were then cultured with a mix of peptides derived from pp65 protein (JPT Peptide Technologies) at a concentration of 3 μg per peptide. Proliferating T cells were expanded for 14 d by addition of recombinant hIL-2 (50 U/ml) at days 4, 7 and 10. At day 14, autologous PBMCs were plated in a 96 well plate at 5 × 105 cells/well in complete medium and loaded with pp65 protein (Miltenyi Biotec) at 50 μg/ml with or without SMIP2.1 (15 μM). After 2 hours, cells were washed twice and co-cultured over-night with the expanded HCMV specific CD8+ T cells population in a ratio of 1:1 in the presence of Brefeldin A (Sigma). Cells were then fixed, stained for intracellular cytokine production with anti-CD3 V450, anti-CD8 APC, anti-IFNγ PerCP-Cy5.5 and CMV-specific tetramer PE, and analyzed by flow cytometry.
Statistical analysis
All data are presented as mean +/− SD. Where not present, samples were analyzed in pool. Student's t test was used for statistical evaluation and a p-value below 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by the Transformational Medical Technologies program contract HDTRA1-07-9-0001 from the Department of Defense Chemical and Biological Defense program through the Defense Threat Reduction Agency (DTRA), European Union's seventh framework program grant code 280873 Advanced Immunization Technologies (ADITEC) and CTN01_00177_962865 grant from Ministero dell'Università e delle Ricerca (MIUR). M. S. was supported by an EMBO short term fellowship.
References
- Rappuoli R, Mandl CW, Black S, De Gregorio E. Vaccines for the twenty-first century society. Nature reviews Immunology 2011; 11:865-72; PMID:22051890
- Wiesel M, Walton S, Richter K, Oxenius A. Virus-specific CD8 T cells: activation, differentiation and memory formation. APMIS 2009; 117:356-81; PMID:19400862; http://dx.doi.org/10.1111/j.1600-0463.2009.02459.x
- Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. The Journal of experimental medicine 1976; 143:1283-8; PMID:1083422; http://dx.doi.org/10.1084/jem.143.5.1283
- Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunological reviews 2005; 207:166-83; PMID:16181335; http://dx.doi.org/10.1111/j.0105-2896.2005.00301.x
- O'Neill D, Bhardwaj N. Exploiting dendritic cells for active immunotherapy of cancer and chronic infection. Methods Mol Med 2005; 109:1-18; PMID:15585909
- Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med 2002; 196:1627-38; PMID:12486105; http://dx.doi.org/10.1084/jem.20021598
- Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol 2003; 4:1223-9; PMID:14625548; http://dx.doi.org/10.1038/ni1010
- Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, DiDonato J, Dziarski R, Akira S, Schoenberger SP, et al. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol 2003; 170:4102-10; PMID:12682240; http://dx.doi.org/10.4049/jimmunol.170.8.4102
- Ishii KJ, Akira S. Toll or toll-free adjuvant path toward the optimal vaccine development. J Clin Immunol 2007; 27:363-71; PMID:17370119; http://dx.doi.org/10.1007/s10875-007-9087-x
- Caproni E, Tritto E, Cortese M, Muzzi A, Mosca F, Monaci E, Baudner B, Seubert A, De Gregorio E. MF59 and Pam3CSK4 Boost Adaptive Responses to Influenza Subunit Vaccine through an IFN Type I-Independent Mechanism of Action. J Immunol 2012; PMID:22351935; http://dx.doi.org/10.4049/jimmunol.1101764
- Steinhagen F, Kinjo T, Bode C, Klinman DM. TLR-based immune adjuvants. Vaccine 2011; 29:3341-55; PMID:20713100; http://dx.doi.org/10.1016/j.vaccine.2010.08.002
- Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, Reis e Sousa C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005; 433:887-92; PMID:15711573; http://dx.doi.org/10.1038/nature03326
- Oh JZ, Kedl RM. The capacity to induce cross-presentation dictates the success of a TLR7 agonist-conjugate vaccine for eliciting cellular immunity. J Immunol 2010; 185:4602-8; PMID:20844205; http://dx.doi.org/10.4049/jimmunol.1001892
- Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini JC, Romero P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest 2005; 115:739-46; PMID:15696196; http://dx.doi.org/10.1172/JCI23373
- Garcon N, Van Mechelen M. Recent clinical experience with vaccines using MPL- and QS-21-containing adjuvant systems. Exp Rev Vaccines 2011; 10:471-86; PMID:21506645; http://dx.doi.org/10.1586/erv.11.29
- Prajeeth CK, Jirmo AC, Krishnaswamy JK, Ebensen T, Guzman CA, Weiss S, Constabel H, Schmidt RE, Behrens GM. The synthetic TLR2 agonist BPPcysMPEG leads to efficient cross-priming against co-administered and linked antigens. Eur J Immunol 2010; 40:1272-83; PMID:20213735; http://dx.doi.org/10.1002/eji.200939790
- Chua BY, Pejoski D, Turner SJ, Zeng W, Jackson DC. Soluble proteins induce strong CD8+ T cell and antibody responses through electrostatic association with simple cationic or anionic lipopeptides that target TLR2. J Immunol 2011; 187:1692-701; PMID:21742967; http://dx.doi.org/10.4049/jimmunol.1100486
- Shen KY, Song YC, Chen IH, Leng CH, Chen HW, Li HJ, Chong P, Liu SJ. Molecular mechanisms of TLR2-mediated antigen cross-presentation in dendritic cells. J Immunol 2014; 192:4233-41; PMID:24683188; http://dx.doi.org/10.4049/jimmunol.1302850
- de Wit J, Souwer Y, Jorritsma T, Klaasse Bos H, ten Brinke A, Neefjes J, van Ham SM. Antigen-specific B cells reactivate an effective cytotoxic T cell response against phagocytosed Salmonella through cross-presentation. PloS One 2010; 5:e13016; PMID:20885961; http://dx.doi.org/10.1371/journal.pone.0013016
- Jiang W, Lederman MM, Harding CV, Sieg SF. Presentation of soluble antigens to CD8+ T cells by CpG oligodeoxynucleotide-primed human naive B cells. J Immunol 2011; 186:2080-6; PMID:21239717; http://dx.doi.org/10.4049/jimmunol.1001869
- Ritchie DS, Yang J, Hermans IF, Ronchese F. B-Lymphocytes activated by CD40 ligand induce an antigen-specific anti-tumour immune response by direct and indirect activation of CD8(+) T-cells. Scandinavian J Immunol 2004; 60:543-51; PMID:15584965; http://dx.doi.org/10.1111/j.0300-9475.2004.01517.x
- Baschang G. Muramylpeptides and lipopeptides: Studies towards immunostimulants. Tetrahedron 1989; 45:6331-60; PMID:NOT_FOUND; http://dx.doi.org/10.1016/S0040-4020(01)89512-0
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007; 130:1071-82; PMID:17889651; http://dx.doi.org/10.1016/j.cell.2007.09.008
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994; 76:17-27; PMID:8287475; http://dx.doi.org/10.1016/0092-8674(94)90169-4
- Calabro S, Tortoli M, Baudner BC, Pacitto A, Cortese M, O'Hagan DT, De Gregorio E, Seubert A, Wack A. Vaccine adjuvants alum and MF59 induce rapid recruitment of neutrophils and monocytes that participate in antigen transport to draining lymph nodes. Vaccine 2011; 29:1812-23; PMID:21215831; http://dx.doi.org/10.1016/j.vaccine.2010.12.090
- Belz GT, Smith CM, Eichner D, Shortman K, Karupiah G, Carbone FR, Heath WR. Cutting edge: conventional CD8 alpha+ dendritic cells are generally involved in priming CTL immunity to viruses. J Immunol 2004; 172:1996-2000; PMID:14764661; http://dx.doi.org/10.4049/jimmunol.172.4.1996
- Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunological reviews 2010; 234:18-31; PMID:20193009; http://dx.doi.org/10.1111/j.0105-2896.2009.00870.x
- Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 2001; 19:47-64; PMID:11244030; http://dx.doi.org/10.1146/annurev.immunol.19.1.47
- Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 2010; 207:1273-81; PMID:20479115; http://dx.doi.org/10.1084/jem.20100348
- Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 2010; 207:1247-60; PMID:20479116; http://dx.doi.org/10.1084/jem.20092140
- den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(−) dendritic cells in vivo. J Exp Med 2002; 196:817-27; PMID:12235214; http://dx.doi.org/10.1084/jem.20020295
- Moron G, Rueda P, Casal I, Leclerc C. CD8alpha- CD11b+ dendritic cells present exogenous virus-like particles to CD8+ T cells and subsequently express CD8alpha and CD205 molecules. J Exp Med 2002; 195:1233-45; PMID:12021304; http://dx.doi.org/10.1084/jem.20011930
- Billetta R, Filaci G, Zanetti M. Major histocompatibility complex class I-restricted presentation of influenza virus nucleoprotein peptide by B lymphoma cells harboring an antibody gene antigenized with the virus peptide. EurJ Immunol 1995; 25:776-83; PMID:7705408; http://dx.doi.org/10.1002/eji.1830250323
- Zhong G, Reis e Sousa C, Germain RN. Antigen-unspecific B cells and lymphoid dendritic cells both show extensive surface expression of processed antigen-major histocompatibility complex class II complexes after soluble protein exposure in vivo or in vitro. J Exp Med 1997; 186:673-82; PMID:9271583; http://dx.doi.org/10.1084/jem.186.5.673
- Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J Immunol 1999; 162:5695-703; PMID:10229801
- Evans DE, Munks MW, Purkerson JM, Parker DC. Resting B lymphocytes as APC for naive T lymphocytes: dependence on CD40 ligand/CD40. J Immunol 2000; 164:688-97; PMID:10623811; http://dx.doi.org/10.4049/jimmunol.164.2.688
- Dougan SK, Ogata S, Hu CC, Grotenbreg GM, Guillen E, Jaenisch R, Ploegh HL. IgG1+ ovalbumin-specific B-cell transnuclear mice show class switch recombination in rare allelically included B cells. Proc Natl Acad Sci U S A 2012; 109:13739-44.
- Harari A, Zimmerli SC, Pantaleo G. Cytomegalovirus (CMV)-specific cellular immune responses. Hum Immunol 2004; 65:500-6; PMID:15172450; http://dx.doi.org/10.1016/j.humimm.2004.02.012
- Schulz O, Reis e Sousa C. Cross-presentation of cell-associated antigens by CD8alpha+ dendritic cells is attributable to their ability to internalize dead cells. Immunology 2002; 107:183-9; PMID:12383197; http://dx.doi.org/10.1046/j.1365-2567.2002.01513.x
- Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y, Takahara K, Steinman RM, Inaba K. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med 2002; 195:1289-302; PMID:12021309; http://dx.doi.org/10.1084/jem.20020161
- Crespo MI, Zacca ER, Nunez NG, Ranocchia RP, Maccioni M, Maletto BA, Pistoresi-Palencia MC, Moron G. TLR7 triggering with polyuridylic acid promotes cross-presentation in CD8alpha+ conventional dendritic cells by enhancing antigen preservation and MHC class I antigen permanence on the dendritic cell surface. J Immunol 2013; 190:948-60; PMID:23284054; http://dx.doi.org/10.4049/jimmunol.1102725
- Browne EP. Regulation of B-cell responses by Toll-like receptors. Immunology 2012; 136:370-9; PMID:22444240; http://dx.doi.org/10.1111/j.1365-2567.2012.03587.x
- Schreibelt G, Klinkenberg LJ, Cruz LJ, Tacken PJ, Tel J, Kreutz M, Adema GJ, Brown GD, Figdor CG, de Vries IJ. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 2012; 119:2284-92; PMID:22234694; http://dx.doi.org/10.1182/blood-2011-08-373944
- Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, Brimnes MK, Moltedo B, Moran TM, Steinman RM. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med 2004; 199:815-24; PMID:15024047; http://dx.doi.org/10.1084/jem.20032220
- Idoyaga J, Lubkin A, Fiorese C, Lahoud MH, Caminschi I, Huang Y, Rodriguez A, Clausen BE, Park CG, Trumpfheller C, et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proceedings of the National Academy of Sciences of the United States of America 2011; 108:2384-9.
- Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med 2010; 207:1261-71; PMID:20479117; http://dx.doi.org/10.1084/jem.20092618
- Cho HJ, Takabayashi K, Cheng PM, Nguyen MD, Corr M, Tuck S, Raz E. Immunostimulatory DNA-based vaccines induce cytotoxic lymphocyte activity by a T-helper cell-independent mechanism. Nat Biotechnol 2000; 18:509-14; PMID:10802617; http://dx.doi.org/10.1038/75365
- Shirota H, Sano K, Hirasawa N, Terui T, Ohuchi K, Hattori T, Shirato K, Tamura G. Novel roles of CpG oligodeoxynucleotides as a leader for the sampling and presentation of CpG-tagged antigen by dendritic cells. J Immunol 2001; 167:66-74; PMID:11418633; http://dx.doi.org/10.4049/jimmunol.167.1.66
- Ingulli E. Tracing tolerance and immunity in vivo by CFSE-labeling of administered cells. Methods Mol Biol 2007; 380:365-76; PMID:17876106; http://dx.doi.org/10.1007/978-1-59745-395-0_23