
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Interleukin 10 (IL-10) is a cytokine that is able to downregulate inflammation. Its overexpression is directly associated with the difficulty in the clearance of chronic viral infections, such as chronic hepatitis B, hepatitis C and HIV infection, and infection-related cancer. IL-10 signaling blockade has been proposed as a promising way of clearing chronic viral infection and preventing tumor growth in animal models. Recently, we have reported that peptides with a helical repeating pattern of hydrophobic and hydrophilic residues are able to inhibit IL-10 significantly both in vitro and in vivo.Citation1 In this work, we seek to further study the inhibiting mechanism of these peptides using sequence-modified peptides. As evidenced by both experimental and molecular dynamics simulation in concert the N-terminal hydrophobic peptide constructed with repeating hydrophobic and hydrophilic pattern of residues is more likely to inhibit IL10. In addition, the sequence length and the ability of protonation are also important for inhibition activity.
Introduction
Interleukin 10 (IL-10) is a cytokine with pleiotropic effects in immune regulation and inflammation. IL-10 is also known as human cytokine synthesis inhibitory factor (CSIF), and is encoded by the IL-10 gene. It is produced by many cells of the adaptive immune system, including T helper subtype 1 (Th1), Th2 and Th17 cell subsets, T Reg cells, CD8+ T cells and B cells, and also expressed by cells of the innate immune system, including dendritic cells (DCs), macrophages, mast cells, natural killer (NK) cells, eosinophils and neutrophils.Citation2 IL-10 plays a central role in infection by limiting the immune response to pathogens and thereby preventing excessive damage to the host.Citation3
IL-10 can abort T cell responses when present during priming stage of an immune response and inhibit ongoing T cell activity to viral infections.Citation4 Also, IL-10 acts directly on T cells to limit proliferation, functional differentiation, and effector activity.Citation5,6 Remarkable over-expression of IL-10 has been found in the serum of patients with chronic viral infections and cancers, which can diminish immune responses to the viruses and thus result in detrimental consequences, while lower IL-10 production are associated with clearance of viral infection and enhanced viral control during chronic HCV, HBV, HIV and EBV infections.Citation7
The IL-10/IL-10R pathway has been identified as a key regulation check point of viral persistence. Recent reports have shown that inhibition of IL-10 signaling by anti-IL-10 monoclonal antibodies clears the chronic lymphocytic choriomeningitis virus (LCMV) infection in the mouse model.Citation4,8,9 Moreover, inhibition of IL-10 at the time of therapeutic vaccine immunization further improves the clearance of chronic viral infection.Citation10 It has been demonstrated that IL-10 inhibition and immunization simultaneously prevent tumor growth in mouse tumor models.Citation11,12 Blockade of IL-10 combined with vaccination may provide a promising alternative to increase the efficacy of therapeutic vaccines for better control of chronic viral infection, and chronic viral infection-related cancers, such as cervical cancer and HBV relevant liver cancer.Citation13 Therefore, a clinical grade IL-10 signaling inhibitor is urgently needed to investigate whether blocking IL-10 signaling at the time of immunization can improve the efficacy of a therapeutic vaccine against chronic viral infection or tumor in clinical trial.
IL-10 signals via an interaction with its IL-10 receptor (IL-10R). IL-10R is a class II cytokine family member composed of 2 subunits: IL-10R1 is the unique ligand-binding subunit and IL-10R2 is the signaling subunit that is shared with other family member cytokines, including IL-22, IL-26, IL-28, and IL-29.Citation14-16 Thus, if the binding sites important for IL-10/IL-10R1 signaling can be determined, it will then be possible to develop drugs (e.g., small organic molecules, peptides, proteins and DNA segments) to block the IL-10 signaling and therefore inhibit the effect of IL-10 overexpression. However, very few clinically useful molecules are suitable for this purpose; one possible reason is that classical small molecules are not always ideal for disrupting protein–protein interactions.Citation17,18 Additionally, endogenous proteins or humanized monoclonal antibodies are difficult to produce and are high cost.
There are no IL-10 signaling inhibitors currently available for clinical use. Previously, a monoclonal antibody against IL-10 or an IL-10 receptor antibody was used to block IL-10 signaling in vivo using an animal model. Many attempts have been made to develop clinical grade anti-IL-10 anti-bodies, anti-IL-10 receptor antibodies, or IL-10 signaling inhibitors, using different techniques such as recombinant DNA technology, phage display and screening, and selection of short oligonucleotide aptamers.Citation3 However, these techniques are time consuming and labor intensive.
Structure-based biofunctional peptide design in drug and vaccine development supported by computational modeling is becoming increasingly successful.Citation19-23 A recent report used a structure-based approach involving rational defining of the protein interfaces of non-continuous and unstructured nature, to design peptides that bind IL-10R1 in vitro.Citation24 We proposed a way for designing IL-10 inhibiting peptides based on a helix segment of IL-10 within IL-10/IL-10R1 interaction region, and demonstrated that peptides with repeating pattern of hydrophobic and hydrophilic residues are able to inhibit IL-10 both in vitro and in vivo.Citation1 We have confirmed that the peptides are able to bind IL-10R, so as to inhibit the signaling of IL-10 with high efficiency supported by the following assaysCitation1:- i) Surface Plasmon Resonance (SPR) spectral analysis suggested the peptides displayed a level of binding affinity at sub micro-molar level with IL-10R, and in addition their binding competed with IL-10; ii) an MTT [3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] assay of mouse mast cell MC/9 cells indicated that the peptides counteracted the effect of IL-10 stimulation and limited cell proliferation, iii) the peptides inhibited IL-10 secretion, confirmed by ELISA using LPS-stimulated human PBMCs and macrophage U937 cell lines; iv) the peptides competed with anti-IL-10R antibodies for binding with IL-10R1 while showing negligible cytotoxicity; and v) one peptide enhanced vaccine induced antigen specific CD8+ T cell responses. These results indicated that the peptides are functional, with in vivo bio-activity, and potentially may block IL-10 signaling in human.
Though verified experimentally, the underlying inhibiting mechanism behind our design warrants further investigation, to help optimize the design method. In this study, using the sequences of 2 designed IL-10 inhibiting peptides,Citation1 i.e., P1 (FFKKFFKKFFKKFFKK-OH) and P2 (FFRRFFRRFFRRFFRR-OH), as models, a number of synthetic peptides with different sequence modifications (including sequence length, N-/C- terminal hydrophobicity, protonation capability, chirality and secondary structure variation) were introduced, and the key structural characteristics affecting IL-10 inhibition were clarified.
Results
Rational structure-based introducing sequence modifications
As shown in Fig. S1,Citation1 the design of P1 and P2 employed the hydrophobic and hydrophilic pattern of the helix segment of IL-10 (PDB 1J7V), which bends at residue Val33. Val33 is surrounded by repeated hydrophilic and hydrophobic residues. To strengthen the helical conformation, P1 or P2 don't have the breaking pattern observed at Val33. It has been noted that the biophysical properties of protein-binding peptides, such as main chain hydrogen bond forming ability with target protein, sequence length, secondary structures, side-chain/main-chain, salt bridge,Citation25-29 affect its binding activity directly. In this study, we thus synthesized sequence-modified peptides (P1.1 to 1.5, P2.1 to 2.3 listed in ) based on P1 and P2, to further clarify the sequential characteristics that could influence IL-10 inhibitory activity. In addition, to maintain the helical confirmation propensity, we followed the α-helical folding principle when carrying out the substitution, to ensure a regular turn every 3.6 amino acids formed by the peptide carbonyl O atom and amide proton between the ith and (i+4)th amino acid positions.Citation30 Briefly, the modifications to the peptides include:- i) C-terminal truncation (P1.1 and 1.2), ii) all D-form amino acid substitutions (P1.3) and iii) residue substitution to change the hydrophobicity, helix propensity and sequence pattern (P1.4, 1.5, 2.1, 2.2 and 2.3). The IL-10 inhibiting activity of these peptides was evaluated using IL-10 ELISA of LPS stimulated human U937 cells, in comparison with anti-IL-10 (aIL10) and anti-IL-10R1 (aIL10R) as positive controls.
Table 1. The sequences of P1, P2 and peptides modified based on P1 or P2.
Modulation of P1 and P2 secondary structures using 2,2,2-Trifluoroethanol
In a previous study,Citation1 it was shown that P1 and P2 compete with IL-10 in binding with IL-10R1 (Fig. S2). The conformations of these peptides, especially when they approach a near-membrane environment, could shed light on their interaction mechanism with IL-10R1. Therefore the membrane mimetic 2,2,2-trifluoroethanol (TFE) was used to modulate the secondary structures of P1 and P2.
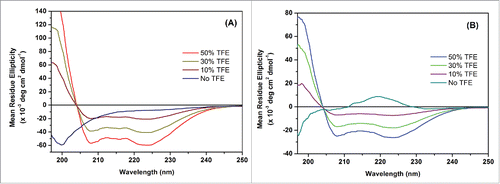
The CD spectra of the P1 and P2 in the presence of TFE at different concentrations are shown in . In buffered aqueous solution, P1 exhibits a far-UV CD spectrum characteristic of random coil structure, however, addition of TFE (0-50 % v/v) to native P1 showed that an α-helical structure can be induced, as evidenced by the characteristic negative ellipticity maxima at approximately 210 and 220 nm (). The far-UV CD spectra demonstrate that in 10 and 30% v/v TFE, there is a relative low degree of α-helical secondary structure induced, which increases upon addition to 50% v/v TFE. A similar scenario can be observed for P2 (). In addition, at the same concentration of TFE, P1 exhibited a higher helical propensity than P2 at a concentration of 4.5 μM.
Figure 1. Effect of different TFE concentrations on the conformation of P1 and P2. Far-UV CD spectra of P1 (A) and P2 (B). Representative data are shown from 3 independent experiments performed in triplicate.
P1 or P2 binds to IL-10R1 with different affinity: formation of a (P1/IL-10R1 or P2/IL-10R1) 1:1 complex
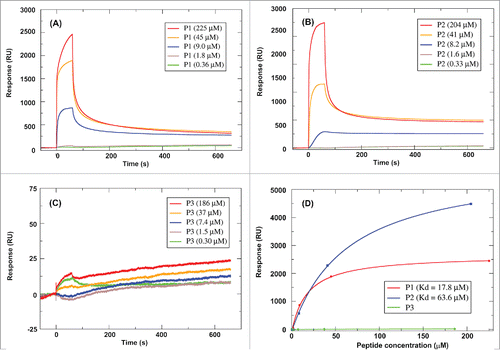
shows the binding affinity analysis of P1/P2 and P3 (control peptide) with IL-10R1 by SPR spectroscopy. The sensorgrams of the concentration of each peptide in relation to time are recorded in , from which it can be observed that P1 and P2 forms complexes with IL-10R1, while P3 cannot find a stable complex with IL-10R1 up to a concentration of 186 μM. The speed of complex formation of P1 is higher than that of P2 at a similar concentration. At relatively low concentration, P1 seemed more sensitive in binding IL-10R1. Overall, P1 and P2 displayed a level of binding affinity at 17.8 and 63.6 μM to IL-10R1, respectively, as shown in Fig. 2D. No binding to P3 was observed.
Figure 2. Interactions between P1/P2/P3 and IL-10R1 using Surface Plasmon Resonance Spectroscopy. Sensorgrams of the surface binding affinities of peptide P1 (A), P2 (B) and P3 (C) at different concentrations in relation with time, and (D) Binding affinity measurement of P1, P2 and P3 with IL-10R1.
In our previous studies, positive-ion MALDI mass spectrometry of a P1/IL-10R1 or P2/IL-10R1 mixture showed the presence of a small but reproducible peak corresponding to an [P1/IL-10R1]H+ or [P2/IL-10R1]H+ complex.Citation1 No multiply-charged adduct peaks, formed between P1/P2 and IL-10R1, were detected using MALDI mass spectrometry, and no corresponding peak was identified in the case of P3 (negative control). Thus, to further study the potential for the interplay of bioactive peptides P1 and P2 with IL-10R1, molecular dynamics (MD) simulation was used to model the complexes formed, and compared to the negative control peptide P3. After individually docking P1, P2 and P3 around the defined binding zone of IL-10/IL-10R1 (Fig. S1), the MD simulation confirmed that P1 can equilibrate to a stable 1:1 complex with IL-10R1 within about 80 ns after being heated to 325K, with its potential energy stabilized at about 90 kcal/mol (Fig. S3). In terms of the P2/IL-10R1 complex, it stabilized within a much shorter duration as the root-mean-squared-deviation (rmsd) was below 2Å during the entire equilibration after the heating process, and the potential energy showed a smaller variation of ∼50 kcal/mol less (Fig. S4). Though P3 was initially docked with a similar orientation as those for P1 and P2, it gradually moved away from IL-10R1 at a relatively rapid pace (it started leaving after 2,000 ps, and had a distance of ∼132.19 Å at 4,500 ps, see Fig. S5), suggesting that it is unlikely to bind to IL-10R1 under the experimental conditions (see Methods). This was in accordance with our previous report that P1 or P2 can prevent IL10 from binding to immobilized IL10-R1, and no competitive binding was observed for P3.Citation1
MD simulation confirms P1 or P2 binds to IL-10R1 with higher affinity than IL-10
The representative structures of 1:1 complexes P1/IL-10R1 and P2/IL-10R1 (shown in Fig. S3C and S4C) were selected to evaluate the interaction mechanism. For the representative structure of the P1/IL-10R1 complex, there were 9 H-bonds, as listed in ; the involvement of P1 in forming these H-bonds only included the Lys residues of the second and the third ‘FFKK’ units. In the case of the representative structure of P2/IL-10R1 complex (Fig. S4), the hydrophilic interaction was stronger, involving 15 H-bonds, contributed by nearly every Arg residue of the 4 ‘FFRR’ units (Table S1). Almost all these H-bonds involved the hydrogen of side chain amide of Arg residues of P2, except for one (Phe14: O-Arg95: HN). Basically all Phe residues were involved in the hydrophobic interaction with either IL-10R1 or the peptides themselves.
Table 2. Hydrogen bonds between P1 and IL-10R1 complex [side chain NH (HN), hydroxyl hydrogen (HO); backbone amide nitrogen (H), backbone carbonyl oxygen (O), carboxylate oxygen (OX) and hydroxyl oxygen (or amide-connected oxygen on side chain, OH).
It was of interest to compare the stability of complexes formed by IL-10/IL-10R1, P1/IL-10R1 and P2/IL-10R1 theoretically. Both MM/PBSA and MM/GBSA methods have been proven to provide relatively accurate binding free energies for protein/ligand interactions,Citation31-34 though the accuracy largely depends on a few parameters, including simulation length, solute dielectric constant and the number of snapshots.Citation35
shows the ΔG0D,solv of each complex. The different ΔG0D,solv values (both PBSA and GBSA methods) of the 3 complexes IL-10R1/IL-10, IL-10R1/P1 and IL-10R1/P2, suggested that the complex involving P1 or P2 is much more stable by 7.07 or 22.93 kcal/mol respectively, using PBSA method. This is consistent with the results obtained through the GBSA method (for details of calculation see Table S2-S4). These results meant that IL-10R1 would bind preferentially to P1 or P2 rather than IL-10, further supporting the SPR assay result that P1 and P2 compete with IL10 in binding to immobilized IL10-R1.Citation1
Table 3. The dissociation free energy (ΔG0D,solv, in kcal/mol) of IL-10R with IL-10, P1 or P2 (the full calculation results were listed in S6 Table).
In vitro inhibition of IL-10 activity by truncated and mutated P1 and P2 peptides: the key structural characteristics of inhibiting peptides
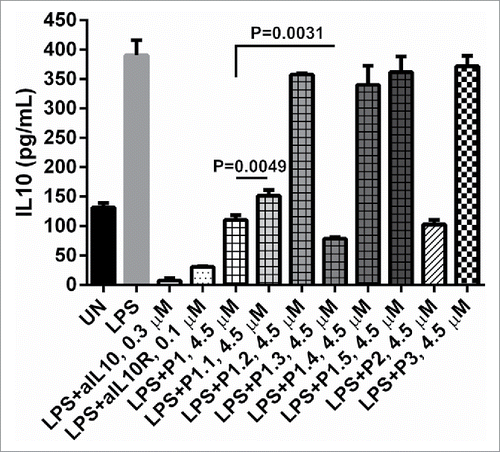
IL-10 is known to negatively regulate antigen presentation cells secreting IL-12; if IL-10 signaling is blocked, antigen presentation cells will produce more IL-12, which is critical to mount a T helper 1 response. Strong Th1 responses correlate with viral clearance and tumor rejection.Citation3 Using U937 cells as a model, displays the IL-10 ELISA results of the IL-10 inhibition activity of the P1 modified peptides. Compared to the inhibitory activity of P1, it can be clearly seen that, at same concentration, C-terminal truncation reduced the inhibiting magnitude. P1.1 (C-terminal Lys-Lys truncated) reduced inhibition by about 30% (P = 0.0049) and the inhibition was negligible for P1.2 (C-terminal Phe-Phe-Lys-Lys truncated). The D-form P1 (P1.3) was able to increase the inhibitory effect by approximately 40% (P = 0.0031). To unveil the influence of the sequence pattern, the ‘FFKK’ unit of P1 was substituted by ‘FFKA’ or ‘FRKK’ in P1.4 or P1.5, where Ala and Arg represent hydrophobic and hydrophilic substitutions, respectively. It was observed that neither P1.4 nor P1.5 could inhibit the IL-10 (). Similar truncations and D-form substitution of P2 showed consistent results with P1 (data not shown).
Figure 3. The effects of truncated and mutated P1 peptides on IL-10 level in U937 cells stimulated by LPS. Supernatants were measured for the presence of IL-10 by ELISA. The concentration of LPS is 4 × 10−3 µM: 3 × 105 human U937 were either left unstimulated (UN) or stimulated with LPS, LPS + 0.1 µM of anti-IL10R, LPS + 0.3 µM of aIL10, LPS + 0.1 µM of aIL10R, L+P1, P1.1, P1.2, P1.3, P1.4, P1.5, P2 and P3 at 4.50 µM overnight, respectively. P values were calculated using 2 tailed Student's t-test.
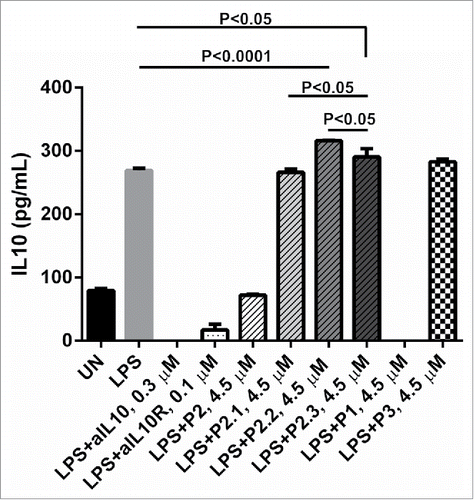
Previous results indicated only P2 (not P1) promoted LPS mediated IL-12 production by human PBMCs and enhanced CD8 T cell responses induced by an E7 peptide based vaccine for human papillomavirus 16.Citation1 Therefore, more sequence modifications were introduced to P2. The first consideration was N-terminal hydrophobicity, i.e., the sequence starting with PhePhe. To investigate this highly hydrophilic residue Glu was substituted for Phe in P2.1. Moreover, to evaluate the importance of helix and sequence flexibility, helix stabilized residues were introduced in P2.2 and P2.3. Their activities on the IL-10 secretion of LPS-stimulated U937 cells are displayed in , together with aIL10 and aIL10R as positive controls and P3 as the negative control. It can be observed that aIL10, aIL10R and P1 inhibited IL-10 secretion significantly; the inhibition magnitudes of aIL10 and P1 were close to 100%, respectively. They also seemed to eliminate any IL10 that should have been produced by untreated cells. The addition of P2 counteracted the stimulatory effects of LPS, while the hydrophilication of its N-terminus, by substituting the first Phe of every ‘FFRR’ unit by Glu in sequence P2.1, eliminated the inhibitiory activity completely. P2.2 and P2.3, in contrary, increased the secretion of IL-10 by U937 cells, by approximately 14% (P < 0.0001) and 7% (P < 0.05) compared to LPS stimulation, respectively.
Figure 4. The effects of mutated P2 peptides on IL-10 secretion in PBMCs stimulated by LPS. Supernatants were measured for the presence of IL-10 by ELISA. The concentration of LPS is 4 × 10−3 µM (100 ng): 1 × 106 human U937 were either left unstimulated (UN) or stimulated with LPS, LPS + 0.3 µM of anti-IL10, LPS+0.1 µM of anti-IL10 receptor (aIL10R), LPS + P1, P2, P2.1, P2.2, P2.3 and P3 at 4.50 µM overnight, respectively. P values were calculated using 2 tailed Student's t-test.
Discussion
Peptide based, clinical grade IL-10 signaling inhibitors
It has been shown that blocking IL-10 clears chronic viral infections in animal models. Furthermore, IL-10 signaling blockade combined with immunization improves the clearance of chronic viral infections, and controls tumor growth in vivo. Therefore, it is worth investigating whether this strategy will improve the efficacy of therapeutic vaccines against chronic viral infection and cancer, especially pathogen related cancers. However, there is no available clinical grade IL-10 signaling inhibitor. The many attempts to develop a human IL-10 signaling inhibitor have met with little success. Recently, we have demonstrated that, by using a structure-based method, IL-10 signaling inhibiting peptides, P1 and P2 have the ability to inhibit IL-10 signaling in vitro, and P2 can increase a vaccine induced T cell response in vivo. Therefore, P2 has the potential to be used in the clinic. Similarly, a report by Ruiz-Gómez et al. made the use of a regular expression syntax to define minimal descriptors of geometric and functional constraints in IL-10/IL-10R1, to help design IL-10R1 mimetics, which were shown to interfere IL-10/IL-10R1 interaction in vitro.Citation24 It is now possible to investigate whether blocking IL-10 at the time of immunization can better control chronic viral infection, and increase the efficacy of a therapeutic vaccine against cancer. However, understanding how P1 and P2 inhibit IL-10 signaling is a necessity.
The importance of α-helical structure, hydrogen bonding and length for inhibiting activity
TFE is capable of destabilizing hydrophobic interactions within polypeptide chains but is able to stabilize local hydrogen bonds between proximal residues in a polypeptide. Consequently, as a cosolvent it can stabilize the formation of any secondary structures.Citation36,37 From the data of CD spectra, it can be concluded that the degree of α-helical structure present in P1 and P2, plays a key role in the interactions with IL-10R1 and the effect of the flexibility of the helical structures are worthy of further study. Given that the protein adopts a predominantly α-helical structure in the presence of TFE, it is likely that the different near-membrane environment affects the IL-10 inhibiting activity of P1 and P2.
We have previously shown that the interaction between IL-10 and its receptor mainly involves 5 H-bonds and 3 hydrophobic interaction regions related to the peptide segment DLDRDAFSRVKTFFQM, based on MD simulation (Fig. S1c.f. Citation1). It can be seen that several Ser residues of IL-10R1 contribute significantly to these hydrophilic interactions, including Ser191 that forms an H-bond with IL-10. A previous study has found that LPS-induced production of IL-10 is promoted by the serine/threonine kinaseCitation38; in addition, it has been demonstrated that the short strong H-bond formed between enzyme and targets are important for enzymatic reactions.Citation38 The strong hydrogen bonding network can increase the enzyme-resistance of a protein.Citation39-41 In the cases of IL-10/IL-10R1 interaction, the participation of these Ser residues in forming stable H-bonds would reduce their propensity in enzymatic reactions catalyzed by the kinases, so as to decrease the secretion of IL-10 compared to LPS stimulation only. These results meant that a peptide constructed with amino acids that are relatively easier to be protonated, in combination with strong hydrophobic residues, would favor the interaction with IL-10R1. This was further confirmed by the inhibitory activity of P2.1; the Glu substitution neutralized the sequence (net charge 0), which reduced the propensity for H-bond formation between P2.1 and IL-10R1, as there are more electron negative oxygen atoms exposed within the binding-zone of IL-10R1.Citation1 Although residue Glu is able to form intra-helical salt bridges (Glu-…Arg+) to stabilize the helical conformation of P2.1,Citation42 hydrogen-bond forming capability plays a more significant role for the inhibiting activity.
Additionally, the length of the peptide also plays a key role in the inhibition activity, as evidenced in . It was also of interest to find that all D-amino acid substitution increased the inhibiting activity. There is a report that partial D-amino acid substitutions of membrane-active peptides can improve in vivo activity, which may be attributed to its higher stability while keeping the secondary structure.Citation43 The toxicity of this peptide ex vivo needs further clarification.
Balanced hydrophobic and hydrophilic residue repeating sequence pattern
The change of sequence pattern introduced in P1.4 and P1.5 eliminated inhibiting activity. Furthermore, the amino acid substitutions might also change the total hydrophobicity of these peptides, as Ala is more hydrophobic than Lys, while Arg is more hydrophilic than Phe. In terms of the structure of P1.4, the substitution of ‘Lys’ by ‘Ala’ would largely stabilize the helix conformation,Citation44,45 especially when placing ‘Ala’ near the C-terminus of designed helices,Citation46 thus reducing the flexibility of the sequence; meanwhile, it would decrease the net charges (from +8 to +4), leading to the reduction in its capacity in forming hydrogen bond. In contrast, the introduction of ‘Arg’ in replacing ‘Phe’ of P1.5 increases the net charge of the sequence to +12, which could reduce the stability of the peptide with certain secondary structure due to stronger repulsion, thus decrease its binding with IL-10R1. It has been found that Arg side chains may regulate the biological functions of membrane proteins, including sodium channels,Citation47 acetylcholine receptors,Citation48 and integrins,Citation49 via charge-charge interactions, which are implicated for regulating the voltage gating of cation channels.Citation50 Recently, it has been found that Arg directly associates with the opening and closing of voltage-gated proton channels, which play important roles in various physiological processes, including the innate immune response.Citation51 Thus, promoting the effect by having more Arg may be counteracted by simultaneously introducing structure instability.
The biological hydropathy scale devised predicts that Trp and Tyr would be the least energetically favored of the hydrophobics, and they are able to participate in both hydrophobic and polar interactions with proximal residues.Citation52,53 In P2.2 and P2.3, we used ‘WY’ and ‘FY’ to substitute ‘FF’ in the first and second ‘FFRR’ units, respectively, to make the N-terminus less hydrophobic (but still hydrophobic overall). Additionally, stabilizing the last helix was facilitated by 2 ‘AH’ substitutions of ‘FF’, as Ala is able to significantly stabilize helices in short peptidesCitation54-56 and His has been found to increase the stability of helices by H-bonding.Citation57,58 Moreover, in P2.3, we replaced ‘RR’ by ‘HH’ in the first 2 ‘XXRR’ (X represents an amino acid) units, which would also stabilize the first helix at physiological pHCitation57; additionally, it has been reported that solvent-exposed intra-helical Trp and His bridges could increase the helical conformation.Citation59 In contrast to P2, the ELISA results show that the secretion of IL-10 was increased in the presence of P2.2 or P2.3, These results suggest that the repeating sequence pattern with balanced hydrophobicity similar to ‘HHPP’ was crucial to the activity.
In summary, besides the primary considerations regarding the sequence modifications described in , there could be potential “side-effects” raised by these changes a, contributing to negligible activity overall, if too much effort was advocated to improving one biophysical property.
Conclusions
In this work, we have further clarified the key structural characteristics of the 2 peptides (P1 and P2) that were shown to inhibit IL-10 secretion in vitro and in vivo.Citation1 The IL-10 inhibiting peptides have the following features:- (i) a repeated hydrophobic and hydrophilic pattern with preferred helix-forming propensity; (ii) a length that is crucial to the biological function, whereby a minimum length of 14 amino acids is suggested; (iii) the chirality of amino acids were less relevant; and (iv) high N-terminal hydrophobicity appears to be vital. Finally, the likelihood of being protonated is another important factor for IL-10 inhibition yet the repulsion caused by being over-positively charged results in activity abolition. The results from this study combine insight from both experimental and theoretical bases into the development of an IL-10 inhibiting peptide, and the inhibiting characteristics of these peptides may provide clues for designing other peptides with optimal activity. We are currently working on the design of peptides with stronger IL-10 signaling inhibition ability. If successful, these peptides can be used as immune regulators to inhibit IL-10 bioactivity in vitro, and in vivo, to disrupt the tumor micro-environment, and to enhance vaccine induced T cell responses against chronic viral infection and cancers.
Materials and methods
Peptide synthesis and sequence confirmation by LCMS
The sequences of the peptides studied are listed in , together with descriptions of different modifications. All designed peptides were synthesized by GenicBio Biotech (Hongkong, China) with the purity >95% obtained by analytical reverse phase high performance liquid chromatography (RP-HPLC). Their purity was confirmed by RP-HPLC and LC-MS/MS on a Shimadzu Prominance Nano HPLC (Japan) coupled to a Triple-ToF 5600 mass spectrometer (ABSCIEX, Canada) equipped with a nano electrospray ion source. The protocol has been provided in detail previously.Citation60 Briefly, peptides were re-suspended in 0.1% formic acid to the concentration of 4.5 µM. Aliquots (6 µL) of each peptide solution were de-salted on a trap column (Agilent Technologies, Australia), placed in-line with the analytical nanoHPLC column (Agilent Technologies, Australia). Peptide elution used a linear gradient of 1-40% solvent [90:10 acetonitrile: 0.1% formic acid (aq)]. Peptide was sequenced based on the b and y-ions of the mass spectra.
Surface plasmon resonance (SPR) spectroscopy
The binding affinities of the peptides were determined at 25°C on a Biacore T100 SPR instrument, using an IL10R-immobilized CM5 sensor chip prepared with a GE Amine Coupling Kit. A channel treated following the same procedure but without IL10R immobilization was employed as a blank reference. The running buffer was 1X phosphate buffered saline, pH 7.4. Peptides diluted with the running buffer at various concentrations were injected at a flow rate of 10 μL/min for 1 min, followed by 5 min dissociation. Sensorgrams from each cycle was substracted by the corresponding blank run. Steady-state affinity analysis was performed using Biacore T100 Evaluation Software v2.0.3.
Circular dichroism (CD) spectroscopy
CD experiments were performed in a Chirascan CD spectrophotometer (Applied Photophysics, Leatherhead, UK). A quartz cuvette with a 10 mm path length was used for the recording of spectra over a wavelength range of 190-260 nm with a 1 nm bandwidth, 1 nm step size and time of 0.5 s per point. A buffer (5 mM Na2HPO4) baseline was collected in the same cuvette and was subtracted from the sample spectra. For the CD experiment of different peptides, different concentration (0, 30% and 50%, v/v) of 2,2,2-Trifluoroethanol was used as the mimics of near-membrane environment.
Antibodies
Anti-IL-10 (Cat. 506802), Anti-IL-10R antibodies (Cat. 308802) were purchased from BioLedgend.
Cell line
U937 cell line (CRL1593.2) was purchased from ATCC (USA), and their culture was followed the protocols in the product sheets.
Human IL10 ELISA
Human IL-10 ELISA MAX™ Deluxe 5 plates (Cat. 430604), were purchased from Biolegend, and performed following the manufacturer's instruction.
Molecular dynamics simulation
The structure of the complex between IL-10 and IL-10R1 was built starting from the crystal structure, PDB entry 1J7V. It was first subjected to a 10,000 steps of minimization in generalized Born (GB) solvent modelCitation61,62 (igb = 5) to reach a local minimum using of AMBER version 12-13.07,Citation63 and the energy of the complex and the binding zone were obtained and analyzed. From the minimal complex conformation, the structures of IL-10 and IL-10R were extracted and subjected to a further 10,000 steps minimization in GB solution, and then free energies were calculated based on these structures, respectively.
The linear conformations of the peptides were built using AMBER. The initial conformation of the IL-10R/peptide complex was built by docking the peptide above the binding zone in such a way, that its residues align approximately with those in the helice of IL-10 (-DLDRDAFSRVKTFFQM-), from which P1 and P2 were designed, and it was located about 5 Å from the corresponding binding segment of IL-10R. The LEAP module of AMBER was used in the model building process. The ff13 force field was employed, and the structure was initially optimized to a nearby minimum, which was used as the restarting point for subsequent heating simulation. Each complex structure was heated to 325K over 100 ps to avoid being kinetically trapped in local minima, and then subjected to simulation at 325K for the purpose of complex equilibration, with a weak restraint at a force constant of 1.0 kcal mol-1 angstrom-2 positioned on IL-10R. The simulations were continued until the root mean square deviation (RMSD) of structures in reference to the lowest energy structure within a reasonable time range is ≤∼2Å, then the lowest energy structure can be considered as the representative conformation for the complex simulated over this particular period. H-bonds between P1, P2 and IL-10R1 and RMSD were evaluated using PTRAJ and CPPTRAJ modules implemented in AMBER. Visualization of the systems was done using via VMD software version 1.9.1.Citation64,65
Then, both Molecular Mechanics/Poisson Boltzmann Surface Area (MM/PBSA) and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) methodsCitation66 were performed to evaluate the binding affinity of IL-10/IL-10R1, IL-10R1/P1 and IL-10R1/P2, and dissociation free energy in solution, ΔG0D,solv, was used here to indicate the stability of each complex. The MM-PB(GB)SA method was used to compare the free energy between 2 states, often representing the bound and unbound states of 2 solvated molecules or alternatively 2 different solvated conformations of the same molecule.Citation67-71
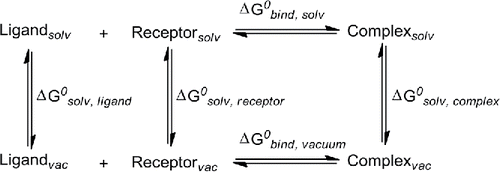
In terms of dissociation of peptide/protein complex, we introduced the following definition.where
and
are the dissociation free energy and binding free energy in water solution, respectively. The calculation of
is performed according to the following thermodynamic cycleCitation33:
The energy minimization of solvated complexes (in TIP5BOX model) were firstly carried out to a local minima within a short cycle, followed by a 500 ps of heating to 300 K and then 500 ps of density equilibration with weak restraints on the complexes. The equilibration was under constant pressure at 300K for 500 ps. A total of 50 ns of production simulation was performed for each complex, recording the coordinates every 10 ps. All simulations were run with shake on hydrogen atoms, a 2 fs time step and langevin dynamics for temperature control. The snapshots extracted were 400 for IL-10/IL-10R1 and 500 for P1/IL-10R1 and P2/IL-10R1, respectively.
For the simulation, the complexes obtained by MD simulation in GB solution model were initially dissolved in TIP5PBOX water model, subjected to 2500 steps minimization and followed by heating process to 325K during 50 ps. Then, they were equilibration at 325 K for more than 12000 ps. The snapshots extracted were 400 for IL-10/IL-10R1 and 500 for P1/IL-10R1 and P2/IL-10R1, respectively. Energy decomposition per residue and binding free energy post-processing analysis of the trajectories were performed in implicit solvent using the MM-PBSA method as implemented in AMBER.
Statistical analysis
Statistical analysis was performed by the 2 tailed Student's t-test by using Prism 5.0 (Graphpad Software, San Diego). Results are considered as significant if P value is less than 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Files
Download Zip (1 MB)Acknowledgments
We gratefully thank Dr Jian Zhan (The Institute for Glycomics and School of Information and Communication Technology, Griffith University) for advice and assistance with the SPR experiment, and Dr Shu Chen (Cancer Research Institute, Foshan First People's Hospital, China) for assistance with circular dichroism spectroscopy. This research was undertaken with the assistance of resources from the National Computational Infrastructure (NCI), which is supported by the Australian Government.
Funding
We thank the University of the Sunshine Coast and Sunshine Coast City Council and National Natural Science Foundation of China (81472451) for funding this research. We also thank Foshan municipal Government (2012AA100461; 2015AG1003) and Guangdong Provincial Government (2016A020213001) for funding support.
References
- Ni G, Chen S, Yang Y, Cummins SF, Zhan J, Li Z, Zhu B, Mounsey K, Walton S, Wei MQ, et al. Investigation the Possibility of Using Peptides with a Helical Repeating Pattern of Hydro-Phobic and Hydrophilic Residues to Inhibit IL-10. PLoS One 2016; 11:e0153939; PMID:27100390; http://dx.doi.org/10.1371/journal.pone.0153939
- Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001; 19:683-765; PMID:11244051; http://dx.doi.org/10.1146/annurev.immunol.19.1.683
- Ni G, Wang T, Walton S, Zhu B, Chen S, Wu X, Wang Y, Wei MQ, Liu X. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol 2015; 293:126-9; PMID:25596475; http://dx.doi.org/10.1016/j.cellimm.2014.12.012
- Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, von Herrath MG. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med 2006; 203:2461-72; PMID:17030951; http://dx.doi.org/10.1084/jem.20061462
- Brooks DG, Walsh KB, Elsaesser H, Oldstone MB. IL-10 directly suppresses CD4 but not CD8 T cell effector and memory responses following acute viral infection. Proc Natl Acad Sci 2010; 107:3018-23; PMID:20133700; http://dx.doi.org/10.1073/pnas.0914500107
- Maynard CL, Weaver CT. Diversity in the contribution of interleukin‐10 to T‐cell‐mediated immune regulation. Immunol Rev 2008; 226:219-33; PMID:19161427; http://dx.doi.org/10.1111/j.1600-065X.2008.00711.x
- Mocellin S, Marincola FM, Young HA. Interleukin-10 and the immune response against cancer: a counterpoint. J Leukocyte Biol 2005; 78:1043-51; PMID:16204623; http://dx.doi.org/10.1189/jlb.0705358
- Ejrnaes M, von Herrath MG. Cure of chronic viral infection by neutralizing antibody treatment. Autoimmun Rev 2007; 6:267-71; PMID:17412296; http://dx.doi.org/10.1016/j.autrev.2006.09.002
- Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 2006; 12:1301-9; PMID:17041596; http://dx.doi.org/10.1038/nm1492
- Brooks DG, Lee AM, Elsaesser H, McGavern DB, Oldstone MB. IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. J Exp Med 2008; 205:533-41; PMID:18332180; http://dx.doi.org/10.1084/jem.20071948
- Chen S, Wang X, Wu X, Wei MQ, Zhang B, Liu X, Wang Y. IL-10 signalling blockade at the time of immunization inhibits Human papillomavirus 16 E7 transformed TC-1 tumour cells growth in mice. Cell Immunol 2014; 290:145-51; PMID:24983823; http://dx.doi.org/10.1016/j.cellimm.2014.06.002
- Chard LS, Lemoine NR, Wang Y. New role of Interleukin-10 in enhancing the antitumor efficacy of oncolytic vaccinia virus for treatment of pancreatic cancer. Oncoimmunology 2015; 4:e1038689; PMID:26405610; http://dx.doi.org/10.1080/2162402X.2015.1038689
- Ha SJ, West EE, Araki K, Smith KA, Ahmed R. Manipulating both the inhibitory and stimulatory immune system towards the success of therapeutic vaccination against chronic viral infections. Immunol Rev 2008; 223:317-33; PMID:18613845; http://dx.doi.org/10.1111/j.1600-065X.2008.00638.x
- Tan JC, Braun S, Rong H, DiGiacomo R, Dolphin E, Baldwin S, Narula SK, Zavodny PJ, Chou CC. Characterization of recombinant extracellular domain of human interleukin-10 receptor. J Biol Chem 1995; 270:12906-11; PMID:7759550; http://dx.doi.org/10.1074/jbc.270.21.12906
- Josephson K, Logsdon NJ, Walter MR. Crystal structure of the IL-10/IL-10R1 complex reveals a shared receptor binding site. Immunity 2001; 15:35-46; PMID:11485736; http://dx.doi.org/10.1016/S1074-7613(01)00169-8
- Josephson K, McPherson DT, Walter MR. Purification, crystallization and preliminary X-ray diffraction of a complex between IL-10 and soluble IL-10R1. Acta Crystallogr D Biol Crystallogr 2001; 57:1908-11; PMID:11717514; http://dx.doi.org/10.1107/S0907444901016249
- Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 2004; 3:301-17; PMID:15060526; http://dx.doi.org/10.1038/nrd1343
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007; 450:1001-9; PMID:18075579; http://dx.doi.org/10.1038/nature06526
- Dhanda SK, Usmani SS, Agrawal P, Nagpal G, Gautam A, Raghava GP. Novel in silico tools for designing peptide-based subunit vaccines and immunotherapeutics. Brief Bioinform 2016; 1-12; http://dx.doi.org/10.1093/bib/bbw025; PMID:27016393
- Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angewandte Chemie (International ed in English) 2015; 54:8896-927; PMID:26119925; http://dx.doi.org/10.1002/anie.201412070
- Bhattacharjya S. De novo designed lipopolysaccharide binding peptides: structure based development of antiendotoxic and antimicrobial drugs. Curr Med Chem 2010; 17:3080-93; PMID:20629624; http://dx.doi.org/10.2174/092986710791959756
- Dings RP, Mayo KH. A journey in structure-based drug discovery: from designed peptides to protein surface topomimetics as antibiotic and antiangiogenic agents. Acc Chem Res 2007; 40:1057-65; PMID:17661438; http://dx.doi.org/10.1021/ar700086k
- Luque I, Freire E. Structure-based prediction of binding affinities and molecular design of peptide ligands. Method Enzymol 1998; 295:100-27; http://dx.doi.org/10.1016/S0076-6879(98)95037-6
- Ruiz-Gomez G, Hawkins JC, Philipp J, Kunze G, Wodtke R, Loser R, Fahmy K, Pisabarro MT. Rational structure-based rescaffolding approach to De Novo design of interleukin 10 (IL-10) receptor-1 mimetics. PloS one 2016; 11:e0154046; PMID:27123592; http://dx.doi.org/10.1371/journal.pone.0154046
- London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure (London, England : 1993) 2010; 18:188-99; PMID:20159464; http://dx.doi.org/10.1016/j.str.2009.11.012
- Springer S, Doring K, Skipper JC, Townsend AR, Cerundolo V. Fast association rates suggest a conformational change in the MHC class I molecule H-2Db upon peptide binding. Biochemistry 1998; 37:3001-12; PMID:9485452; http://dx.doi.org/10.1021/bi9717441
- Zarutskie JA, Sato AK, Rushe MM, Chan IC, Lomakin A, Benedek GB, Stern LJ. A conformational change in the human major histocompatibility complex protein HLA-DR1 induced by peptide binding. Biochemistry 1999; 38:5878-87; PMID:10231540; http://dx.doi.org/10.1021/bi983048m
- Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science 1995; 267:383-6; PMID:7529940; http://dx.doi.org/10.1126/science.7529940
- Sood VD, Baker D. Recapitulation and design of protein binding peptide structures and sequences. J Mol Biol 2006; 357:917-27; PMID:16473368; http://dx.doi.org/10.1016/j.jmb.2006.01.045
- Pauling L, Corey RB. Compound helical configurations of polypeptide chains: structure of proteins of the alpha-keratin type. Nature 1953; 171:59-61; PMID:13025480; http://dx.doi.org/10.1038/171059a0
- Liu F, Lu J, Hu W, Wang SY, Cui SJ, Chi M, Yan Q, Wang XR, Song HD, Xu XN, et al. New perspectives on host-parasite interplay by comparative transcriptomic and proteomic analyses of Schistosoma japonicum. Plos Pathogens 2006; 2:268-81; http://dx.doi.org/10.1371/journal.ppat.0020029
- Wang W, Donini O, Reyes CM, Kollman PA. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Biophy Biomol Struct 2001; 30:211-43; http://dx.doi.org/10.1146/annurev.biophys.30.1.211
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 2000; 33:889-97; PMID:11123888; http://dx.doi.org/10.1021/ar000033j
- Kuhn B, Gerber P, Schulz-Gasch T, Stahl M. Validation and use of the MM-PBSA approach for drug discovery. J Med Chem 2005; 48:4040-8; PMID:15943477; http://dx.doi.org/10.1021/jm049081q
- Rastelli G, Rio AD, Degliesposti G, Sgobba M. Fast and accurate predictions of binding free energies using MM‐PBSA and MM‐GBSA. J Comput Chem 2010; 31:797-810; PMID:19569205
- Hong DP, Hoshino M, Kuboi R, Goto Y. Clustering of fluorine-substituted alcohols as a factor responsible for their marked effects on proteins and peptides. J Am Chem Soc 1999; 121:8427-33; http://dx.doi.org/10.1021/ja990833t
- Buck M. Trifluoroethanol and colleagues: Cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys 1998; 31:297-355; PMID:10384688; http://dx.doi.org/10.1017/S003358359800345X
- Kim KS, Kim D, Lee JY, Tarakeshwar P, Oh KS. Catalytic mechanism of enzymes: preorganization, short strong hydrogen bond, and charge buffering. Biochemistry 2002; 41:5300-6; PMID:11955080; http://dx.doi.org/10.1021/bi0255118
- Nishiyama Y, Langan P, Chanzy H. Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 2002; 124:9074-82; PMID:12149011; http://dx.doi.org/10.1021/ja0257319
- Wang C-C, Tsong T-Y, Hsu Y-H, Marszalek PE. Inhibitor Binding Increases the Mechanical Stability of Staphylococcal Nuclease. Biophys J 2011; 100:1094-9; PMID:21320455; http://dx.doi.org/10.1016/j.bpj.2011.01.011
- Hassani L, Ranjbar B, Khajeh K, Naderi-Manesh H, Naderi-Manesh M, Sadeghi M. Horseradish peroxidase thermostabilization: the combinatorial effects of the surface modification and the polyols. Enzyme Microbial Technol 2006; 38:118-25; http://dx.doi.org/10.1016/j.enzmictec.2005.05.006
- Meier M, Lustig A, Aebi U, Burkhard P. Removing an interhelical salt bridge abolishes coiled-coil formation in a de novo designed peptide. J Struct Biol 2002; 137:65-72; PMID:12064934; http://dx.doi.org/10.1006/jsbi.2002.4467
- Hong SY, Oh JE, Lee K-H. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem Pharmacol 1999; 58:1775-80; PMID:10571252; http://dx.doi.org/10.1016/S0006-2952(99)00259-2
- Gregoret LM, Sauer RT. Tolerance of a protein helix to multiple alanine and valine substitutions. Fold Design 1998; 3:119-26; http://dx.doi.org/10.1016/S1359-0278(98)00017-0
- Rohl CA, Fiori W, Baldwin RL. Alanine is helix-stabilizing in both template-nucleated and standard peptide helices. Proc Natl Acad Sci U S A 1999; 96:3682-7; PMID:10097097; http://dx.doi.org/10.1073/pnas.96.7.3682
- Song K, Stewart JM, Fesinmeyer RM, Andersen NH, Simmerling C. Structural insights for designed alanine-rich helices: comparing NMR helicity measures and conformational ensembles from molecular dynamics simulation. Biopolymers 2008; 89:747-60; PMID:18428207; http://dx.doi.org/10.1002/bip.21004
- Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 2010; 67:915-28; PMID:20869590; http://dx.doi.org/10.1016/j.neuron.2010.08.021
- Cymes GD, Grosman C. Tunable pKa values and the basis of opposite charge selectivities in nicotinic-type receptors. Nature 2011; 474:526-30; PMID:21602825; http://dx.doi.org/10.1038/nature10015
- Kim C, Schmidt T, Cho EG, Ye F, Ulmer TS, Ginsberg MH. Basic amino-acid side chains regulate transmembrane integrin signalling. Nature 2012; 481:209-13; http://dx.doi.org/10.1038/nature10697
- DeCaen PG, Yarov-Yarovoy V, Scheuer T, Catterall WA. Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci U S A 2011; 108:18825-30; PMID:22042870; http://dx.doi.org/10.1073/pnas.1116449108
- Chamberlin A, Qiu F, Rebolledo S, Wang Y, Noskov SY, Larsson HP. Hydrophobic plug functions as a gate in voltage-gated proton channels. Proc Natl Acad Sci U S A 2014; 111:E273-82; PMID:24379371; http://dx.doi.org/10.1073/pnas.1318018111
- Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfeld R. C-H…pi-interactions in proteins. J Mol Biol 2001; 307:357-77; PMID:11243825; http://dx.doi.org/10.1006/jmbi.2000.4473
- Chakrabarti P, Chakrabarti S. C–H…O hydrogen bond involving proline residues in alpha-helices. J Mol Biol 1998; 284:867-73; PMID:9837710; http://dx.doi.org/10.1006/jmbi.1998.2199
- Marqusee S, Robbins VH, Baldwin RL. Unusually stable helix formation in short alanine-based peptides. Proc Natl Acad Sci U S A 1989; 86:5286-90; PMID:2748584; http://dx.doi.org/10.1073/pnas.86.14.5286
- Lopez MM, Chin DH, Baldwin RL, Makhatadze GI. The enthalpy of the alanine peptide helix measured by isothermal titration calorimetry using metal-binding to induce helix formation. Proc Natl Acad Sci U S A 2002; 99:1298-302; PMID:11818561; http://dx.doi.org/10.1073/pnas.032665199
- Padmanabhan S, Marqusee S, Ridgeway T, Laue TM, Baldwin RL. Relative helix-forming tendencies of nonpolar amino acids. Nature 1990; 344:268-70; PMID:2314462; http://dx.doi.org/10.1038/344268a0
- Armstrong KM, Baldwin RL. Charged histidine affects alpha-helix stability at all positions in the helix by interacting with the backbone charges. Proc Natl Acad Sci U S A 1993; 90:11337-40; PMID:8248249; http://dx.doi.org/10.1073/pnas.90.23.11337
- Armstrong KM, Baldwin RL. Charged histidine affects alpha-helix stability at all positions in the helix by interacting with the backbone charges. Proc Natl Acad Sci U S A 1993; 90:11337-40; PMID:8248249; http://dx.doi.org/10.1073/pnas.90.23.11337
- Fernandez-Recio J, Vazquez A, Civera C, Sevilla P, Sancho J. The tryptophan/histidine interaction in alpha-helices. J Mol Biol 1997; 267:184-97; PMID:9096217; http://dx.doi.org/10.1006/jmbi.1996.0831
- Adamson KJ, Wang T, Rotgans BA, Kuballa AV, Storey KB, Cummins SF. Differential peptide expression in the central nervous system of the land snail Theba pisana, between active and aestivated. Peptides 2015; 80:61-71; PMID:26303007
- Xia B, Tsui V, Case DA, Dyson HJ, Wright PE. Comparison of protein solution structures refined by molecular dynamics simulation in vacuum, with a generalized Born model, and with explicit water. J Biomol NMR 2002; 22:317-31; PMID:12018480; http://dx.doi.org/10.1023/A:1014929925008
- Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2000; 56:275-91; PMID:11754341; http://dx.doi.org/10.1002/1097-0282(2000)56:4%3c275::AID-BIP10024%3e3.0.CO;2-E
- Case DA, Darden TD, Cheatham, TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M.-J., Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. (2012), AMBER 12, University of California, San Francisco. AMBER 12, University of California, San Francisco, 2012.
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Grap Model 1996; 14:33-8; http://dx.doi.org/10.1016/0263-7855(96)00018-5
- http://www.ks.uiuc.edu/Research/vmd/. http://www.ks.uiuc.edu/Research/vmd/.
- Rastelli G, Del Rio A, Degliesposti G, Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 2010; 31:797-810; PMID:19569205
- Jiang GJ, Wang K, Miao DQ, Guo L, Hou Y, Schatten H, Sun QY. Protein profile changes during porcine oocyte aging and effects of caffeine on protein expression patterns. PloS one 2011; 6:e28996; PMID:22194971; http://dx.doi.org/10.1371/journal.pone.0028996
- Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Dis Design 2000; 18:113-35; http://dx.doi.org/10.1023/A:1008763014207
- Rastelli G, Del Rio A, Degliesposti G, Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 2010; 31:797-810; PMID:19569205
- Miller BR, 3rd, McGee TD, Jr., Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An efficient program for end-state free energy calculations. J Chem Theor Comput 2012; 8:3314-21; http://dx.doi.org/10.1021/ct300418h
- Wang J, Morin P, Wang W, Kollman PA. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J Am Chem Soc 2001; 123:5221-30; PMID:11457384; http://dx.doi.org/10.1021/ja003834q