
Abstract
Recent experimental studies and pathological analyses of patient brain tissue samples with refractory epilepsy suggest that inflammatory processes and neuroinflammation plays a key-role in the etiopathology of epilepsy and convulsive disorders. These inflammatory processes lead to the secretion of pro-inflammatory cytokines responsible for blood-brain-barrier disruption and involvement of resident immune cells in the inflammation pathway, occurring within the Central Nervous System (CNS). These elements are produced through activation of Toll-Like Receptors (TLRs) by exogenous and endogenous ligands thereby increasing expression of cytokines and co-stimulatory molecules through the activation of TLRs 2, 3, 4, and 9 as reported in murine studies.It has been demonstrated that IL-1β intracellular signaling and cascade is able to alter the neuronal excitability without cell loss. The activation of the IL-1β/ IL-1β R axis is strictly linked to the secretion of the intracellular protein MyD88, which interacts with other cell surface receptors, such as TLR4 during pathogenic recognition. Furthermore, TLR-signaling pathways are able to recognize molecules released from damaged tissues, such as damage-associated molecular patterns/proteins (DAMPs). Among these molecules, High-mobility group box-1 (HMGB1) is a component of chromatin that is passively released from necrotic cells and actively released by cells that are subject to profound stress. Moreover, recent studies have described models of epilepsy induced by the administration of bicuculline and kainic acid that highlight the nature of HMGB1-TLR4 interactions, their intracellular signaling pathway as well as their role in ictiogenesis and epileptic recurrence.The aim of our review is to focus on different branches of innate immunity and their role in epilepsy, emphasizing the role of immune related molecules in epileptogenesis and highlighting the research implications for novel therapeutic strategies.
Introduction
Recent experimental studies and pathological analyses of brain tissues of patients with refractory epilepsy suggest that inflammatory processes and neuroinflammation play a key-role in the etiopathology of epilepsy and convulsive disorders. These inflammatory processes are responsible for the secretion of pro-inflammatory cytokines, which cause blood brain barrier disruption as well as involvement in the inflammatory pathway of resident immune cells in the brain. Furthermore animal models used to identify inflammatory triggers in epileptogenesis have shown an increased susceptibility to seizures with inflammation in the developing rat brain, with pharmacological blockade of the IL-1beta/IL-1 receptor type 1 axis during epileptogenesis providing neuroprotection in 2 rat models of temporal lobe epilepsy.Citation1 More recently IL-1β and High-mobility group box-1 (HMBG-1), along with their receptors, have been studied as potentially important pro-convulsant molecules due to increased expression in the sera of experimental models and brain tissue of epileptic patients. In addition experimental models have suggested that neural damage and the onset of spontaneous recurrent seizures are modulated via complex interactions between innate and adaptive immunity.Citation2-3
The aim of our current review is to focus on different branches of innate immunity and their role in epilepsy, emphasizing the role of immune-related molecules in epileptogenesis and highlighting the research implications for novel therapeutic strategies
The Burden of Epilepsy: A Contemporary Look at its Epidemiology
Epilepsy is a disease characterized by recurrent unprovoked epileptic seizures due to aberrant, hyperexcitability and synchronized firing of groups of neurons, along with cognitive, neurobiological and psychosocial consequences.Citation1,2 While epilepsy is one of the most common neurologic diseases worldwide, its etiology is not completely understood and pharmacological therapies fail in about one-third of patients.Citation3 Epilepsy occurs in all age groups with around 50 million people estimated to be affected worldwide,Citation4 the majority of whom are left untreated particularly in developing countries. Furthermore, the World Health Organization (WHO) reported in 2001 that neurological conditions account for 30.8% of all years of life lived with disability (YLDs) and epilepsy accounts for 0.5% of the total burden of diseases.Citation8,9 Epilepsy is thus considered a global challenge.
Despite appropriate therapy, seizures in about 30% of patients remain uncontrolled. More than 50% of patients respond to the first anti-epileptic drug (AED) and of the remaining, only 20% respond to alternative or additional anti-convulsants. In addition, some patients develop resistance during the course of disease although the exact mechanism of drug-resistant epilepsy is not understood.Citation5 Whereas AEDs tend to provide symptomatic relief, they do not modulate the underlying disease mechanisms. Several mechanisms have been proposed for drug-resistance, and more recently the pro-inflammatory hypothesis is finding traction as a potential new explanation.
The Role of the Innate Immune System
The mammalian immune system includes 2 main branches: Innate immunity and adaptive (acquired) immunity. Innate immunity is the first line of defense, developing at an early stage of life and is followed by the development and activation of adaptive immunity as the later phase of host defense. While adaptive immunity is confined to vertebrates and responds specifically to antigens on different pathogens as well as incorporating immunological memory through a process of gene rearrangement, innate immunity is an evolutionarily conserved mechanism shared with plants and invertebrates.Citation10
Epithelial surfaces along the skin, respiratory tract, and gastrointestinal tract as well as antigen presenting cells including neutrophils and macrophages, along with the complement system and preformed plasma proteins are the major components of innate immunity. Moreover, cytokines are the main immunomodulators of innate immunity and include tumor necrosis factor (TNF), interleukins and interferons.Citation10-12 The Innate immune system recognizes conserved molecular patterns in pathogens via germ-line encoded receptors, called pattern recognition receptors (PRR). Common molecular patterns recognized are classified together as pathogen-assisted molecular patterns (PAMPs) and include bacterial cell wall molecules, as well as viral and fungal products. In addition, important PRRs include Toll-like Receptors (TLR), Nod-like receptors (NLR), and retinoic acid-inducible gene 1-like receptors (RLR).Citation6
Toll-like receptors, endogenous and exogenous ligands
Toll-like receptors are the most studied members of the PRRs. The Toll gene was first identified in 1984 in drosophila melanogaster embryo as an essential gene for dorso-ventral axis development in these flies. Later it was discovered that Toll protein is an essential receptor for initiating the innate immune response against fungal infections.Citation14 A mammalian ortholog receptor inducing inflammatory genes was found one year later, named the Toll-like receptor.Citation15 Toll-like receptors are transmembrane receptors composed of 3 domains: The transmembrane, intracellular and extracellular domains. The extracellular domain is designed for ligand recognition and is characterized by a leucine-rich repeats (LRRs) region. The intracellular domain, called the Toll/interleukin-1 receptor (TIR) domain due to its similarity to IL-1 receptor family, initiates a signaling cascade leading to induction of inflammatory genes.Citation15
Eleven toll-like receptors have been identified in humans. TLRs 3, 7–9 are located in the cellular endolysosomal compartment, while TLRs 1, 2, 4–6 and 11 are expressed in the plasma membrane. Several of these TLRs have been shown to respond to exogenous ligands. TLR 2 recognizes microbial components like bacterial peptidoglycans, teichoic acid, lipoproteins, lipopeptides, lipoarabinomannan, glycosylphosphatidylinositol, porins and zymosan from the yeast cell wall, either alone or in combination with TLRs 1 and 6. Furthermore TLR1/6 heterodimer recognizes diacylated lipopeptide whereas TLR1/2 recognizes triacylated lipopeptides.Citation7 Moreover TLR4 binds to the lipopolysaccharide (LPS) of gram negative bacteria, some viral products and endogenous ligands.Citation8,9 TLR5 recognizes bacterial flagellinCitation10 and TLRs 3, 7, 8 and 9 bind to viral PAMPs. TLR3 recognizes double stranded RNA, TLR7 and TLR8 recognize viral and non-viral single stranded RNAs and TLR9 recognizes unmethylated CpG dinucleotides found in the viral and bacterial genome.Citation11,12 The ligand for TLR10 has not yet been identified.Citation13 Finally TLR11 binds to uro-pathogenic antigens and a profiling-like molecule in toxoplasma gondii.Citation22-26
Among the major molecules contributing to innate immunity, damage-associated molecular patterns (DAMPs) are a major group of endogenous ligands for TLRs released from damaged cells. DAMPs include heat-shock proteins (HSP), hyaluronan, nucleic acids, heparan sulfate, surfactant protein-A, fibrinogen and high mobility group box 1 (HMGB1).Citation14–16 Furthermore, fibrinogen, heparan sulfate, hyaluronan, β defensin 2, surfactant protein-A, fibronectin extra domain A are endogenous ligands of TLR4Citation13 whereas Heat-shock proteins like HSP60, HSP70, HSP90 and GP96 activate TLR 2 and 4.Citation13,16,25-27,36,38 In addition HMGB1 is an endogenous ligand for TLR2 and 4Citation28,29 and mRNA is an endogenous ligand for TLR3.Citation30
TLR signaling pathways
TLR signaling cascade begins with adaptor proteins being recruited through TLR activation. There are 6 known adaptor proteins: myeloid differentiation factor 88 (MyD88), Toll IL-1 receptor domain containing adaptor protein (TIRAP), also known as myeloid differentiation factor 88 adapter like (MAL) protein, Toll-interleukin 1 receptor domain containing adapter-inducing interferon-β (TRIF) or TIR-containing adapter molecule-1 (TICAM-1), TRIF-related adapter molecule (TRAM) and Sterile-α and Armadillo motif containing protein (SARM).Citation31,32 In addition, TLRs signaling pathways are divided into MyD88-dependent and MyD88-independent pathways.Citation42,43 With the exception of TLR3, all TLRs recruit MyD88. The TIR domain of the TLR interacts with the TIR domain of MyD88, recruiting interleukin-1 receptor-associated kinase 4 (IRAK4), which results in IRAK1 phosphorylation. Subsequently, the TNF receptor-associated factor 6 (TRAF6), activated by IRAK1, stimulates transforming growth factor-b (TGF-b)-activated kinase 1 (TAK1). At this point 2 different pathways are initiated. One pathway includes degradation of inhibitor of nuclear factor κB kinase (IKK) that leads to translocation of NFκB as a transcription factor to the nucleus. The other pathway involves activation of mitogen-activated protein (MAP) kinases, subsequently triggering the activator protein-1 (AP-1) transcription factors.Citation42,43 These transcription factors induce the production of pro-inflammatory mediators.
The signaling cascade of TLR2 and 4 depends upon TIRAP/MAL in addition to MyD88. For example data from TIRAP/Mal deficient mice shows they were not able to induce inflammatory response when TLRs 2 and 4 were triggered.Citation44,33 TLR4 uses both MyD88 dependent pathway and TRIF (MyD88 independent) pathway; however TLR3 is exclusively dependent on TRIF (also named TICAM-1) pathway.Citation34-36 In the TRIF pathway, NFκB and interferon-regulatory factor 3 (IRF3) are activated, leading to induction of interferon-β (IFN-β).Citation37 When the TRIF pathway is triggered by TLRs, TRAM is also recruited as an intermediate protein.Citation38
The fifth member of this family, SARM, is a negative regulator of the TRIF pathwayCitation39 and of basal MAPK activity.Citation34
Brain cells with immune capabilities
Microglia are the macrophage-like antigen presenting resident cells in the brain originated from myeloid progenitor cells.Citation40,41 Other brain cells originate from the neuroepithelial cell linage and include neurons and glia, with glia differentiating into astrocytes, oligodendrocytes and polydendrocytes. Expression of TLRs and associated proteins is observed in astrocytes, and microglia as well as neurons.Citation42 Moreover there is evidence that these cells mediate inflammation that may contribute to epileptogenesis.
Data from human samples has further demonstrated TLR expression on glial cells as well as limited evidence of neuronal expression. Human microglial cells express mRNA for TLRs 1–9.Citation57 In particular high cell-surface TLR2 expression, as well as intracellular TLR3 expression, was shown in human microglia (ref). In addition astrocytes with a restricted repertoire of TLRs, showed high levels of TLR3 expression along with low levels of TLRs 1, 4, 5 and 9. Furthermore, high levels of IL-12, TNF-α, IL-6, CXCL−10, IL-10 and IFN-β were reported in response to TLR3 induction in human microglia and IL-6, CXCL−10 and IFN-β were produced in response to TLR3 activation in astrocytes.Citation43 Furthermore an in vitro study of cultured astrocytes showed preferential expression of TLR3,.Citation44,45 In vitro and in vivo study examining the expression of TLRs by microglia and astrocytes therefore show a wide range of TLRs that are differentially expressed in these cells with astrocytes mainly expressing TLR 2 and 3. Finally, there is some evidence that neurons also express TLRs, particularly intracellular TLRs 3, 7, 8 and 9.Citation42,46,47
There is evidence from experimental models that stimulation of TLRs expressed in the brain can mediate inflammation. Firstly, TLRs expression has been observed within in vitro studies in mice brain cells using real-time PCR demonstrating that murine microglia expresses TLRs 1–9. The same study also reported increased microglial expression of TLRs 3, 6 and 9 mRNA in response to IFN-γ as well as increased expression of TLRs 2, 4, 6, 8 and 9 in response to lipopolysaccharide (LPS) and increased expression of cytokines and co-stimulatory molecules through activation of TLRs 2, 3, 4, and 9.Citation48 A further experimental study in mice showed a TLR2-mediated response in glial cells in response to brain injury.Citation49 Pro-inflammatory cytokines were also produced in response to the TLR9 agonist unmethylated CpGDNA in murine microglia.Citation50 Finally, Poly I: C treated murine microglial cells showed increased production of pro-inflammatory cytokines through TLR3 activation.Citation51 Data from inflamed CNS tissue from neurodegenerative patients also shows higher levels of TLR3 and 4 expression.Citation52
Activated glial and microglial cells produce pro-inflammatory cytokines such as IL-1β, a proconvulsant cytokine, within minutes the onset of seizureCitation53,54 as well as up-regulating its receptor (IL-1R1), and increasing nuclear expression and translocation of HMGB1 (endogenous ligand of TLR4 and 2) into the cytoplasm, consequently decreasing the seizure threshold. In addition activation of microglial and glial cells by precipitating factors such as infections, injuries and stroke, cause the release of pro-inflammatory molecules, and any prolonged trigger leads to the excessive production of inflammatory molecules. Subsequent disruptions in the BBB as well as dysregulation in ion transport and inhibitory neurotransmitters can lead to epileptogenesis.Citation55
Toll-like Receptors Activation and Epileptogenesis
Increased levels of pro-inflammatory cytokines in the CSF and sera of patients with chronic epilepsy provides evidence for the involvement of neuroinflammation in epileptogenesis.Citation56-58 These cytokines are produced through activation of TLRs by exogenous and endogenous ligands. Experimental studies on animal models demonstrate that intracerebral injection of LPS, a TLR4 ligand, is associated with decreased seizure threshold. LPS-treated rats (during a specific period of postnatal development) had decreased seizure threshold when stimulated with pro-convulsant agents including lithium–pilocarpine, kainic acid, or pentylenetetrazole.Citation56-58 Moreover increased levels of pro-inflammatory cytokines as well as neurodegeneration were detected in these rats.Citation59 Furthermore electrophysiological studies in rats showed increased epileptiform discharges in response to cortical application of LPS which could be inhibited through the effects of an IL-1 receptor antagonist (IL-1 Ra). Additionally, brain injury and CNS infections trigger immune responses via LPS and DAMPs in glial cells, leading to increased IL-1 production and increased neuronal excitability.Citation60
Another TLR ligand demonstrated to be involved in epileptogenesis is HMGN1, the TLR4 endogenous ligand. HMGB1 is released from glia and neurons in mice models of seizures and use of the HMGB1 antagonist BoxA (a fragment of HMGB1 with receptor antagonist activity) as well as a TLR4 antagonist were associated with reduced frequency of seizures triggered by kainate or bicuculline. The same study also showed that TLR4-knockout mice were resistant to seizures.Citation61 In addition, an ex vivo study on surgical specimens from patients with focal cortical dysplasia, turberous sclerosis and ganglogliomas showed increased expression of TLR2- and TLR4- mRNAs, RAGE mRNA and HMGB1 in tissues from epileptic patients compared to controls. TLR2 was expressed by activated microglia and TLR4 was expressed by astrocytes and neurons whereas RAGE was also expressed in glial cells and neurons. HMGB1 was expressed in the nuclei of neurons and glial cells in normal brain specimens, while in specimens from patients with focal cortical dysplasia, turberous sclerosis and ganglogliomas, it was expressed in the cytoplasm of activated astrocytes and microglia. Moreover it was observed that IL-1β was responsible for the translocation of HMGB1 from nuclei to cytoplasm in human cultured astrocytes.Citation62
Increased expression of HMGB1 and TLR4 in tissue specimens from patients with refractory epilepsy constitutes evidence for their role in human epilepsy. HMGB1 acts through ifenprodil-sensitive N-methyl-d-aspartate (NMDA) receptors, as well as IL-1β. When HMGB-1 and IL-1β are released from astrocytes, the IL-1 receptor (IL-1 R)/TLR is activated. This leads to initiation of TLR signaling pathway that finally activates NF-κB. As a result, one of the subunits of N-methyl D-aspartate acid (NMDA) receptor is phosphorylated leading to calcium influx into the neuron, thus increasing the cellular excitability.Citation55 Moreover, IL-1β inhibits glutamate reuptake by astrocytes and induces its release by glia and this glutamate imbalance further increases neuronal hyperexcitability.Citation55
Finally, TLR3 seems to play a pivotal role in seizure activity. Involvement of TLR3 in seizures is supported by evidence in febrile seizures that occur as a result of viral infection with viral PAMPs interacting with TLR3. Intra-cerebroventricular administration of poly I: C (TLR3 ligand) in 14-day old rats caused fever and increased amounts of IL-1β in rat brains. These animal models were also more susceptible to lithium-pilocarpine and pentylenetetrazol-induced seizures, showing increased amounts of NMDA and AMPA receptor subunit mRNA expression later in life.Citation63
An Explanatory Model for TLR4-mediated Epilepsies
The chronic secretion of IL-1β in activated atrocytes during epileptogenesis suggests a keyrole for these cells in sustaining activation of inflammatory cascades before the onset of spontaneous seizuresCitation81 (). IL-1β activates a signaling pathway involving the IL-1R1, IL-1R accessory protein complex and MyD88 complex. The latter by stimulation of the Src family kinases, leading to NMDA receptor-2B phosphorylation and subsequent enhancement of NMDA-dependent Ca2+ influx. This increased Ca2+ influx in neurons facilitates ictiogenesis.Citation82
Figure 1. Model of inflammatory epileptogenesis.
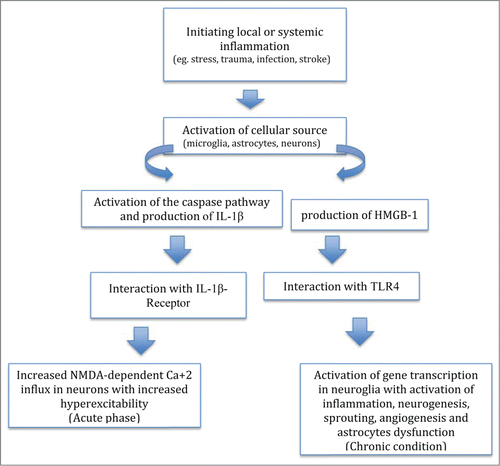
It has been demonstrated that IL-1β intracellular signaling cascade is able to alter the neuronal excitability without cell loss. Dubé et al. showed that hippocampal IL-1β levels are chronically elevated in murine activated astrocytes affected by spontaneous seizures after febrile status epilepticus at postnatal day 11.Citation83 These increased IL-1β levels and spontaneous seizures were not linked to any cell loss, T2 MRI hippocampal abnormalities or interictal EEG activity. Experimental febrile seizure duration in these rats influenced both the probability of developing limbic epilepsy as well as the severity and duration of spontaneous seizures. However, the relationship between hippocampal IL-1β levels and the duration of initial neural activity is still not clearly defined.Citation83
The activation of the IL-1β/ IL-1β R axis is strictly linked to the secretion of the intracellular protein MyD88, which interacts with other cell surface receptors, such as TLR4 during pathogen recognition. However the role of MyD88 proteins remains unclear as few studies have investigated the implications of this pathway for epileptogenesis. In addition, TLR-signaling pathways are able to recognize molecules released from damaged tissues, such as DAMPs. Among these molecules, HMGB1 is a component of chromatin that is passively secreted from necrotic cells and actively released by cells that are subject to profound stress. Maroso et al. used chemical epilepsy models developed by the administration of bicuculline and kainic acid to highlight the nature of HMGB1-TLR4 interactions, their intracellular signaling pathway, as well as their role in ictiogenesis and epileptic recurrence. These authors observed increased TLR4 and HMGB1 expression in neurons, astrocytes and microglia in the hippocampus of murine models following intrahippocampal injection of kainic acid and bicuculline. The same result was found in human hippocampal tissue from intractable temporal lobe epilepsy patients. The injection of recombinant HMGB1 to murine hippocampi treated with kainic acid had a proconvulsant effect, which was not observed in mice defective in TLR4 signaling. The authors therefore suggested that HMGB1–TLR4 signaling is intimately involved in epileptogenesis. Nevertheless, the authors were not able to show both the origin of HMGB1 secretion and the triggers for HMGB1 and TLR4 expression in glia or neurons during seizures in these models.Citation66,74 However they did show that ictal activity itself as well as hypoxia and interactions with neurons may determine the rate of synthesis and release of HMGB1 from rat glial populations. Furthermore, HMGB1-TLR4 antagonists decreased the number of seizures, their duration as well as increasing the onset latency period in the bicuculline-induced non-lesional model of seizures. In addition, Moroso's study shows the extent to which HMGB1-TLR4 signaling pathway influences chronic epilepsy. They showed that HMGB1 and TLR4 receptor antagonists were able to block both acute seizures induced by the injection of bicuculline and kainic acid as well as recurrence of seizures in the C57BL/6 mouse chronic model of epilepsy.Citation66,74 Thus, these data suggest that ictal activity in neurons is sufficient to facilitate HMGB1 synthesis and subsequent release outside the cell.Citation66,74 However Maroso's studies have not provided evidence for continuous HMGB1 synthesis and activity and also did not distinguish the role of the IL-1β pathway in these processes. Nevertheless, given the recognized role of proinflammatory cytokines in astrocyte synapses and neurodevelopmental disorders associated with epileptogenesis, including Fragile × syndrome and autism, developmental apoptosis or neural activity-induced cell stress from any process may contribute to the activation of the HMGB1-TLR4 axis, with a consequent induction of aberrant synaptic connectivity, neuronal excitability, and epileptogenesis.Citation84-86
The role of HMGB1–TLR4 signaling in ictogenesis remains to be clarified. HMGB1–TLR4 signaling may interact with only a part of the IL-1β pathway and therefore act with a distinct pathophysiology. HMGB1 may control the IL-1β pathway through modifications of intracellular signaling components or primary regulation of IL-1β, itself. The lack of cell death as a requirement and the presence of common signaling molecules for the pro-epileptogenic actions of IL-1β and HMGB1 suggest that pharmacologic interventions along this pathway may yield effective treatments for current drug-resistant epilepsies, particularly intractable pediatric epilepsies.
Therapeutic Perspectives
Given its potential role in mediating the pathophysiology of epilepsy, targeting the immune system, in particular the pro-convulsant cytokines, has been suggested as a potential therapeutic strategy in drug resistant epilepsies.Citation64 The benefits of targeting inflammation for the treatment of epilepsy arise from targeting the underlying molecular mechanism of pathogenesis rather than symptoms control which in turn could be beneficial as a preventative measure in multiple neurological disorders that lead to increased risk of seizures including post-trauma, stroke and infections.Citation65 presents a summary of new immunotherapeutic approaches currently in the experimental phase to treat drug-resistant epilepsies.
Table 1. Experimental immunotherapy, in course of study, to be used as immunotherapeutic approach in drug-resistant epilepsies
IL-1β inhibitors
Among these potential therapeutic agents, the interleukin converting enzyme (ICE)/caspase-1 has been considered a possible target for the treatment of drug-resistant epilepsies. This enzyme inhibits the conversion of pro-IL-1β to the pro-convulsant IL-1β. In particular Pralnacasan and Belnacasan (a pro-drug of VRT-043198) are inhibitors of the IL-1β converting enzyme as well as the selective inhibitor of caspases from the ICE/caspase-1 family respectively. Belnacasan is currently undergoing phase III clinical trial as a treatment for inflammatory disorders.Citation66 Moreover, this drug seems to decrease the production of IL-18 and IL-1β in vitro and in vivo.Citation67 Furthermore Ravizza T, et al showed that the release of IL-1β in LPS-ATP treated murine organotypic hippocampal slice cultures decreased with caspase-1 inhibition. Moreover, intracerebroventricular administration of pralnacasan and intraperitoneal administration of the experimental compound VX765, a selective inhibitor of ICE, in these mice led to a 50% reduction in seizure duration and a twofold delay in seizure onset. The same study also evaluated seizures frequency in mice with caspase-1 gene deletion and showed a 70% reduction in seizures along with a fourfold delay in their onset.Citation68 In addition, the selective inhibition of ICE via VX765 decreased IL-1β release in astrocytes leading to the blockade of kindling epileptogenesis in rats.Citation69 Furthermore VX765 is under study in patients affected by drug resistant epilepsy. A recent phase 2b double-blind randomized controlled trial investigated its safety in 60 patients with drug-resistant partial epilepsy. Results were evaluated after 6 weeks of treatment with 900 mg of VX765 3 times daily in 48 patients, in comparison to 12 patients who received placebo. The authors showed that the percentage responder-rate (defined as the percentage of patients with a 50 percent or greater reduction in seizure frequency), the percentage of patients who were seizure-free for 2 weeks, and the percentage of reduction in seizure rates ranged from 13% to 19% in patients who received VX765.Citation70
TLRs modulators
Manipulation of HMGB1-TLR4 axis is also a potential novel anti-convulsive strategy since seizure recurrence is decreased with HMGB1 and TLR4 antagonists.Citation61,71,72 An example is Resveratrol, a type of natural phenol produced as a protective factor in response to bacteria and fungi in plants that is found in grape skin, peanuts and red wine. It has shown anti-inflammatory as well as anti-cancer properties and is considered a neuroprotective agent.Citation73–76 Studying the effects of Resveratrol in TLR-mediated pathways showed that Resveratrol suppresses NFκB induced by TLRs 3 and 4 and this property was specifically shown to be mediated through inhibition of TRIF. Expression of IFN-β induced by LPS and poly I: C was also inhibited by Resveratrol.Citation77 In addition, Wu et al. evaluated the anti-epileptic properties of Resveratrol in rats with kainic-acid-induced temporal lobe epilepsy with behavioral monitoring and electroencephalography as well as molecular studies.Citation100 Resveratrol was shown to decrease the frequency of seizures and inhibit epileptiform discharges.102–104 Resveratrol successfully inhibited kainate-induced neuronal cell death in CA1 and CA3 regions of these rat brain.Citation78 Another study described the anti-oxidant effect of Resveratrol against epileptogenic oxidative stress in the brain.Citation79 It has also been shown that Resveratrol inhibits microglial activation and mediates its anti-epileptic effects through neuroprotective, anti-inflammatory strategies.Citation80 One way in which this could be mediated is through inhibition of cyclooxygenases, often involved in epileptogenesis.Citation81,82
NMDA-blockers
The downstream signaling events denoted by presumed activation of Src protein kinases, phosphorylation of NR2B, and increased NMDA-mediated Ca2+ influx can be blocked by ifenprodil, a sensitive blocker of NR2B-containing NMDA receptors. Ifenprodil has been showed to block the proconvulsant effects of HMGB1 in the kainic acid model of acute seizures and decreases seizure recurrence in chronically epileptic mice.Citation74 However, ifenprodil failed to block acute seizures from kainic acid in the absence of the proconvulsant HMGB1 protein, suggesting that inflammatory processes are necessary for the anti-seizure effect of ifenprodil.Citation74
Conclusion
During the past decade different experimental and clinical studies have been designed to identify the exact role of inflammation and immunity in epilepsy. Neurons and immune resident cells have been shown to play important roles in etiopathogenesis of epilepsy. Different immune-mediated cytokines are regarded as inducers of neuroplastic changes, decreased seizure threshold and extension of aberrant neuronal firings among neurons. These findings raise the possibility of developing drugs for current drug-resistant epilepsies that comprise a relatively large number of epilepsies through focusing on the underlying neurobiology and molecular pathologies rather than previous disease-modifying and symptoms control agents. Possible involvement of toll-like receptors and interleukin receptors elucidates the role of innate immunity in triggering seizures and spreading neuronal hyperexcitability. Thus inflammatory mediators are the new therapeutic targets and the point of interest in recent studies as discussed in this review. Several experimental studies on novel immunotherapeutic drugs targeting the IL-1β axis show early promise for the treatment of seizures. However it has to be established whether targeted immunosuppression is effective in treating inflammation predisposing to epilepsy or if a more extensive immunosuppressive approach is needed such as provided by steroids. Moreover it should be established if blocking a partial mechanism of inflammation is sufficient or a more extensive range of pro-inflammatory targets should be included in the treatment (such as TNF-α and other ILs blockers). Finally, new research efforts should be concentrated to establish if immunotherapy alone is sufficient to replace the conventional anticonvulsant therapy or if it should be used as additional therapy to the latter.
The authors think that this topic is of extreme importance not only from a therapeutic point of view, but also as for developing preventive strategies. Knowledge of the immune alterations predisposing to epilepsy represents a new opportunity to develop immuno-modulatory drugs that not only treat but prevent the onset of seizures.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Noe FM, Polascheck N, Frigerio F, Bankstahl M, Ravizza T, Marchini S, Beltrame L, Banderó CR, Löscher W, Vezzani A. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis 2013; 59:183-93; PMID:23938763; http://dx.doi.org/10.1016/j.nbd.2013.07.015
- Vitaliti G. Pavone P. Mahmood F, Nunnari G, Falsaperla R. Targeting inflammation as a therapeutic strategy for drug-resistant epilepsies. An update of new immunomodulating approaches. Hum Vaccine Immunother 2014; 10:41-8; http://dx.doi.org/10.4161/hv.28400
- Falsaperla R, Pavone P, Miceli-Sopo S, Mahmood F, Scalia F, Corsello G, Lubrano R, Vitaliti G. Epileptic seizures as a manifestation of cow's milk allergy: a studied relationship and description of our pediatric experience. Expert Rev Clin Immunol 2014; 10:1597-609; PMID:25394911; http://dx.doi.org/10.1586/1744666X.2014.977259
- Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J Jr, Forsgren L, French JA, Glynn M, et al. ILAE Official Report: A practical clinical definition of epilepsy. Epilepsia 2014; 55:475-82; PMID:24730690; http://dx.doi.org/10.1111/epi.12550
- de Boer HM, Mula M, Sander JW. The global burden and stigma of epilepsy. Epilepsy Behav 2008; 12:540-6; PMID:18280210
- Sander JW. The epidemiology of epilepsy revisited. Curr Opin Neurol 2003; 16:165-70
- Banerjee PN, Filippi D, Allen Hauser W. The descriptive epidemiology of epilepsy—a review. Epilepsy Res 2009; 85:31-45; PMID:19369037; http://dx.doi.org/10.1016/j.eplepsyres.2009.03.003
- Leonardi M, Ustun TB. The global burden of epilepsy. Epilepsia 2002; 43:21-5; PMID:12190974; http://dx.doi.org/10.1046/j.1528-1157.43.s.6.11.x
- Sirven JI, Pedley TA, Wilterdink JL. Evaluation and management of drug-resistant epilepsy 2011.
- Lodish H, Berk A, Zipursky S. Molecular Cell Biology 4th edition 2000.
- Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J 2009; 420:1-16; PMID:19382893; http://dx.doi.org/10.1042/BJ20090272
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol 2011; 30:16-34; PMID:21235323; http://dx.doi.org/10.3109/08830185.2010.529976
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124:783-801; PMID:16497588; http://dx.doi.org/10.1016/j.cell.2006.02.015
- Lemaitre B, Nicolas E, Michaut L, Reichhart J-M, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996; 86:973-83; PMID:8808632; http://dx.doi.org/10.1016/S0092-8674(00)80172-5
- Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997; 388:394-7; PMID:9237759; http://dx.doi.org/10.1038/41131
- Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol 2002; 169:10-4; PMID:12077222; http://dx.doi.org/10.4049/jimmunol.169.1.10
- Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel C, Du X, Modlin RL, Akira S. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998; 282:2085-8; PMID:9851930; http://dx.doi.org/10.1126/science.282.5396.2085
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999; 11:443-51; PMID:10549626; http://dx.doi.org/10.1016/S1074-7613(00)80119-3
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001; 410:1099-103; PMID:11323673; http://dx.doi.org/10.1038/35074106
- Bowie AG, Haga IR. The role of Toll-like receptors in the host response to viruses. Mol Immunol 2005; 42:859-67; PMID:15829275; http://dx.doi.org/10.1016/j.molimm.2004.11.007
- Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol 2004; 76:514-9; PMID:15178705; http://dx.doi.org/10.1189/jlb.0304127
- Arancibia SA, Beltrán CJ, Aguirre IM, Silva P, Peralta AL, Malinarich F, Hermoso MA. Toll-like receptors are key participants in innate immune responses. Biol Res 2007; 40:97-112; PMID:18064347; http://dx.doi.org/10.4067/S0716-97602007000200001
- Kang SS, Kauls LS, Gaspari AA. Toll-like receptors: applications to dermatologic disease. J Am Acad Dermatol 2006; 54:951-83; PMID:16713451; http://dx.doi.org/10.1016/j.jaad.2005.05.004
- Akira S. Mammalian Toll-like receptors. Curr Opin Immunol 2003; 15:5-11; PMID:12495726; http://dx.doi.org/10.1016/S0952-7915(02)00013-4
- Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 2003; 85:85-95; PMID:12527213; http://dx.doi.org/10.1016/S0165-2478(02)00228-6
- Cai Z, Shi Z, Sanchez A, Zhang T, Liu M, Yang J, Wang F, Zhang D. Transcriptional regulation of Tlr11 gene expression in epithelial (16, 23, 83-85)
- Mallard C. Innate immune regulation by toll-like receptors in the brain. ISRN Neurol 2012; 2012:701950; PMID:23097717; http://dx.doi.org/10.5402/2012/701950
- Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol 2002; 23:509-12; PMID:12401394; http://dx.doi.org/10.1016/S1471-4906(02)02317-7
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF 3rd. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 2001; 276:10229-33; PMID:11150311; http://dx.doi.org/10.1074/jbc.M100099200
- Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol 2001; 167:2887-94; PMID:11509636; http://dx.doi.org/10.4049/jimmunol.167.5.2887
- Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol 2002; 168:5989-92; PMID:12055204; http://dx.doi.org/10.4049/jimmunol.168.12.5989
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med 2002; 195:99-111; PMID:11781369; http://dx.doi.org/10.1084/jem.20001858
- Tsan M-F, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol 2004; 76:514-9; PMID:15178705; http://dx.doi.org/10.1189/jlb.0304127
- Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol 2002; 168:5233-9; PMID:11994480; http://dx.doi.org/10.4049/jimmunol.168.10.5233
- Moghimpour Bijani F, Vallejo JG, Rezaei N. Toll-like receptor signaling pathways in cardiovascular diseases: challenges and opportunities. Int Rev Immunol 2012; 3:379-95; http://dx.doi.org/10.3109/08830185.2012.706761
- Wallin R, Lundqvist A, Moré SH, von Bonin A, Kiessling R, Ljunggren H-G. Heat-shock proteins as activators of the innate immune system. Trends Immunol 2002; 23:130-5; PMID:11864840; http://dx.doi.org/10.1016/S1471-4906(01)02168-8
- Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Häcker H, Wagner H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 2001; 276:31332-9; PMID:11402040; http://dx.doi.org/10.1074/jbc.M103217200
- Dybdahl B, Wahba A, Lien E, Flo TH, Waage A, Qureshi N, Sellevold OF, Espevik T, Sundan A. Inflammatory response after open heart surgery release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 2002; 105:685-90; PMID:11839622; http://dx.doi.org/10.1161/hc0602.103617
- Park JS, Svetkauskaite D, He Q, Kim J-Y, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004; 279:7370-7; PMID:14660645; http://dx.doi.org/10.1074/jbc.M306793200
- Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim J-Y, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol 2006; 290:C917-C24; PMID:16267105
- Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004; 279:12542-50; http://dx.doi.org/10.1074/jbc.M310175200
- Watters TM, Kenny EF, O'Neill LA. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol 2007; 85:411-9; PMID:17667936; http://dx.doi.org/10.1038/sj.icb.7100095
- Belinda LW-C, Wei WX, Hanh BTH, Lei LX, Bow H, Ling DJ. SARM. a novel Toll-like receptor adaptor, is functionally conserved from arthropod to human. Mol Immunol 2008; 45:1732-42; PMID:17980913; http://dx.doi.org/10.1016/j.molimm.2007.09.030
- Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002; 420:329-33; PMID:12447442; http://dx.doi.org/10.1038/nature01180
- Peng J, Yuan Q, Lin B, Panneerselvam P, Wang X, Luan XL, Lim SK, Leung BP, Ho B, Ding JL. SARM inhibits both TRIF-and MyD88-mediated AP-1 activation. Eur J Immunol 2010; 40:1738-47; PMID:20306472; http://dx.doi.org/10.1002/eji.200940034
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 2005; 17:1-14; PMID:15585605; http://dx.doi.org/10.1093/intimm/dxh186
- Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun 2009; 388:621-5; PMID:19686699; http://dx.doi.org/10.1016/j.bbrc.2009.08.062
- Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3–mediated interferon-β induction. Nat Immunol 2003; 4:161-7; PMID:12539043; http://dx.doi.org/10.1038/ni886
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4–mediated MyD88-independent signaling pathway. Nat Immunol 2003; 4:1144-50; PMID:14556004; http://dx.doi.org/10.1038/ni986
- O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007; 7:353-64; PMID:17457343; http://dx.doi.org/10.1038/nri2079
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation 2005; 2:24; http://dx.doi.org/10.1186/1742-2094-2-24
- Carpentier PA, Duncan DAS, Miller SD. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun 2008; 22:140-7; PMID:17920811
- Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci 2011; 121:367-87; http://dx.doi.org/10.1042/CS20110164
- Pelvig D, Pakkenberg H, Stark A, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging 2008; 29:1754-62; PMID:17544173; http://dx.doi.org/10.1016/j.neurobiolaging.2007.04.013
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 2004; 173:3916-24; PMID:15356140; http://dx.doi.org/10.4049/jimmunol.173.6.3916
- Babcock AA, Wirenfeldt M, Holm T, Nielsen HH, Dissing-Olesen L, Toft-Hansen H, Millward JM, Landmann R, Rivest S, Finsen B, et al. Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J Neurosci 2006; 26:12826-37; PMID:17151286; http://dx.doi.org/10.1523/JNEUROSCI.4937-05.2006
- Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9). FASEB J 2004; 18:412-4
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature 2001; 413:732-8; PMID:11607032; http://dx.doi.org/10.1038/35099560
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol 2005; 175:4320-30; PMID:16177072; http://dx.doi.org/10.4049/jimmunol.175.7.4320
- Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol 2005; 159:12-9; PMID:15652398; http://dx.doi.org/10.1016/j.jneuroim.2004.09.009
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol 2007; 28:138-45; PMID:17276138; http://dx.doi.org/10.1016/j.it.2007.01.005
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol 2002; 61:1013-21; PMID:12430718
- Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem 2007; 102:37-50; PMID:17403033; http://dx.doi.org/10.1111/j.1471-4159.2007.04524.x
- Tang S-C, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, et al. Pivotal role for neuronal toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci 2007; 104:13798-803; http://dx.doi.org/10.1073/pnas.0702553104
- Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, Flavell R, Strittmatter SM, Volpe J, Sidman R, et al. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci 2007; 27:13033-41; PMID:18032677; http://dx.doi.org/10.1523/JNEUROSCI.4290-06.2007
- Maroso M, Balosso S, Ravizza T, Liu J, Bianchi M, Vezzani A. Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J Internal Med 2011; 270:319-26; PMID:21793950; http://dx.doi.org/10.1111/j.1365-2796.2011.02431.x
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol 2011; 7:31-40; PMID:21135885; http://dx.doi.org/10.1038/nrneurol.2010.178
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci 2013; 36:174-84; PMID:23298414; http://dx.doi.org/10.1016/j.tins.2012.11.008
- Peltola J, Hurme M, Miettinen A, Keränen T. Elevated levels of interleukin-6 may occur in cerebrospinal fluid from patients with recent epileptic seizures. Epilepsy Res 1998; 31:129-33; PMID:9714504; http://dx.doi.org/10.1016/S0920-1211(98)00024-2
- Vezzani A. Inflammation and epilepsy. Epilepsy Curr 2005; 5:1-6; http://dx.doi.org/10.1111/j.1535-7597.2005.05101.x
- Aronica E, Crino PB. Inflammation in epilepsy: clinical observations. Epilepsia 2011; 52:26-32; PMID:21542843; http://dx.doi.org/10.1111/j.1528-1167.2011.03033.x
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 2008; 28:6904-13; PMID:18596165; http://dx.doi.org/10.1523/JNEUROSCI.1901-08.2008
- Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR, Barth DS. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain 2009; 132:2478-86; PMID:19567702; http://dx.doi.org/10.1093/brain/awp177
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 2010; 16:413-9; PMID:20348922; http://dx.doi.org/10.1038/nm.2127
- Zurolo E, Iyer A, Maroso M, Carbonell C, Anink JJ, Ravizza T, Fluiter K, Spliet WG, van Rijen PC, Vezzani A, et al. Activation of Toll-like receptor, RAGE and HMGB1 signalling in malformations of cortical development. Brain 2011; 134:1015-32; PMID:21414994; http://dx.doi.org/10.1093/brain/awr032
- Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends Mol Med 2007; 13:460-9; PMID:18029230; http://dx.doi.org/10.1016/j.molmed.2007.09.002
- Vezzani A. Epilepsy and inflammation in the brain: overview and pathophysiology. Epilepsy Curr 2014; 14:3-7; http://dx.doi.org/10.5698/1535-7511-14.s2.3
- Galic M, Riazi K, Henderson A, Tsutsui S, Pittman Q. Viral-like brain inflammation during development causes increased seizure susceptibility in adult rats. Neurobiol Dis 2009; 36:343-51; PMID:19660546; http://dx.doi.org/10.1016/j.nbd.2009.07.025
- Bo T, Chen Y, Mao D, Li Y, Zhu X. [Long-term effects of neonatal recurrent seizures on gamma-aminobutyric acid A receptor alpha1 and gamma2 subunit expressions in the rat brain]. Beijing Da Xue Xue Bao 2006; 38:628-33; PMID:17173085
- Zhang G, Raol YSH, Hsu FC, Brooks-Kayal AR. Long-term alterations in glutamate receptor and transporter expression following early-life seizures are associated with increased seizure susceptibility. J Neurochem 2004; 88:91-101; PMID:14675153; http://dx.doi.org/10.1046/j.1471-4159.2003.02124.x
- Vezzani A, Balosso S, Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun 2008; 22:797-803; PMID:18495419; http://dx.doi.org/10.1016/j.bbi.2008.03.009
- Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, Vezzani A. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain 2008; 131:3256-65; PMID:18952671; http://dx.doi.org/10.1093/brain/awn271
- Dubé CM, Ravizza T, Hamamura M, Zha Q, Keebaugh A, Fok K, Andres AL, Nalcioglu O, Obenaus A, Vezzani A, Baram TZ. Epileptogenesis provoked by prolonged experimental febrile seizures: mechanisms and biomarkers. J Neurosci 2010; 30:7484-94; http://dx.doi.org/10.1523/JNEUROSCI.0551-10.2010
- Yuskaitis CJ, Beurel E, Jope RS. Evidence of reactive astrocytes but not peripheral immune system activation in a mouse model of Fragile × syndrome. Biochim Biophys 2010; 1082:1006-12; http://dx.doi.org/10.1016/j.bbadis.2010.06.015
- Lee RH, Mills EA, Schwartz N, Bell MR, Deeg KE, Ruthazer ES, Marsh-Armstrong N, Aizenman CD. Neurodevelopmental effects of chronic exposure to elevated levels of pro-inflammatory cytokines in a developing visual system. Neural Dev 2010; 5:1-18; PMID:20047651; http://dx.doi.org/10.1186/1749-8104-5-2
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 2005; 57:67-81; PMID:15546155; http://dx.doi.org/10.1002/ana.20315
- Randle JC, Harding MW, Ku G, Schönharting M, Kurrle R. ICE/Caspase-1 inhibitors as novel anti-inflammatory drugs. Expert Opin Invest Drugs 2001; 10:1207-9; PMID:11772244; http://dx.doi.org/10.1517/13543784.10.7.1207
- Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, Decker C, Charifson P, Weber P, Germann UA, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3, 3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R, 3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1β and IL-18. J Pharmacol Exp Ther 2007; 321:509-16; PMID:17289835; http://dx.doi.org/10.1124/jpet.106.111344
- Ravizza T, Lucas SM, Balosso S, Bernardino L, Ku G, Noé F, Malva J, Randle JC, Allan S, Vezzani A. Inactivation of caspase-1 in rodent brain: a novel anticonvulsive strategy. Epilepsia 2006; 47:1160-8; PMID:16886979; http://dx.doi.org/10.1111/j.1528-1167.2006.00590.x
- Ravizza T, Noé F, Zardoni D, Vaghi V, Sifringer M, Vezzani A. Interleukin converting enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1β production. Neurobiol Dis 2008; 31:327-33; PMID:18632279; http://dx.doi.org/10.1016/j.nbd.2008.05.007
- Incorporated VP. Study of VX-765 in subjects with treatment-resistant partial epilepsy. (Study code-clinicaltrials.gov identifier: NCT01048255)
- Yang QW, Wang J-Z, Li J-C, Zhou Y, Zhong Q, Lu F-L, Xiang J. High-mobility group protein box-1 and its relevance to cerebral ischemia. J Cereb Blood Flow Metab 2009; 30:243-54; PMID:9794402
- Barnes GN. “For whom the bell tolls”: blockade of toll-like receptors may regulate seizure occurrence. Epilepsy Curr 2010; 10:164-5; PMID:21157547; http://dx.doi.org/10.1111/j.1535-7511.2010.01389.x
- Walker L, Sills GJ. Inflammation and epilepsy: the foundations for a new therapeutic approach in epilepsy? Epilepsy Curr 2012; 12:8-12; http://dx.doi.org/10.5698/1535-7511-12.1.8
- Frémont L. Biological effects of resveratrol. Life Sci 2000; 66:663-73; http://dx.doi.org/10.1016/S0024-3205(99)00410-5
- Bhat KP, Kosmeder JW, Pezzuto JM. Biological effects of resveratrol. Antioxid Redox Signal 2001; 3:1041-64; PMID:11813979; http://dx.doi.org/10.1089/152308601317203567
- Clément M-V, Hirpara JL, Chawdhury S-H, Pervaiz S. Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood 1998; 92:996-1002
- Albani D, Polito L, Signorini A, Forloni G. Neuroprotective properties of resveratrol in different neurodegenerative disorders. Biofactors 2010; 36:370-6; PMID:20848560; http://dx.doi.org/10.1002/biof.118
- Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J Immunol 2005; 175:3339-46; PMID:16116226; http://dx.doi.org/10.4049/jimmunol.175.5.3339
- Wu Z, Xu Q, Zhang L, Kong D, Ma R, Wang L. Protective effect of resveratrol against kainate-induced temporal lobe epilepsy in rats. Neurochem Res 2009; 34:1393-400; PMID:19219549; http://dx.doi.org/10.1007/s11064-009-9920-0
- Gupta Y, Briyal S, Chaudhary G. Protective effect of tans-resveratrol against kainic acid-induced seizures and oxidative stress in rats. Pharmacol Biochem Behav 2002; 71:245-9; http://dx.doi.org/10.1016/S0091-3057(01)00663-3
- Zhang F, Liu J, Shi J-S. Anti-inflammatory activities of resveratrol in the brain: role of resveratrol in microglial activation. Eur J Pharmacol 2010; 636:1-7; PMID:20361959; http://dx.doi.org/10.1016/j.ejphar.2010.03.043
- Falchetti R, Fuggetta MP, Lanzilli G, Tricarico M, Ravagnan G. Effects of resveratrol on human immune cell function. Life Sci 2001; 70:81-96; PMID:11764009; http://dx.doi.org/10.1016/S0024-3205(01)01367-4
- Subbaramaiah K, Chung WJ, Michaluart P, Telang N, Tanabe T, Inoue H, Jang M, Pezzuto JM, Dannenberg AJ. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J Biol Chem 1998; 273:21875-82; PMID:9705326; http://dx.doi.org/10.1074/jbc.273.34.21875