
ABSTRACT
Chagas disease, caused by Trypanosoma cruzi, results in an acute febrile illness that progresses to chronic chagasic cardiomyopathy in 30% of patients. Current treatments have significant side effects and poor efficacy during the chronic phase; therefore, there is an urgent need for new treatment modalities. A robust TH1-mediated immune response correlates with favorable clinical outcomes. A therapeutic vaccine administered to infected individuals could bolster the immune response, thereby slowing or stopping the progression of chagasic cardiomyopathy. Prior work in mice has identified an efficacious T. cruzi DNA vaccine encoding Tc24. To elicit a similar protective cell-mediated immune response to a Tc24 recombinant protein, we utilized a poly(lactic-co-glycolic acid) nanoparticle delivery system in conjunction with CpG motif-containing oligodeoxynucleotides as an immunomodulatory adjuvant. In a BALB/c mouse model, the vaccine produced a TH1-biased immune response, as demonstrated by a significant increase in antigen-specific IFNγ-producing splenocytes, IgG2a titers, and proliferative capacity of CD8+ T cells. When tested for therapeutic efficacy, significantly reduced systemic parasitemia was seen during peak parasitemia. Additionally, there was a significant reduction in cardiac parasite burden and inflammatory cell infiltrate. This is the first study demonstrating immunogenicity and efficacy of a therapeutic Chagas vaccine using a nanoparticle delivery system.
Introduction
Chagas disease (American trypanosomiasis) is a neglected tropical disease of great importance in the Americas. Estimates from the Global Burden of Disease Study 2013 indicate that almost 10 million people are living with Chagas disease worldwide.Citation1 Chagas disease results in over $7 billion in lost productivity and health care costs annually.Citation2 The causative protozoan parasite, Trypanosoma cruzi (T. cruzi), is most frequently transmitted by triatomines (kissing bugs), but can also be transmitted vertically, resulting in congenital infection.Citation3 Chagas disease typically results in an acute febrile illness that progresses to chronic cardiomyopathy in 30% of patients.Citation3 Initially, cardiac dysfunction is characterized by conduction disorders, which can then progress to cardiomyopathy and even sudden cardiac death.Citation4 An estimated 1.17 million people suffer from chagasic cardiomyopathy in Latin America,Citation5 and it is the leading cause of cardiovascular death in persons between ages 30–50 living in endemic areas.Citation6
Current pharmacological treatments (nifurtimox and benznidazole) have poor efficacy beyond the acute phase of disease, with serological cure rates of only 5% to 20% in the chronic phase.Citation7 The recent BENEFIT trial of chagasic cardiomyopathy patients showed endpoint PCR conversion in 46.7% of the benznidazole treatment group, compared to 33.1% of the placebo group, far below what is seen if treatment is initiated in the acute phase of disease.Citation8 Both treatments are also associated with side effects in up to 50% of patients, most frequently cutaneous hypersensitivity reactions, gastrointestinal distress, and neurological symptoms,Citation7 and are contraindicated in pregnancy.Citation9 Therefore, there is an urgent need for new treatment modalities. In humans, production of the TH1-associated cytokine IFNγ by CD8+ T cell has been correlated with less severe cardiac disease.Citation10 Depletion of CD8+ T cells in mice results in uncontrolled parasitemia and high mortality.Citation11,12 Therefore, a therapeutic vaccine could bolster the TH1-mediated CD8+ T cell immune response to the infection, thereby slowing or halting the progression of disease.Citation13 A therapeutic vaccine for Chagas disease has potential advantages including reduced adverse effects and the potential to be used as a replacement for current therapies or paired with reduced-dose chemotherapy.Citation14 Economic modeling has shown that a therapeutic vaccine would be highly cost effective.Citation15
One promising target antigen is the 24 kD flagellar Ca2+ binding protein Tc24 (first called FCaBP).Citation16 Tc24 is a calcium-acyl switch protein localized to the flagellar pocket of the parasite, and is expressed in all developmental stages of T. cruzi.Citation17 There is greater than 97% sequence conservation in Tc24 between the T. cruzi strains.Citation18 Early studies in mice have shown that prophylactic immunization with Tc24 is protective in T. cruzi infection.Citation19,20 More recently, studies using a DNA plasmid encoding Tc24 have demonstrated therapeutic benefit.Citation21,22 The vaccine induced high levels of antigen-specific IFNγ+ CD8+ cells, protecting against parasitemia and cardiac pathology.Citation23 While recombinant protein vaccines have been licensed for hepatitis B and human papillomavirus, no DNA vaccines have progressed to licensure. Therefore, we focused on a recombinant protein vaccine strategy, combining the two prior approaches in order to employ a Tc24 recombinant protein antigen in a therapeutic vaccine. This recombinant protein was found to be immunogenic and partially protective as a prophylactic vaccine in mice when formulated with the Toll-like receptor 4 (TLR-4) agonist monophosphoryl lipid A.Citation24 In humans, recombinant Tc24 has been utilized as an antigen for serodiagnosis of Chagas disease and as a tool to monitor treatment success,Citation25,26 and T cell epitopes have been investigated.Citation11
Multiple studies have demonstrated that a TH1-mediated CD8+ T cell response is required for protective immunity to T. cruzi.Citation23,27 In particular, production of IFNγ is critical for control of T. cruzi infection, unlike IL-4, a TH2-associated cytokine.Citation23,28-30 Nanoparticles have been shown to increase antigen uptake by antigen-presenting cells Citation31 and enhance the TH1-mediated CD8+ T cell response Citation32-34 through increased MHC I antigen presentation.Citation35 Nanoparticles can also serve as a depot for antigen,Citation36 allowing prolonged stimulation of the TH1 pathway.Citation37 A common polymer used in nanoparticle synthesis is poly(lactic-co-glycolic acid) (PLGA).Citation38 PLGA is biocompatible and biodegradable. A second component of a nanoparticle-based vaccine designed to produce a protective immune response is the TLR-9 agonist CpG motif-containing oligodeoxynucleotides (CpG ODN) as an immunomodulatory adjuvant. CpG ODNs are potent stimulators of the TH1-mediated CD8+ T cell immune response.Citation39,40 In this study, we investigated the immune response elicited by a Tc24 recombinant protein vaccine in mice when delivered in a PLGA nanoparticle delivery system with CpG ODN as an immunomodulatory adjuvant. We also tested the therapeutic efficacy of this vaccine in a mouse model of Chagas disease. The purpose of this study is to demonstrate that PLGA nanoparticles can be used as a delivery system to improve immunogenicity and efficacy of a protein-based therapeutic vaccine for Chagas disease.
Results
Depot effect of PLGA nanoparticle delivery system
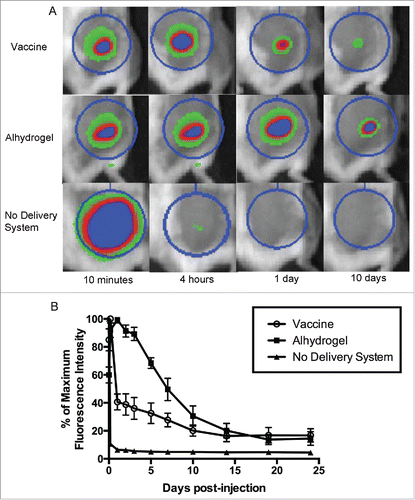
Nanoparticles were produced using a water-oil-water double emulsion method (Tc24) or an oil-water single emulsion method (CpG ODN) as described in the methods section of this paper. Loading efficiency and characterization by SEM () were performed to confirm consistency between batches. To test the depot effect of the PLGA nanoparticle delivery system, we injected mice with fluorescently labeled protein and observed the antigen dispersal over time using an in vivo imaging system. Fluorescently labeled Tc24 protein encapsulated in the PLGA nanoparticle delivery system was dispersed over 14 days after injection (). There was a drop in fluorescence over the first day, likely representing release of the protein on the exterior of the nanoparticles, then a second, slower, decrease in fluorescence through day 14, representing the slow degradation of the nanoparticles and release of encapsulated protein. In comparison, protein that lacked a delivery system was dispersed within one day. Duration of dispersal of encapsulated protein was similar, though somewhat shorter, than protein adsorbed to Alhydrogel®, which continued to show dispersal of protein until 19 days post-injection. The similar duration of antigen dispersal between our vaccine and Alhydrogel® indicates that PLGA nanoparticles exhibit a favorable depot effect for purposes of vaccine delivery. In addition to providing a depot effect for uptake of the antigen and adjuvant by antigen-presenting cells, a successful Chagas disease vaccine must also stimulate a TH1-biased immune response, which Alhydrogel® does not accomplish,Citation41,42 but PLGA nanoparticles have been shown to induce.Citation33-35
Figure 1. Nanoparticle delivery system. SEM images of PLGA nanoparticles containing (A) Tc24 and (B) CpG. Scale bars are 1 μm.

Figure 2. Depot effect of PLGA nanoparticle delivery system. To test the depot effect of the PLGA nanoparticle delivery system, mice were injected subcutaneously with vaccine containing fluorescently labeled protein. Dispersal of the antigen from the site of injection over time was observed using an in vivo imaging system. The nanoparticle delivery system was compared to the common vaccine adjuvant Alhydrogel®, as well as protein with no delivery system. (A) Representative images of selected time points. The regions of interest (circles) are used to calculate total fluorescence intensity. (B) Time course of fluorescence intensity as a percentage of the maximum value. Data are plotted as mean ± SD (n = 8).
Immunogenicity of recombinant protein Tc24 vaccine with CpG ODN in a PLGA nanoparticle delivery system
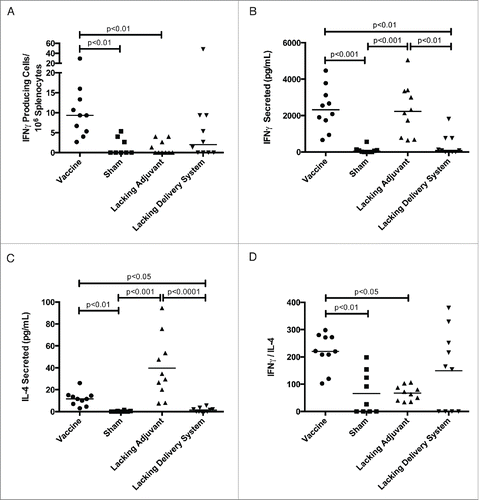
In order to characterize the immune response of our vaccine, we investigated antigen-specific TH1- and TH2-associated cytokine production, antibody titers, and CD8+ T cell proliferation capability. The vaccine produced a nine-fold increase in Tc24-specific IFNγ-secreting cells compared to the lacking adjuvant control () and a six-fold increase in Tc24-specific secreted IFNγ compared to the lacking delivery system control (). The lacking adjuvant control produced a comparable amount of secreted IFNγ to the vaccine (), though from fewer Tc24-specific cells (). However, this group also produced three-fold greater Tc24-specific IL-4 than the vaccine (). Comparing the IFNγ to IL-4 ratio to determine the degree to which the immune system is TH1-biased, the vaccine produced a three-fold larger IFNγ/IL-4 ratio compared to the lacking adjuvant control (). We conclude that the PLGA nanoparticle delivery system produces a robust mixed TH1/TH2 immune response, while the CpG ODN immunomodulatory adjuvant skews this response toward a TH1-bias.
Figure 3. Tc24-specific TH1 and TH2 cytokine response. BALB/c mice were vaccinated in a prime-boost model, with 4 weeks between prime and boost. The vaccine was compared to control groups lacking the CpG ODN (lacking adjuvant control) or the PLGA nanoparticles (lacking delivery system control), as well as a sham vaccine group. Two weeks after the boost vaccination, the antigen-specific cytokine response was assessed. (A) Tc24-specific IFNγ producing lymphocytes as assessed by ELISPOT. Background = 2 spot forming cells/106 splenocytes with no significant difference between groups. (B) Tc24-specific IFNγ secreted by 2 × 106 splenocytes/mL as assessed by ELISA. (C) Tc24-specific IL-4 secreted by 2 × 106 splenocytes/mL as assessed by ELISA. (D) TH1-biasing of the immune response as determined by the ratio of Tc24-specific secreted IFNγ to IL-4. All data shown here is background subtracted. Each point represents an individual animal (n = 10); median values are denoted by horizontal lines. Significance was calculated using Kruskal-Wallis test with Dunn's correction for multiple comparisons.
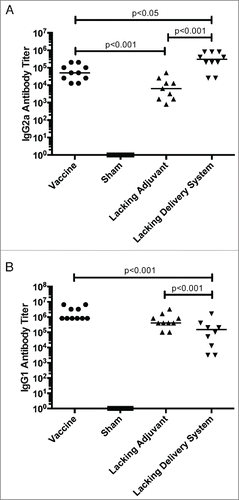
Tc24-specific antibody isotype titers corroborate with the cytokine results. IgG2a titers, a mouse antibody isotype associated with a TH1-bias, is most robust in the vaccine formulations that contain the CpG ODN immunomodulatory adjuvant compared to the lacking adjuvant control (). However, the IgG1 antibody response is most robust in the formulations containing the PLGA nanoparticle delivery system, compared to the lacking delivery system control (). This further supports the findings of an overall immune system stimulation that results from PLGA nanoparticles, and a favorable TH1-biasing with the addition of CpG ODN.
Figure 4. Tc24-specific antibody response. BALB/c mice were vaccinated in a prime boost model, with 4 weeks between prime and boost. The vaccine was compared to control groups lacking the CpG ODN (lacking adjuvant control) or the PLGA nanoparticles (lacking delivery system control), as well as a sham vaccine group. Two weeks after the boost vaccination, the antigen-specific antibody response was assessed by ELISA. (A) Tc24-specific IgG2a antibody titer, a TH1-associated antibody isotype. (B) Tc24-specific IgG1 antibody titer. Titers were recorded as the last positive dilution above a cut-off O.D., which was determined by the average O.D. of serum from naïve mice plus 3x standard deviation. Each point represents an individual animal (n = 10); median values are denoted by horizontal lines. Data was log transformed and significance was calculated by one-way ANOVA with Tukey's correction for multiple comparisons. All groups were significantly greater than the sham vaccine, and so notation was omitted from the graphs for clarity of interpretation.
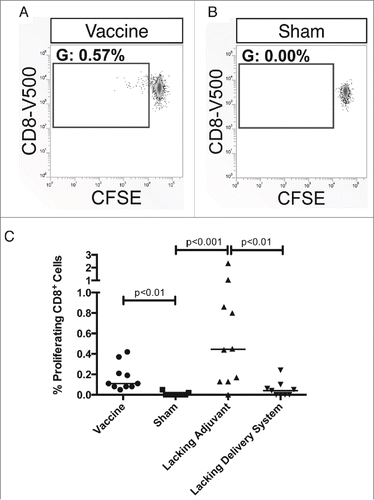
Lastly, we explored the Tc24-specific proliferative capacity of CD8+ T cells. Immunization with both the vaccine and the lacking adjuvant control resulted in a CD8+ T cell population that expanded upon re-stimulation with Tc24 and was statistically significant compared to the sham vaccine (). In comparison, the lacking delivery system control did not produce a statistically significant population. This is consistent with the findings that the nanoparticle delivery system is necessary for a robust immune response to the vaccine.
Figure 5. Tc24-specific proliferative capacity of CD8+ T cells. BALB/c mice were vaccinated in a prime-boost model, with 4 weeks between prime and boost. The vaccine was compared to control groups lacking the CpG ODN (lacking adjuvant control) or the PLGA nanoparticles (lacking delivery system control), as well as a sham vaccine group. Two weeks after the boost vaccination, the antigen-specific CD8+ T cell response was assessed by carboxyfluorescein diacetate succinimidyl ester (CFSE) proliferation assay. (A) Representative gating from a vaccinated mouse with the gate showing the percentage of live, CD19−, CD3+, CD8+ T cells that have undergone division. (B) Representative gating from a mouse in the sham vaccine group with the gate showing the percentage of live, CD19−, CD3+, CD8+ T cells that have undergone division. (C) Comparison of the proliferative capacity of CD8+ T cells between the vaccine and control groups. Data is background subtracted. Background = 0.06% proliferating cells with no significant difference between groups. Each point represents an individual animal (n = 10); median values are denoted by horizontal lines. Significance was calculated using Kruskal-Wallis test with Dunn's correction for multiple comparisons.
Therapeutic efficacy of the Tc24 and CpG ODN vaccine in a nanoparticle delivery system
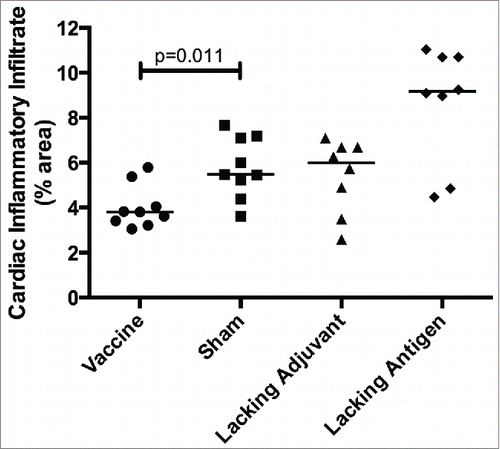
In order to evaluate therapeutic efficacy of the vaccine, mice were infected with T. cruzi then vaccinated. Mice receiving the vaccine had the lowest parasitemia throughout the acute phase of infection (), including a six-fold reduction in parasite burden at the peak of parasitemia compared to the sham vaccine. None of the control groups were able to achieve this reduction in parasitemia. The initial reduction in parasitemia seen in all groups may be due to development of lytic antibodies that have been described in humans, Citation43 and which could become exhausted resulting in the second spike. The significant reduction in parasitemia at the second peak in mice receiving the vaccine, but not in the control groups, is likely due to the TH1-biased immune response generated by the vaccine. Additionally, protection from widespread parasite invasion in the cardiac tissue was also seen in vaccinated mice () compared to mice in the sham vaccine group (). The number of T. cruzi amastigote nests in the cardiac tissue was three-fold lower in vaccinated mice than mice receiving the sham vaccination (). The lacking adjuvant control also showed approximately a three-fold reduction in amastigote nests in the cardiac tissue compared to the sham group, though it was not statistically significant. However, the lacking adjuvant control was unable to achieve the near sterilizing immunity in the cardiac tissue that the vaccine produced in 4/9 mice. Overall, there was a preservation of normal cardiac tissue in the vaccinated mice, whereas mice receiving the sham vaccine had widespread inflammatory infiltrate and destruction of normal cardiac architecture. Mice receiving the vaccine had a 30% reduction in inflammatory infiltrate compared to the sham vaccine group (). The lacking antigen control had a 67% increase in the inflammatory infiltrate compared to the sham vaccine, though it was not statistically significant. This evidence of cardiac protection and reduction in systemic parasitemia at the height of infection are suggestive that this vaccine is not only immunogenic, but also efficacious as a therapeutic vaccine against T. cruzi infection.
Figure 6. Therapeutic efficacy as measured by systemic parasitemia. Mice were infected with T. cruzi, and then immunized at days 7 and 14 post infection, as shown by arrows (↓). The vaccine was compared to controls lacking the CpG ODN (lacking adjuvant control) or the Tc24 protein (lacking antigen control), both delivered in the nanoparticle delivery system, as well as a sham vaccine group. Parasitemia in the blood was measured twice weekly by quantitative real-time PCR throughout the acute phase of infection. Data are plotted as median and interquartile range (n = 10). Significance was calculated using Kruskal-Wallis test with Dunn's correction for multiple comparisons; *p < 0.05 when comparing the vaccine to the sham.
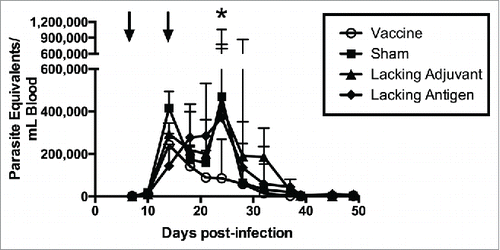
Figure 7. Therapeutic efficacy as measured by cardiac parasite burden. Mice were infected with T. cruzi, and then immunized at days 7 and 14 post infection. At 50 d post infection, the vaccine was compared to controls lacking the CpG ODN (lacking adjuvant control) or the Tc24 protein (lacking antigen control), both delivered in the nanoparticle delivery system, as well as a sham vaccine group. Representative H&E stained section of a heart from (A) a vaccinated mouse, (B) a mouse in the sham vaccine group. Examples of amastigote nests shown with an arrow (→). (C) Quantification of the amastigote nests in the cardiac tissue over 20 representative fields of view (400x) per section. Each point represents an individual animal (n = 8–9); median values are denoted by horizontal lines. Significance was calculated using Kruskal-Wallis test with Dunn's correction for multiple comparisons.
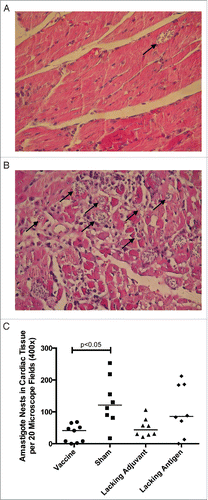
Figure 8. Therapeutic efficacy as measured by cardiac inflammatory infiltrate Mice were infected with T. cruzi, and then immunized at days 7 and 14 post infection. At 50 days post infection, the vaccine was compared to controls lacking the CpG ODN (lacking adjuvant control) or the Tc24 protein (lacking antigen control), both delivered in the nanoparticle delivery system, as well as a sham vaccine group. Quantification of the percentage of area occupied by inflammatory cell nuclei in the cardiac tissue over 5 representative fields of view (100x). Each point represents an individual animal (n = 8–9); median values are denoted by horizontal lines. Significance was assessed by the Kruskal-Wallis test with the post-hoc comparison of all groups to the sham vaccine control using the Mann-Whitney test with Bonferroni adjustment.
Discussion
Our laboratory is working to develop a therapeutic Chagas disease vaccine with a target product profile focused on prevention or delay of onset of chagasic cardiomyopathy in patients who seroconvert to T. cruzi.. Citation15 Chagas disease presents a distinct challenge for vaccine development: while most vaccines rely on eliciting a protective antibody response, a Chagas disease vaccine requires a cell-mediated immune response. Aluminum hydroxide adjuvant (such as Alhydrogel®), the adjuvant utilized in 80% of currently licensed vaccines, produces a predominantly TH2-skewed immune response,Citation41,42 and is unsuitable for Chagas vaccine development. In this study, we employed a novel approach to elicit a protective TH1-biased immune response by combining our protein antigen of interest, Tc24, with CpG ODN as an immunomodulatory adjuvant and encapsulating the antigen and adjuvant in a PLGA nanoparticle delivery system. CpG ODN is already in use as an adjuvant in clinical trials for hepatitis B, malaria, anthrax, and flu vaccines,Citation44 and PLGA is currently utilized as a drug delivery system in several FDA approved therapeutics.Citation45 By utilizing these two proven technologies, we anticipate that this vaccine could transition into clinical trials more quickly.
In this study we show that PLGA nanoparticles provide a depot effect similar to that of Alhydrogel®, allowing for prolonged release of Tc24 antigen. The delivery system also elicits an overall robust immune response that vaccine formulations lacking the PLGA nanoparticle delivery system do not achieve. The CpG ODN immunomodulatory adjuvant assists in biasing the vaccine toward a protective TH1-mediated immune response. This TH1-biasing is primarily due to the pathway activated in binding of CpG ODN by TLR-9 in the endolysosome of antigen-presenting cells,Citation46 but is likely also enhanced by the PLGA nanoparticle delivery system, which enhances antigen uptake by antigen-presenting cells,Citation32 facilitates uptake of CpG ODN into the endolysosome,Citation47 and increases MHC I antigen presentation.Citation36 Overall, the vaccine containing both the CpG ODN immunomodulatory adjuvant and the PLGA nanoparticle delivery system was necessary to produce a robust TH1-baised immune response to the Tc24 protein. Our findings here are similar to our previous observations using this adjuvant and delivery system with a model antigen (ovalbumin).Citation37 They are also consistent with reported studies showing TH1-biased enhancement of other protein antigens when combined with CpG ODN in PLGA nanoparticles.Citation48-50 Other therapeutic vaccines against Chagas disease have been found to induce a protective antigen-specific CD8+ T cell population that expands upon challenge with infection and produces TH1-associated cytokines that include IFNγ and TNFα,Citation51 as well as a CD4+ T cell population capable of IFNγ production.Citation24 Our results are consistent with these findings, though further characterization of the immune response could be pursued to determine the cell types responsible for protection in this model.
When tested for therapeutic efficacy, the vaccine significantly reduced systemic parasitemia during peak parasitemia, whereas none of the control groups were able to achieve this protection. Additionally, the vaccine significantly reduced the number of T. cruzi amastigote nests in the cardiac tissue. While the adjuvant control also showed a similar, though non-significant, reduction in amastigote nests in the cardiac tissue, it was unable to achieve the near-sterilizing immunity that the vaccine did in 40% of mice. Therefore, while a degree of protection is conferred by the Tc24 protein in the PLGA nanoparticle delivery system alone, the complete vaccine is necessary for the greatest reduction in parasitemia. The assessment of parasite burden by amastigote nests is a method that is widely used in the Chagas vaccine development field,Citation31 though future studies could also evaluate cardiac parasitemia by the more sensitive but less standardized PCR method. Parasite persistence is considered a key cause of chagasic cardiomyopathy in humans.Citation52 Therefore, the observed reduction in cardiac parasite burden suggests protection from parasite-driven cardiac damage. This protection will be a key feature of a therapeutic vaccine for Chagas disease. Further supporting the findings of cardiac protection is the reduction in inflammatory infiltrate in the cardiac tissue of mice receiving the vaccine compared to the sham control. The lacking antigen control showed an increase in inflammatory infiltrate in the cardiac tissue compared to the sham control, though it was not statistically significant. While the CpG ODN adjuvant may be non-specifically inducing an inflammatory response, by administering the CpG ODN adjuvant with the Tc24 antigen the immune response is directed toward the appropriate target, preventing widespread inflammation. The vaccine containing both the Tc24 antigen and the CpG ODN adjuvant in the PLGA nanoparticle delivery system is necessary for therapeutic efficacy against parasitemia and cardiac inflammation.
One limitation of this study is that efficacy of the vaccine was tested using the acute murine model of infection. This model utilizes BALB/c mice, a strain that is particularly susceptible to the T. cruzi parasite and demonstrates high parasitemia through the acute phase of infection. Humans similarly show a period of high parasitemia, but symptomatic chagasic cardiomyopathy does not develop until the chronic phase of disease. Further testing of our vaccine in a mouse model of chronic Chagas disease will be important to further confirm therapeutic efficacy. Studies testing efficacy of the Tc24 DNA vaccine showed protection in both an acute and a chronic mouse model of disease.Citation22,24 Therefore, we anticipate that our findings of therapeutic efficacy here will predict efficacy in a chronic model of disease. Ultimately, vaccine development is a stepwise process and using the acute model for screening vaccine candidates can be used as an effective in vivo model to determine whether a vaccine would stimulate a sufficient immune response to control parasitemia, and is frequently used in drug efficacy testing.Citation53-55
Additionally, while a concerted effort has been made to select mouse models that closely mimic human disease, the T-cell response is not directly translatable between the two species. While we predict that our findings of immunogenicity and therapeutic efficacy in mice will be translatable to humans, additional studies in non-human primates will be needed before moving the vaccine to the clinic. Lastly, while the PLGA nanoparticle production method employed in this paper is a common method that has been previously published,Citation37 debate exists as to the ideal size of nanoparticles for vaccine delivery. Some reports suggest that “viral-size” nanoparticles may enhance the immune response, while other studies report that larger particles elicit a better immune response.Citation38 Further refinement of this vaccine could involve exploring alternative production methods that would yield tunable sizes of nanoparticles, to determine the optimum particle size for eliciting the desired immune response.
While PLGA nanoparticles have been widely investigated as a vaccine delivery system, only very limited work has been conducted on the use of PLGA nanoparticles in vaccines against parasites.Citation56,57 This is the first reported study utilizing a nanoparticle delivery system approach in developing a therapeutic vaccine against Chagas disease. Our findings of parasite reduction, but not elimination, are similar to the majority of Chagas disease vaccine studies.Citation24,25,29 Though sterilizing immunity may not be achievable in this model, it is clear that a successful vaccine will need to produce a meaningful reduction in parasitemia. Ultimately, Tc24 may be combined with other T. cruzi antigens, such as TSA1, TcG2, and TcG4 to increase vaccine efficacy.Citation22,24,29,30,58 Additionally, this vaccine could potentially be tested in combination with reduced dose chemotherapy.Citation15 If the vaccine provides a dose-sparing effect on the chemotherapeutic agents, the side effect profile could be minimized while increasing efficacy though multiple parasite killing mechanisms. In conclusion, this vaccine utilizing a PLGA nanoparticle delivery system demonstrates robust TH1-biased immunogenicity and promising therapeutic efficacy in an acute mouse model of Chagas disease. This study represents an important proof of principle that PLGA nanoparticles can be used as a delivery system to improve immunogenicity and efficacy of a protein-based vaccine for Chagas disease.
Materials & methods
Production of recombinant Tc24 protein
Recombinant Tc24 was expressed in E. coli BL21(DE3) and purified with Ni-column chromatography as described previously.Citation25 Briefly, E. coli codon-optimized DNA coding for full-length H1 strain T. cruzi Tc24 was cloned in frame into E. coli expression vector pET41a (EMD Millipore, 70556) with fusion GST deleted (NdeI/XhoI). The recombinant plasmid DNA was transformed into BL21(DE3) (EMD Millipore, 69450) and recombinant protein was induced with IPTG. The recombinant Tc24 was purified with IMAC (GE Healthcare, Little Chalfont, United Kingdom), and with Q column purification (GE Healthcare, Little Chalfont, United Kingdom) to remove endotoxin contamination.
Production and characterization of PLGA nanoparticles
Tc24 protein was encapsulated using water-oil-water double emulsion method.Citation37 Tc24 protein in PBS containing 0.5% PVA and 50 mg/mL D-mannitol was homogenized with PLGA pre-dissolved in chloroform, using an IKA® T25 homogenizer (IKA Works, Wilmington, NC). The primary emulsion was homogenized with 5% PVA solution. The resulting emulsion was poured into a large volume of 5% PVA solution and stirred overnight at room temperature to evaporate the chloroform.
CpG ODN 1826, an agonist for mouse TLR-9 (5′-tccatgacgttcctgacgtt-3′ with phosphorothioate bonds at all bases, Integrated DNA Technologies), was first ion-paired with a cationic lipid DOTAP at a 1:1 molar ratio of nucleic base to DOTAP molecule to create a complex soluble in dichloromethane, prior to encapsulation by oil-in-water single emulsion method.Citation37 The ion-paired CpG was mixed with PLGA to form a homogenous oil phase and then homogenized with 5% PVA solution. After evaporation of organic solvent, nanoparticles were collected by centrifugation, washed, re-suspended, and lyophilized.
Particle morphology was characterized by scanning electronic microscopy (SEM, Jeol JSM-6100, Peabody, MA). Protein encapsulation levels was measured by Micro BCA™ Protein Assay (Life Technologies, 23235) after nanoparticles were digested in a mixture of DMSO, SDS, and NaOH. Citation59 Average encapsulation for Tc24 nanoparticles was 15 μg Tc24 in 1 mg PLGA nanoparticles. To quantify CpG encapsulation, nanoparticles were digested in the same manner, followed by pH adjustment to neutral and measurement of CpG concentration by OliGreen® ssDNA Assay (Life Technologies, O11492).Citation37 Average encapsulation of CpG nanoparticles was 40 μg CpG in 1 mg PLGA nanoparticles.
Kinetics of antigen dispersal
In vivo imaging was utilized to investigate antigen dispersal from the nanoparticle vaccine over time. Mice were injected subcutaneously with 1 μg of Alexa Fluor 660-labeled (Life Technologies, A-20171) Tc24 protein encapsulated in PLGA nanoparticles. Control groups included protein formulated on Alhydrogel® (aluminum hydroxide adjuvant, Brenntag Biosector, 21645-51-2) or protein alone. Fluorescent imaging was performed on an IVIS® Lumina II (Caliper Life Sciences, Hopkinton, MA) in the Small Animal Imaging Facility at Texas Children's Hospital. The imaging settings utilized were: excitation wavelength = 640 nm, emission wavelength = 704 nm, f-stop = 2 or 4, binning = 2, exposure time = 1 s or 2 s. The total radiant efficiency ([photons/sec]/[mW/cm2]) at the injection site was measured using Living Image® software (Caliper Life Sciences, Hopkinton, MA). The total radiant efficiency at each time point was normalized to the maximal reading.
Mice and immunization
Female BALB/c mice (Taconic Biosciences), aged 6–8 weeks, were used in all experiments. Animal experiments were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, 8th edition, under a protocol approved by Baylor College of Medicine's Institutional Animal Care and Use Committee (IACUC). Mice were vaccinated subcutaneously with vaccine and control group formulations (). The vaccine was compared to a group that controlled for the effect of CpG ODN (lacking adjuvant control) as well as a group that controlled for the effect of the PLGA nanoparticles (lacking delivery system control). Outcomes assessed were antigen-specific and so no lacking antigen control was tested. A dose of 30 μg Tc24 protein and 40 μg CpG ODN was administered. All vaccine formulations were resuspended in saline. Four weeks after the prime vaccination, mice were boosted with the same vaccine formulations as the prime vaccination. Two weeks after the boost vaccination, mice were humanely euthanized and serum and spleens were harvested to evaluate antigen-specific immune responses.
Table 1. Treatment groups. Description of the vaccine and control groups utilized in (A) the immunogenicity studies and (B) the therapeutic efficacy studies. The controls have been named based on the vaccine component lacking from the formulation.
Blood samples were allowed to coagulate in Serum-Gel clotting tubes (SARSTEDT, 41.1378.005) at room temperature and centrifuged to separate serum per manufacturer's instructions. Whole spleens were mechanically dissociated using 40 μm strainers (BD Biosciences, 352340). Red cells were lysed with ACK lysis buffer (Lonza, 10-548E). Viable cells were counted using a Cellometer Auto 2000 automated cell counter and AOPI Staining Solution (Nexcelom Biosciences, Lawrence, MA). Cells were diluted to final concentrations in 10% FBS, 1X Pen/Strep in RPMI-1640 (all from Corning, 35-011-CV, 30-002-CI, 10-040-CV) for cellular assays.
IFNγ-producing splenocytes
Antigen-specific IFNγ-producing splenocytes were quantified by ELISpot. Using an ImmunoSpot kit (Cellular Technology Limited, MIFNG-1M), 2.5 × 105 splenocytes were incubated with 50 µg/mL Tc24, 5 µg/mL Concanavalin A (ConA, Sigma-Aldrich, C0412), or media only for 24 hrs, 37°C, 5% CO2. After the cytokine detection process per manufacturer's instructions, spots were counted using a CTL-ImmunoSpot® S6 Macro Analyzer (Cellular Technology Limited, Shaker Heights, OH). The frequencies of antigen-specific IFNγ-producing cells were calculated by background subtraction of the unstimulated cells.
IFNγ and IL-4 levels
Antigen-specific cytokine release from splenocytes was quantified by ELISA. Briefly, 2×106 splenocytes/mL were incubated with 50 µg/mL Tc24, 5 µg/mL ConA, or media only for 72 hrs, 37°C, 5% CO2. To measure IFNγ and IL-4 in the supernatants, a sandwich ELISA method was employed, using Mouse IFNγ and IL-4 ELISA kits (eBioscience, 88–7314, 88–7044) per manufacturer's instructions. Cytokine concentrations produced by antigen-specific cells were calculated by background subtraction of the unstimulated cells.
Serum antibody titers
Serum antibodies to Tc24 were measured by ELISA. Plates were coated with 1.25 µg/mL Tc24 in coating solution (KPL, 50-84-00). Plates were blocked, and two-fold serially diluted serum samples were added, beginning with 1:400. Bound antibody was detected with 1:4,000 HRP-conjugated goat anti-mouse IgG1 or IgG2a secondary antibody (LifeSpan Biosciences, LS-C59107, LS-C59112). The reaction was developed with TMB Substrate (Thermo Scientific). Titers were recorded as the last positive dilution above a cut-off O.D., as determined by the O.D.Avg+ 3SD of serum from naïve mice. Mice that did not have a detectable titer were assigned a titer of 1.
Lymphocyte proliferation
Antigen-specific lymphocyte proliferation was measured by carboxyfluorescein diacetate succinimidyl ester (CFSE) as described previously.Citation60 Briefly, 5×106 splenocytes were stained with CFSE (Life Technologies, C34554) and then incubated with 50 µg/mL Tc24, 5 µg/mL ConA, or media only, 72 hrs, 37°C, 5% CO2. Cells were stained with LIVE/DEAD™ Fixable Blue Stain (Life Technologies, L-34961), and then incubated with Purified Rat Anti-Mouse CD16/CD32 (BD Biosciences, 553142). Surface markers were assessed with PE-Cy7-conjugated anti-CD19, PE-conjugated anti-CD3, V500-conjugated anti-CD8 (all BD Biosciences, 552854, 553064, 560776), and APC-Alexa Fluor 750-conjugated anti-CD4 (Life Technologies, MCD0427). Cells were fixed with Cytofix solution (BD Biosciences, 554655). Samples were acquired on a LSR Fortessa (BD Biosciences, San Jose, CA) at the Baylor College of Medicine Cytometry and Cell Sorting Core. Data analysis was performed using VenturiOne 5.0 (AppliedCytometry, Dinnington, Sheffield, United Kingdom).
Parasites and infection
T. cruzi H1 strain parasites, previously isolated from a human case in Yucatan, Mexico,Citation22 were maintained by serial passage in mice. To test therapeutic efficacy of the vaccine, naïve mice were infected intraperitoneally with 500 trypomastigotes. Seven days post-infection (dpi) the mice were immunized subcutaneously with vaccine and control group formulations (). Given that the nanoparticle delivery system was necessary to produce a robust immune response in the immunogenicity experiment, no lacking delivery system control was tested for therapeutic efficacy. Instead, the vaccine was compared to a group that controlled for the effect of CpG ODN (lacking adjuvant control), as in the immunogenicity study, as well as a group that controlled for the effect of the Tc24 protein (lacking antigen control), as there is parasite-derived Tc24 in natural infection. A sham vaccine group was also tested to assess the natural progression of disease. A dose of 30 μg Tc24 protein and 40 μg CpG ODN was used. In the cardiac parasite burden experiment, 15 μg Tc24 protein and 8 μg CpG ODN was utilized. A boost vaccination was administered at 14 dpi. Blood was collected twice weekly for quantification of parasitemia. At 50 dpi, all remaining mice were humanely euthanized.
Evaluation of parasitemia
Total DNA was isolated from blood using a DNEasy blood and tissue kit (Qiagen, 69506) and 10 ng DNA was used in quantitative real-time PCR using TaqMan® Fast Advanced Master Mix (Life Technologies, 4444557) and oligonucleotides specific for the satellite region of T. cruzi nuclear DNA (primers 5′ ASTCGGCTGATCGTTTTCGA 3′ and 5′ AATTCCTCCAAGCAGCGGATA 3′, probe 5′ 6-FAM CACACACTGGACACCAA MGB 3′, Life Technologies, 4304972, 4316032).Citation61,62 Data were normalized to GAPDH (primers 5′ CAATGTGTCCGTCGTGGATCT 3′ and 5′ GTCCTCAGTGTAGCCCAAGATG 3′, probe 5′ 6-FAM CGTGCCGCCTGGAGAAACCTGCC MGB 3′, Life Technologies, 4304972, 4316032),Citation63 and parasite equivalents were calculated based on a standard curve.Citation64,65 Treatment groups were compared at peak parasitemia to evaluate parasite burdens.
Cardiac parasite burden and inflammation
Heart tissue was removed from euthanized animals and fixed in 10% formaldehyde for histopathological analysis. Samples were embedded in paraffin, cut into 5 μm sections, and stained with hematoxylin and eosin. For each mouse, a representative section was identified and amastigote nests were quantified over 20 fields of view at 400x magnification in a blinded manner. To assess inflammation, 5 representative images were acquired per mouse with a Nikon Eclipse E600 optical microscope at 100x magnification in a blinded manner and image analysis was performed using ImageJ software 1.48 v (National Institutes of Health, Bethesda, MD). Pixels corresponding to inflammatory cell nuclei were quantified and normalized to total pixels of the sample to assess the percentage of inflammatory infiltrate area in the muscle fiber.
Statistical analysis
Results for cellular assays, parasitemia, and cardiac parasite burden are shown as individual mouse data points with median values and were analyzed by the Kruskal-Wallis test with Dunn's correction for multiple comparisons. Results for serum antibody are shown as individual mouse data points with median values. Results were log-transformed and analyzed by one-way ANOVA with Tukey's correction for multiple comparisons. Results for inflammatory infiltrate are shown as individual mouse data points with median values. Results were analyzed by the Kruskal-Wallis test with the post-hoc comparison of all groups to the sham vaccine control using the Mann-Whitney test with Bonferroni adjustment. Differences between treatment groups were considered significant if the p-value was less than 0.05. Statistical analysis was performed using Prism software 6.0 (GraphPad Software, La Jolla, CA).
Ethical statement
Animal experiments were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, 8th edition, under a protocol approved by Baylor College of Medicine's Institutional Animal Care and Use Committee (IACUC).
Disclosure of potential conflicts of interest
The Sabin Vaccine Institute and Texas Children's Hospital Center for Vaccine Development has funding to develop and test vaccines for neglected tropical diseases, including Chagas disease. Since the completion of this study, M.J.H. has changed positions and now works as a Principal for Fannin Innovation Studio, an early-stage life sciences commercialization group. The authors do not have any financial or personal interests that might pose a conflict of interest.
Acknowledgments
We would like to thank Texas Children's Hospital for the use of the Small Animal Imaging Facility and the Department of Pathology Electron Microscopy Core Facility.
Funding
This work was supported by the Sabin Vaccine Institute and Texas Children's Hospital Center for Vaccine Development, as well as Consejo Nacional de Ciencia y Tecnologia (CONACYT), Mexico (CB-2010-01-156513). Additional support was provided by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the National Institutes of Health (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M. Sederstrom. Munir Buhaya participated in this project via the Summer Medical and Research Training (SMART) program and was funded through a National Heart, Lung, and Blood Institute educational award (R25 HL108853).
References
- Global Burden of Disease Study C. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015; 386(9995):743-800; PMID:26063472; http://dx.doi.org/10.1016/S0140-6736(15)60692-4
- Lee BY, Bacon KM, Bottazzi ME, Hotez PJ. Global economic burden of Chagas disease: a computational simulation model. Lancet Infect Dis 2013; 13(4):342-8
- Rassi A, Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet 2010; 375:1388-402; PMID:20399979; http://dx.doi.org/10.1016/S0140-6736(10)60061-X
- Rassi A, Jr, Rassi A, Marin-Neto JA. Chagas heart disease: pathophysiologic mechanisms, prognostic factors and risk stratification. Memorias do Instituto Oswaldo Cruz 2009; 104 Suppl 1:152-8; http://dx.doi.org/10.1590/S0074-02762009000900021
- Chagas disease in Latin America: an epidemiological update based on 2010 estimates. Wkly Epidemiol Rec 2015; 90:33-43; PMID:25671846
- Rassi A, Jr, Rassi A, Little WC. Chagas' heart disease. Clin Cardiol 2000; 23:883-9; PMID:11129673; http://dx.doi.org/10.1002/clc.4960231205
- Perez-Molina JA, Norman F, Lopez-Velez R. Chagas disease in non-endemic countries: epidemiology, clinical presentation and treatment. Curr Infect Dis Reports 2012; 14:263-74; http://dx.doi.org/10.1007/s11908-012-0259-3
- Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A, Jr, Rosas F, Villena E, Quiroz R, Bonilla R, Britto C, et al. Randomized Trial of Benznidazole for Chronic Chagas' Cardiomyopathy. N Eng J Med 2015; 373:1295-306; http://dx.doi.org/10.1056/NEJMoa1507574
- Bern C, Montgomery SP, Herwaldt BL, Rassi A, Jr, Marin-Neto JA, Dantas RO, Maguire JH, Acquatella H, Morillo C, Kirchhoff LV, et al. Evaluation and treatment of chagas disease in the United States: a systematic review. JAMA 2007; 298:2171-81; PMID:18000201; http://dx.doi.org/10.1001/jama.298.18.2171
- Laucella SA, Postan M, Martin D, Hubby Fralish B, Albareda MC, Alvarez MG, Lococo B, Barbieri G, Viotti RJ, Tarleton RL. Frequency of interferon- gamma -producing T cells specific for Trypanosoma cruzi inversely correlates with disease severity in chronic human Chagas disease. J Infect Dis 2004; 189:909-18; PMID:14976609; http://dx.doi.org/10.1086/381682
- Tarleton RL. Depletion of CD8+ T cells increases susceptibility and reverses vaccine-induced immunity in mice infected with Trypanosoma cruzi. J Immunol 1990; 144:717-24; PMID:2104903
- Tarleton RL, Sun J, Zhang L, Postan M. Depletion of T-cell subpopulations results in exacerbation of myocarditis and parasitism in experimental Chagas' disease. Infect Immun 1994; 62:1820-9; PMID:8168945
- Autran B, Carcelain G, Combadiere B, Debre P. Therapeutic vaccines for chronic infections. Science 2004; 305:205-8; PMID:15247470; http://dx.doi.org/10.1126/science.1100600
- Dumonteil E, Bottazzi ME, Zhan B, Heffernan MJ, Jones K, Valenzuela JG, Kamhawi S, Ortega J, Rosales SP, Lee BY, et al. Accelerating the development of a therapeutic vaccine for human Chagas disease: rationale and prospects. Expert Rev Vaccines 2012; 11:1043-55; PMID:23151163; http://dx.doi.org/10.1586/erv.12.85
- Lee BY, Bacon KM, Wateska AR, Bottazzi ME, Dumonteil E, Hotez PJ. Modeling the economic value of a Chagas' disease therapeutic vaccine. Hum Vacc Immunotherapeutics 2012; 8:1293-301
- Engman DM, Krause KH, Blumin JH, Kim KS, Kirchhoff LV, Donelson JE. A novel flagellar Ca2+-binding protein in trypanosomes. J Biol Chem 1989; 264:18627-31; PMID:2681200
- Krautz GM, Kissinger JC, Krettli AU. The targets of the lytic antibody response against Trypanosoma cruzi. Parasitol Today 2000; 16:31-4; PMID:10637586
- Maldonado RA, Linss J, Thomaz N, Olson CL, Engman DM, Goldenberg S. Homologues of the 24-kDa flagellar Ca(2+)-binding protein gene of Trypanosoma cruzi are present in other members of the Trypanosomatidae family. Exp Parasitol 1997; 86:200-5
- Taibi A, Plumas-Marty B, Guevara-Espinoza A, Schoneck R, Pessoa H, Loyens M, Piras R, Aguirre T, Gras-Masse H, Bossus M, et al. Trypanosoma cruzi: immunity-induced in mice and rats by trypomastigote excretory-secretory antigens and identification of a peptide sequence containing a T cell epitope with protective activity. J Immunol 1993; 151:2676-89; PMID:7689612
- Taibi A, Espinoza AG, Ouaissi A. Trypanosoma cruzi: analysis of cellular and humoral response against a protective recombinant antigen during experimental Chagas' disease. Immunol Letters 1995; 48:193-200; PMID:8867851
- Dumonteil E, Escobedo-Ortegon J, Reyes-Rodriguez N, Arjona-Torres A, Ramirez-Sierra MJ. Immunotherapy of Trypanosoma cruzi infection with DNA vaccines in mice. Infect Immun 2004; 72:46-53; PMID:14688079
- Sanchez-Burgos G, Mezquita-Vega RG, Escobedo-Ortegon J, Ramirez-Sierra MJ, Arjona-Torres A, Ouaissi A, Rodrigues MM, Dumonteil E. Comparative evaluation of therapeutic DNA vaccines against Trypanosoma cruzi in mice. FEMS Immunol Medical Microbiol 2007; 50:333-41
- Limon-Flores AY, Cervera-Cetina R, Tzec-Arjona JL, Ek-Macias L, Sanchez-Burgos G, Ramirez-Sierra MJ, Cruz-Chan JV, VanWynsberghe NR, Dumonteil E. Effect of a combination DNA vaccine for the prevention and therapy of Trypanosoma cruzi infection in mice: role of CD4+ and CD8+ T cells. Vaccine 2010; 28:7414-9; PMID:20850536
- Martinez-Campos V, Martinez-Vega P, Ramirez-Sierra MJ, Rosado-Vallado M, Seid CA, Hudspeth EM, Wei J, Liu Z, Kwityn C, Hammond M, et al. Expression, purification, immunogenicity, and protective efficacy of a recombinant Tc24 antigen as a vaccine against Trypanosoma cruzi infection in mice. Vaccine 2015; 33:4505-12; PMID:26192358
- Godsel LM, Tibbetts RS, Olson CL, Chaudoir BM, Engman DM. Utility of recombinant flagellar calcium-binding protein for serodiagnosis of Trypanosoma cruzi infection. J Clin Microbiol 1995; 33:2082-5; PMID:7559952
- Guevara AG, Taibi A, Alava J, Guderian RH, Ouaissi A. Use of a recombinant Trypanosoma cruzi protein antigen to monitor cure of Chagas disease. Transactions Royal Society Tropical Med Hygiene 1995; 89:447-8
- Tzelepis F, de Alencar BC, Penido ML, Gazzinelli RT, Persechini PM, Rodrigues MM. Distinct kinetics of effector CD8+ cytotoxic T cells after infection with Trypanosoma cruzi in naive or vaccinated mice. Infect Immun 2006; 74:2477-81; PMID:16552083
- Gupta S, Garg NJ. Prophylactic efficacy of TcVac2 against Trypanosoma cruzi in mice. PLoS Neglected tropical Dis 2010; 4:e797; PMID:20706586
- Gupta S, Garg NJ. Delivery of antigenic candidates by a DNA/MVA heterologous approach elicits effector CD8(+)T cell mediated immunity against Trypanosoma cruzi. Vaccine 2012; 30:7179-86; PMID:23079191
- Pereira IR, Vilar-Pereira G, Marques V, da Silva AA, Caetano B, Moreira OC, Machado AV, Bruna-Romero O, Rodrigues MM, Gazzinelli RT, et al. A human type 5 adenovirus-based Trypanosoma cruzi therapeutic vaccine re-programs immune response and reverses chronic cardiomyopathy. PLoS Pathog 2015; 11:e1004594; PMID:25617628; http://dx.doi.org/10.1371/journal.ppat.1004594
- Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Controlled Release 2006; 112:26-34; http://dx.doi.org/10.1016/j.jconrel.2006.01.006
- Conway MA, Madrigal-Estebas L, McClean S, Brayden DJ, Mills KH. Protection against Bordetella pertussis infection following parenteral or oral immunization with antigens entrapped in biodegradable particles: effect of formulation and route of immunization on induction of Th1 and Th2 cells. Vaccine 2001; 19:1940-50; PMID:11228364; http://dx.doi.org/10.1016/S0264-410X(00)00433-3
- Audran R, Peter K, Dannull J, Men Y, Scandella E, Groettrup M, Gander B, Corradin G. Encapsulation of peptides in biodegradable microspheres prolongs their MHC class-I presentation by dendritic cells and macrophages in vitro. Vaccine 2003; 21:1250-5; PMID:12559806; http://dx.doi.org/10.1016/S0264-410X(02)00521-2
- Evans JT, Ward JR, Kern J, Johnson ME. A single vaccination with protein-microspheres elicits a strong CD8 T-cell-mediated immune response against Mycobacterium tuberculosis antigen Mtb8.4. Vaccine 2004; 22:1964-72; PMID:15121309; http://dx.doi.org/10.1016/j.vaccine.2003.10.035
- Fifis T, Gamvrellis A, Crimeen-Irwin B, Pietersz GA, Li J, Mottram PL, McKenzie IF, Plebanski M. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. J Immunol 2004; 173:3148-54; PMID:15322175; http://dx.doi.org/10.4049/jimmunol.173.5.3148
- Wang Q, Tan MT, Keegan BP, Barry MA, Heffernan MJ. Time course study of the antigen-specific immune response to a PLGA microparticle vaccine formulation. Biomaterials 2014; 35:8385-93; PMID:24986256; http://dx.doi.org/10.1016/j.biomaterials.2014.05.067
- Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Rev Vacc 2010; 9:1095-107; http://dx.doi.org/10.1586/erv.10.89
- Vabulas RM, Pircher H, Lipford GB, Hacker H, Wagner H. CpG-DNA activates in vivo T cell epitope presenting dendritic cells to trigger protective antiviral cytotoxic T cell responses. J Immunol 2000; 164:2372-8; PMID:10679072; http://dx.doi.org/10.4049/jimmunol.164.5.2372
- Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini JC, Romero P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest 2005; 115:739-46; PMID:15696196; http://dx.doi.org/10.1172/JCI23373
- Lindblad EB. Aluminium compounds for use in vaccines. Immunol Cell Biol 2004; 82:497-505; PMID:15479435; http://dx.doi.org/10.1111/j.0818-9641.2004.01286.x
- Hedayat M, Netea MG, Rezaei N. Targeting of Toll-like receptors: a decade of progress in combating infectious diseases. Lancet Infect Dis 2011; 11:702-12; PMID:21719349; http://dx.doi.org/10.1016/S1473-3099(11)70099-8
- Almeida IC, Milani SR, Gorin PA, Travassos LR. Complement-mediated lysis of Trypanosoma cruzi trypomastigotes by human anti-α-galactosyl antibodies. J Immunol 1991; 146:2394-400; PMID:1706399
- US. National Institutes of Health. ClinicalTrials.gov.Search term: “CpG7909.”
- Lu JM, Wang X, Marin-Muller C, Wang H, Lin PH, Yao Q, Chen C. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn 2009; 9:325-41; PMID:19435455; http://dx.doi.org/10.1586/erm.09.15
- Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov 2006; 5:471-84; PMID:16763660; http://dx.doi.org/10.1038/nrd2059
- Krishnamachari Y, Salem AK. Innovative strategies for co-delivering antigens and CpG oligonucleotides. Adv Drug Deliv Rev 2009; 61:205-17; PMID:19272328; http://dx.doi.org/10.1016/j.addr.2008.12.013
- Diwan M, Elamanchili P, Cao M, Samuel J. Dose sparing of CpG oligodeoxynucleotide vaccine adjuvants by nanoparticle delivery. Curr Drug Deliv 2004; 1:405-12; PMID:16305402; http://dx.doi.org/10.2174/1567201043334597
- Diwan M, Tafaghodi M, Samuel J. Enhancement of immune responses by co-delivery of a CpG oligodeoxynucleotide and tetanus toxoid in biodegradable nanospheres. J Controlled Release 2002; 85:247-62; http://dx.doi.org/10.1016/S0168-3659(02)00275-4
- Malyala P, Chesko J, Ugozzoli M, Goodsell A, Zhou F, Vajdy M, O'Hagan DT, Singh M. The potency of the adjuvant, CpG oligos, is enhanced by encapsulation in PLG microparticles. J Pharm Sci 2008; 97:1155-64; PMID:17683059; http://dx.doi.org/10.1002/jps.21065
- Gupta S, Garg NJ. TcVac3 induced control of Trypanosoma cruzi infection and chronic myocarditis in mice. PLoS One 2013; 8:e59434; PMID:23555672; http://dx.doi.org/10.1371/journal.pone.0059434
- Zhang L, Tarleton RL. Parasite persistence correlates with disease severity and localization in chronic Chagas' disease. J Infect Dis 1999; 180:480-6; PMID:10395865; http://dx.doi.org/10.1086/314889
- Cencig S, Coltel N, Truyens C, Carlier Y. Evaluation of benznidazole treatment combined with nifurtimox, posaconazole or AmBisome(R) in mice infected with Trypanosoma cruzi strains. Int J Antimicrob Agents 2012; 40:527-32; PMID:23063742; http://dx.doi.org/10.1016/j.ijantimicag.2012.08.002
- Bustamante JM, Craft JM, Crowe BD, Ketchie SA, Tarleton RL. New, combined, and reduced dosing treatment protocols cure Trypanosoma cruzi infection in mice. J Infect Dis 2014; 209:150-62; PMID:23945371; http://dx.doi.org/10.1093/infdis/jit420
- Olivieri BP, Molina JT, de Castro SL, Pereira MC, Calvet CM, Urbina JA, Araujo-Jorge TC. A comparative study of posaconazole and benznidazole in the prevention of heart damage and promotion of trypanocidal immune response in a murine model of Chagas disease. Int J Antimicrob Agents 2010; 36:79-83; PMID:20452188; http://dx.doi.org/10.1016/j.ijantimicag.2010.03.006
- Santos DM, Carneiro MW, de Moura TR, Soto M, Luz NF, Prates DB, Irache JM, Brodskyn C, Barral A, Barral-Netto M, et al. PLGA nanoparticles loaded with KMP-11 stimulate innate immunity and induce the killing of Leishmania. Nanomedicine 2013; 9:985-95; PMID:23603355; http://dx.doi.org/10.1016/j.nano.2013.04.003
- Tafaghodi M, Eskandari M, Kharazizadeh M, Khamesipour A, Jaafari MR. Immunization against leishmaniasis by PLGA nanospheres loaded with an experimental autoclaved Leishmania major (ALM) and Quillaja saponins. Trop Biomed 2010; 27:639-50; PMID:21399606
- Gupta S, Garg NJ. A Two-Component DNA-Prime/Protein-Boost Vaccination Strategy for Eliciting Long-Term, Protective T Cell Immunity against Trypanosoma cruzi. PLoS Pathog 2015; 11:e1004828; PMID:25951312; http://dx.doi.org/10.1371/journal.ppat.1004828
- Sah H. A new strategy to determine the actual protein content of poly(lactide-co-glycolide) microspheres. J Pharm Sci 1997; 86:1315-8; PMID:9383747; http://dx.doi.org/10.1021/js960363q
- Quah BJ, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc 2007; 2:2049-56; PMID:17853860; http://dx.doi.org/10.1038/nprot.2007.296
- Melo MF, Moreira OC, Tenorio P, Lorena V, Lorena-Rezende I, Junior WO, Gomes Y, Britto C. Usefulness of real time PCR to quantify parasite load in serum samples from chronic Chagas disease patients. Parasit Vectors 2015; 8:154; PMID:25890282; http://dx.doi.org/10.1186/s13071-015-0770-0
- Piron M, Fisa R, Casamitjana N, Lopez-Chejade P, Puig L, Verges M, Gascon J, Gomez i Prat J, Portus M, Sauleda S. Development of a real-time PCR assay for Trypanosoma cruzi detection in blood samples. Acta Trop 2007; 103:195-200; PMID:17662227; http://dx.doi.org/10.1016/j.actatropica.2007.05.019
- Gangisetty O, Reddy DS. The optimization of TaqMan real-time RT-PCR assay for transcriptional profiling of GABA-A receptor subunit plasticity. J Neurosci Methods 2009; 181:58-66; PMID:19406150; http://dx.doi.org/10.1016/j.jneumeth.2009.04.016
- Caldas S, Caldas IS, Diniz Lde F, Lima WG, Oliveira Rde P, Cecilio AB, Ribeiro I, Talvani A, Bahia MT. Real-time PCR strategy for parasite quantification in blood and tissue samples of experimental Trypanosoma cruzi infection. Acta Trop 2012; 123:170-7; PMID:22609548; http://dx.doi.org/10.1016/j.actatropica.2012.05.002
- Cencig S, Coltel N, Truyens C, Carlier Y. Parasitic loads in tissues of mice infected with Trypanosoma cruzi and treated with AmBisome. PLoS Neglected Tropical Dis 2011; 5:e1216; PMID:21738811; http://dx.doi.org/10.1371/journal.pntd.0001216