
ABSTRACT
We conducted a randomized, double-blind, placebo-controlled dose-escalation study in healthy adults to evaluate the safety and immunogenicity of recombinant Staphylococcus aureus candidate vaccine antigens, recombinant α-toxoid (rAT) and a sub-unit of Panton-Valentine leukocidin (rLukS-PV). 176 subjects were enrolled and randomized within 1 of 11 treatment cohorts: monovalent rAT or rLukS-PV dosages of 10, 25, 50, and 100 μg; bivalent rAT:rLukS dosages of 10:10, 25:25, and 50:50 μg; and alum or saline placebo. All subjects were assessed at Days 0, 7, 14, 28, and 84. Subjects in the 50:50 μg bivalent cohort received a second injection on Day 84 and were assessed on Days 98 and 112. Incidence and severity of reactogenicity and adverse events (AEs) were compared. Geometric mean serum concentrations (GMC) and neutralizing activity of anti-rAT and anti-rLukS-PV IgG were assessed. Reactogenicity incidence was significantly higher in vaccine than placebo recipients (77% versus 55%, respectively; p = 0.006). However, 77% of reactogenicity events were mild and 19% were moderate in severity. The AE incidence and severity were similar between the cohorts. All monovalent and bivalent rAT dosages resulted in a significant increase in the anti-rAT IgG and anti- rLukS-PV GMCs between day 0 and 28 compared with placebo, and persisted through Day 84. Exploratory subgroup analyses suggested a higher GMC and neutralizing antibody titers for the 50 μg monovalent or bivalent rAT and rLukS-PV dose as compared to the other doses. No booster effect was observed after administration of the second dose. We conclude that the rAT and rLukS-PV vaccine formulations were well-tolerated and had a favorable immunogenicity profile, producing antibody with neutralizing activity through day 84. There was no benefit observed with a booster dose of the vaccine.
Introduction
Staphylococcus aureus is a highly versatile pathogen that causes a wide range of nosocomial and community-acquired infections. Outbreaks of community acquired skin and soft-tissue infections (SSTIs) due to methicillin-resistant S. aureus have been observed in prisoners, athletes, military personnel, and other risk groups.Citation1-5 S. aureus SSTIs have become a significant public health issue for the US military over the last decade, affecting service members during training and overseas deployment.Citation4,6-8 Cumulative SSTI rates during training range between 4-6%,Citation4,9,10 and SSTIs were estimated to be the cause of 41,951 ambulatory visits and 1,054 hospital admissions for active duty military members in 2014.Citation11,12 An evaluation of Department of Defense (DoD) beneficiaries (2005-2010) reported overall adjusted incidence rates (per 100,000 person-years) of 4.3 for S. aureus bacteremia and 144.5 for S. aureus SSTIs. Among active-duty personnel, CA-MRSA SSTI incidence rate was 280.6, as compared to 165.8 for community-associated methicillin-susceptible S. aureus SSTIs.Citation8
With the growing burden of S. aureus disease, increasing antibiotic resistance, and limited efficacy of decolonization protocols,Citation9,10 there is substantial interest in development of a S. aureus vaccine for use in high risk populations. Although an effective S. aureus vaccine remains elusive,Citation13-18 there is general consensus on a multi-antigen approach to vaccine development, which targets T and B cell responses to S. aureus cell surface components, virulence factors and toxins.Citation19 α-toxin and Panton-Valentine leukocidin (PVL) are 2 toxins of interest for a toxoid based approach that have shown efficacy in animal models.Citation20-22 α- toxin is a highly conserved toxin that causes tissue barrier disruption at host interfaces lined by epithelial or endothelial cells, and undermines the host immune response.Citation23 Reduced skin lesion size and dermonecrosis were observed in mice immunized with a nontoxigenic form of α-toxin, following infection with USA300.Citation24 In observational studies, higher levels of IgG antibody to α-toxin have been associated with reduced risk of sepsis in adult patients with invasive staphylococcal infections.Citation25 In addition, long-term follow-up of children with invasive S. aureus disease reveals a protective association between anti-α-toxin antibody titers and recurrent infection.Citation26
PVL is a pore-forming cytotoxin consisting of 2 subunits, LukS-PV and LukF-PV, which cause leukocyte destruction and tissue necrosis.Citation27 Although produced by <5% of S. aureus strains, a large proportion of methicillin-resistant (MRSA) strains that cause invasive community-acquired infections such as necrotic skin and soft-tissue infections and necrotizing pneumonia are positive for the PVL gene.Citation28 Studies evaluating the role of PVL as a virulence factor reveal a complex interaction between the virulence factor and the host immune response. In vitro data suggest that in the early stages of infection or with a lower pathogen inoculum, sub-cytolytic concentrations of PVL activate the innate immune system, thereby exerting a protective effect against infection.Citation29-31 Anti-PVL antibodies enhance the virulence of PVL producing strains by neutralizing this pro-inflammatory effect at sub-cytolytic concentrations, thus limiting their role in prevention of disease.Citation32 In observational studies, individuals infected with PVL-positive S. aureus strains have higher levels of anti-PVL antibodies compared to patients with PVL- negative S. aureus infections or no S. aureus infection,Citation33 but a protective role of this immune response in humans has not been established. In contrast, some murine models suggest that anti-PVL antibodies may play a role in reducing the severity or protecting against infection. Subcutaneous injection with either rLukS-PV or rLukF-PV significantly improved survival following MRSA USA 300 pulmonary infection, and reduced morbidity following USA 300 dermal infection.Citation34 Protection against USA300 sepsis was demonstrated in a murine model using a vaccine composed of highly attenuated forms of PVL sub-units (rLukS-PV or rLukF-PV).Citation21
Results
A total of 369 subjects were enrolled, of which 176 were randomized and injected with investigational vaccine or placebo (). The demographic characteristics of screen failures were not dissimilar from those who passed. The most common reason for failing screening was the presence of an abnormal baseline laboratory test.
Figure 1. Flow diagram of subject enrollment and completion. Subjects were ineligible to receive second injection if they experienced a grade 3 adverse event or reactogenicity event after the first injection (all events determined probably unrelated to the first injection). For subjects in the 50 µg bivalent cohort (active vaccine, saline placebo, or alum placebo recipients) who received a second injection, the follow-up phone call and study completion occurred on Day 252.
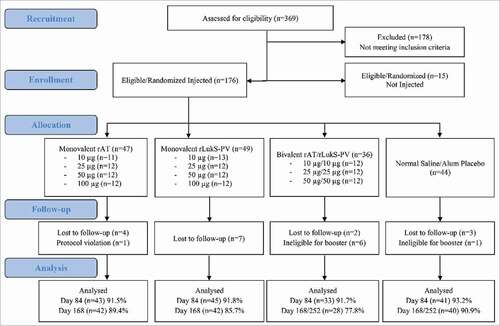
Baseline characteristics for the randomized and injected subjects were similar among those receiving vaccine or placebo (Table S2). No subject had a history of S. aureus infection or SSTI within 3 months prior to baseline. There were 162 (92%) subjects who completed all visits through Day 84, and 152 (86%) completed the follow-up phone call. Of the 24 subjects not completing all study visits, 16 were lost to follow-up due to military deployment or change of duty location, one was removed from immunogenicity analyses but followed for safety due to receiving a steroid injection after study injection (for a non-vaccine related condition), and 7 subjects (6 active vaccine and 1 placebo) in the 50 μg bivalent cohort were unable to receive the second study injection due to experiencing an unrelated grade 3 event after the first injection.
Reactogenicity and adverse events
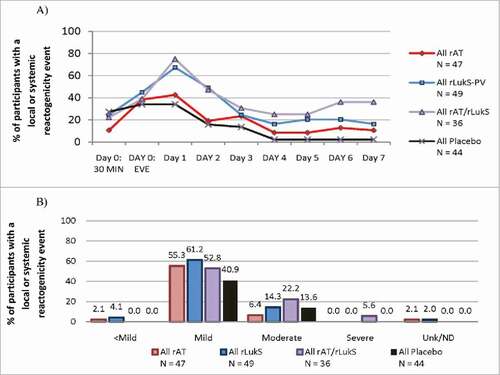
After the first vaccination, 101 (77%) vaccine recipients experienced ≥1 reactogenicity event compared with 24 (55%) placebo recipients (p = 0.006) (see supplementary material for definition). Specifically, the number of vaccine subjects experiencing at least one local or systemic reactogenicity event following the first injection was 31 (66%) for monovalent rAT, 41 (84%) for monovalent rLukS-PV, and 29 (81%) for bivalent recipients. All local reactogenicity events were considered due to vaccination, whereas the incidence of treatment-related systemic reactogenicity was 27% for placebo, 17% for monovalent rAT, 31% for monovalent rLukS-PV, and 42% for bivalent rAT/rLukS-PV groups. Reactogenicity incidence peaked on Day 1, and was higher for subjects in the monovalent rLukS-PV and bivalent arms, as compared to the monovalent rAT arm (). The most common local and systemic reactogenicity events for either antigen were tenderness and headache (Tables S3 and S4). No relationship was observed between reactogenicity severity and antigen dosage or preparation (mono- or bivalent) (). Two reactogenicity events (myalgias and fatigue/general discomfort) occurred after administration of 50 μg rAT/rLukS-PV and were assessed as severe. Following the second injection, local and systemic reactogenicity events occurred in 5/6 vaccine and 2/3 placebo recipients.
Figure 2. Local or systemic reactogenicity (A) incidence and (B) severity during the first week following first vaccination in all treated subjects (n = 176).
There was no significant difference in the incidence of overall or treatment-related AE's among the 4 main treatment groups (). Seventy percent of AEs were mild, and the incidence and severity were similar for vaccine and placebo recipients. The most common type of AE was development of an abnormal asymptomatic laboratory test. Four serious adverse events (SAEs) assessed to be likely not related to vaccination occurred (i.e., malignant astrocytoma, non-cardiac chest pain, pneumonia, and alcoholism relapse). In addition, one SAE occurred in a 50 μg bivalent cohort subject and was judged to have an unknown relationship to vaccination. The subject had a history of antinuclear antibody positivity in 2002 and developed thrombocytopenia after injection. Detailed evaluation, including bone marrow biopsy, was consistent with idiopathic thrombocytopenic purpura. The platelet count returned to the normal range without specific treatment and the event was considered resolved by 4 months after injection. Among subjects receiving the second injection, 3/6 vaccine and 3/3 placebo recipients experienced an AE.
Table 1. Summary of adverse events following first vaccination during follow-up in the safety population, No. (%). There was no significant difference in the incidence of overall AEs or AE's assessed to be treatment-related among the four main treatment groups, except subjects with ≥ 1 severe AE (p<0.01). p-values were calculated using a Wald test statistic for group in a generalized linear model for count outcomes
Immunogenicity
A high proportion of subjects were seropositive for neutralizing antibody against rAT (70.8%) and rLukS-PV (75%) prior to vaccination. All monovalent and bivalent rAT dosages resulted in a significant increase in the anti-rAT IgG GMCs between day 0 and 28 compared with placebo, and persisted through Day 84 (). The 50 µg monovalent rAT dose provided the highest anti-rAT IgG GMC and corresponding increase in neutralizing activity (; Tables S5, S7). Anti-rAT GMCs and neutralizing activity was significantly lower in the 50 µg rAT/rLukS-PV cohort compared to the 50 µg monovalent rAT cohort, although this effect was not observed at the 25 µg rAT dose. Two-fold increases in serum neutralizing antibodies from baseline were noted in 10 of 12 (83%) of subjects in both the rAT 50 µg monovalent and the 50 µg rAT/rLukS-PV cohort at day 84. No booster effect was observed following the second dose ().
Figure 3. Antibody responses for (A) monovalent rAT; (B) bivalent rAT; (C) monovalent rLukS-PV; and (D) bivalent rLukS-PV formulations following first and second vaccination in the completer population. Visits are based on pre-defined windows for serology sample collection and not on the nominal visit. All out-of-window sample collections are excluded). After Day 84, the number of subjects who received the booster dose in the bivalent 50 µg cohorts was six.
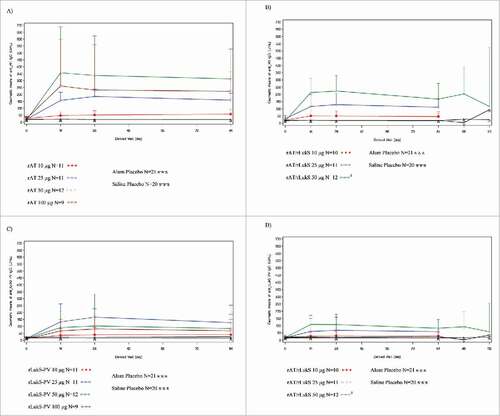
All monovalent and bivalent rLukS-PV dosages resulted in increased GMC of anti-rLukS-PV IgG and GMT of neutralizing antibody through Day 84 (). Following initial vaccination, the 25 µg monovalent and 50 μg bivalent formulations provided the highest anti-rLukS-PV IgG GMC (; Table S6). Overall, geometric mean fold increase (GMFI) values for rLukS-PV neutralizing antibodies were lower than GMFIs for rAT neutralizing antibodies (Table S7). The highest GMFI's were noted in the 50 μg bivalent cohort at Day 28 (5.87 [95% CI: 3.02-11.41]) and 84 (5.81 [95% CI: 3.11-10.85]). A 2-fold increase in serum neutralizing antibodies from baseline was noted in 10 of 12 (83%) of subjects in both the rLukS-PV 50 µg monovalent and the 50 µg bivalent cohort at Day 84. No booster effect was observed following the second dose ().
Correlation of IgG antibody concentration with neutralizing activity
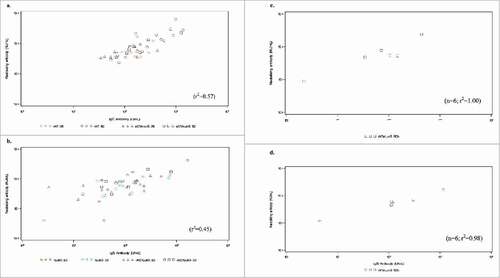
Prior to vaccination, there was poor linear correlation between rAT IgG levels and neutralization of α-toxin induced hemolysis (r2 = 0.37) (Fig. S1). Similarly, rLukS-PV IgG concentration and inhibition of PVL-induced lysis were poorly correlated (r2 = 0.52) (Fig. S2). The linear correlation improved for rAT antibodies following vaccination, (Day 28 r2 = 0.73, Day 84 r2 = 0.57 and Day 112 r2 = 1.00) indicating that the vaccine had induced significant levels of high quality and functionality anti-rAT antibody compared to naturally occurring antibodies (). A weaker correlation was observed between rLukS-PV IgG concentration and inhibition of PVL-induced lysis following vaccination at day 28 (r2 = 0.50), and day 84 (r2 = 0.45) and increased at day 112 (r2 = 0.98).
Figure 4. a) Geometric mean titer of rAT neutralizing antibody b) Geometric mean titer of rLukS-PV neutralizing antibody following vaccination. Volunteers were vaccinated with 25 µg rLukS-PV, 50 µg rLukS-PV, 25µg rAT/rLukS-PV or 50 µg rAT/rLukS-PV. The 50 µg rAT/rLukS-PV cohort received a booster dose on day 84, and evaluation of neutralizing antibodies at day 112.
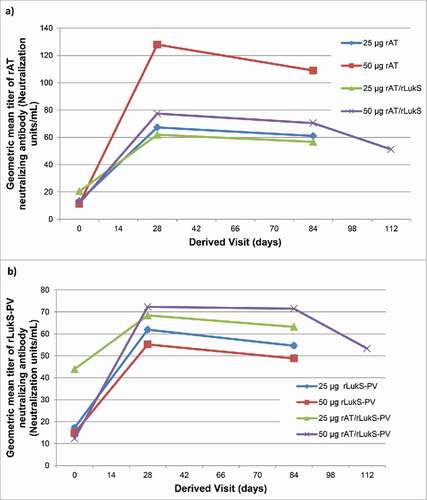
Figure 5. Scatterplot of total IgG levels and neutralizing activity at Day 84 post-vaccination against rAT for the following doses: 25 µg rAT, 50 µg rAT, 25 µg rAT/rLukS-PV or 50 µg rAT/rLukS-PV cohorts (5a), rLukS-PV for the 25 µg rLukS-PV, 50 rLukS-PV, 25 µg rAT/rLukS-PV or 50 µg rAT/rLukS-PV cohorts (5b). Scatterplot of total IgG levels and neutralizing activity against rAT (5c) and rLukS-PV (5d) at Day 112 following receipt of a booster dose of vaccine in patients who received the 50 µg rAT/rLukS-PV dose at baseline.
Discussion
In this vaccine trial, both rAT and rLukS-PV exhibited a favorable safety profile when administered either alone or in combination to young, healthy adults. The antigens were well tolerated, reactogenicity events were mild and resolved within 48-72 hours post-vaccination, and similar treatment-related adverse events were observed between active vaccine and placebo groups. Only the 100 μg rLukS-PV and 50 μg rAT/rLukS-PV formulations had a significantly higher incidence of reactogenicity events when compared with placebo, and no local reactogenicity events were characterized as worse than moderate severity. Furthermore, of the total 105 systemic reactogenicity events, only 2 were assessed as severe. Five SAEs were reported during the trial, 4 of which were assessed to be unrelated to vaccination, and one was judged to have an unknown relationship to vaccination and resolved by the study end. The safety profile is comparable to that reported in other S. aureus toxoid vaccine trials. SA3Ag, comprised of capsular polysaccharides (CPS) 5 and 8 and recombinant clumping factor A (rClfA), had an acceptable safety profile with an rClfA dose of upto 200µg.Citation35 A phase I trial of a 4 component staphylococcal vaccine, utilizing a lower dose of mutant α-toxoid and ClfA (i.e. 10 or 30 µg) in combination with CPS5 and 8 conjugated to tetanus toxoid), also showed a clinically acceptable safety profile with a low incidence of grade 3 reactogenicity events and no SAEs related to vaccine administration.Citation18
Although a large proportion (>70%) of subjects were seropositive for α-toxin and PVL-neutralizing antibodies at baseline, the titer of pre-existing antibodies was low. These antibodies were likely in response to carriage of S. aureus strains (very few patients had a history of SSTI in our cohort) expressing PVL, α-toxin or other cross reactive leukocidins. When given separately or in combination, both rAT and rLukS-PV antigens elicited significant increases in anti-rAT and anti-rLukS-PV GMCs and neutralizing antibody titers following injection, and persisted through day 84. In addition, the linear correlation between IgG antibody concentration and neutralizing activity improved following vaccination, indicating the high functionality of toxin-specific antibodies produced by vaccination. The 50 µg bivalent formulation was associated with good antibody seroresponse rates to α-toxin and PVL at day 28 and 84, and would therefore be a reasonable choice as part of a multicomponent vaccine. There was no booster effect on GMCs or neutralizing titers for either antigen following the second dose, and although the linear correlation between anti-rAT/anti-rLukS-PV IgG concentration and neutralizing activity improved following the booster dose, the small sample size of subjects (n = 6) and lack of seroresponse limits our ability to draw conclusions regarding the benefit of booster doses of these antigens. This booster-like response to the initial dose and lack of effect of additional doses has been observed in other S. aureus vaccine trials. A robust increase in the GMT of α-toxin neutralizing antibodies was observed following the initial dose of the 4 component staphylococcal vaccine (mutant α-toxoid, ClfA, CPS5, CPS8), followed by a lack of response to dose 2 and 3.Citation18 The peak antibody levels observed after the second dose of StapVAX™, a CPS5/CPS8 conjugate vaccine, were lower than the levels achieved following the initial vaccination.Citation36 These findings were attributed to the fact that adult humans were previously primed for these antigens as evident by presence of specific pre-existing antibodies, and therefore one immunization is considered a booster immunization.
Several studies have shown the importance of α-toxin in modulating the adaptive immune response by stimulating secretion of IL-17A, leading to induction of Th17 cells that both respond to and express IL-17A and contribute toward protection against infection.Citation37-39 We did not evaluate the α-toxin specific T-cell responses, which is a limitation of this study, and instead focused on evaluating the functionality of the anti-rAT antibodies generated by vaccination. Good seroresponse rates to the rAT vaccine were observed, with higher neutralizing antibody titers noted at the 50 µg rAT dose as compared to the 25 rAT µg dose in both monovalent and bivalent doses. Interestingly, while the 50 µg monovalent rAT dose provided the highest anti-rAT IgG GMC and corresponding increase in neutralizing activity, the anti-rAT GMCs and neutralizing activity in the 50 µg rAT/rLukS-PV cohort was approximately 50% lower than the monovalent cohort. This was an unexpected finding, and suggests that antibody response to rAT was impaired with the addition rLukS-PV, although a similar effect was not observed between the 25 µg rAT monovalent and bivalent doses. Results from the 4 component staphylococcal vaccine that utilized mutant α-toxoid in combination with ClfA, also showed a robust antibody response to both antigens, although the investigators did not incorporate a monovalent α-toxoid arm in the study design.Citation18
We observed a higher incidence of reactogenicity events in subjects receiving the rLukS-PV antigen (alone or in the bivalent formulation) compared to the subjects receiving rAT alone. Neutralizing antibody titers for rLukS-PV were lower than the titers observed for rAT antibodies, and a weaker correlation was observed between rLukS-PV IgG concentration and inhibition of PVL-induced lysis. Our findings suggest that rAT maybe a more immunogenic vaccine candidate than LukS-PV, although cautious interpretation is required given the poor correlation between serologic biomarkers and protection against infection. The role of PVL as a virulence factorCitation40-42 and the effectiveness of neutralizing antibodies in disease prevention are not fully understood.Citation30,31 PVL is associated with a pro-inflammatory effect in mouse models that is independent of its pore-forming properties.Citation43 However, the species-specific interaction between toxins and complement receptors have called into question the applicability of mouse models for evaluating potential virulence factors as vaccine targets.Citation44,45 Despite these limitations, PVL could play a role as a target for active and passive immunization, due to the structural homology of subunits among members of the bi-component cytotoxin family.Citation46 This structural homology allows for cross-neutralization of several virulence factors with antibodies generated against a single virulence factor. For example, LukS shares approximately 80% amino acid identity with leukocidin component LukE and gamma hemolysin component HlgA, while LukF is highly related to LukD and HlgB. In addition, while only 5-10% of S. aureus strains carry the LukSF gene, almost all strains express the HlgAB cytotoxin and approximately 50-75% also carry LukED, resulting in improved potency of antibodies due to cross-neutralization of multiple leukocidins that play an integral role in staphylococcal pathogenesis.Citation47 Rabbit models of S. aureus infection maybe helpful in evaluating the effectiveness of anti-PVL antibodies generated in such Phase I immunogenicity studies for passive immunization.
In summary, the rAT and/or rLukS-PV based vaccine formulations tested in our study were well-tolerated and induced a robust antibody response with functional antibodies following the first dose of vaccine. Exploratory analysis suggests that the 50 µg bivalent formulation was associated with a good neutralizing antibody seroresponse rate against α-toxin and PVL. Future research is necessary to answer whether a multicomponent vaccine utilizing in addition to rAT and/or rLukS-PV other protective antigens such as capsular polysaccharide conjugates can prevent S. aureus disease.
Patients and methods
Study design
This randomized, placebo controlled dose-escalation vaccine trial was conducted at the San Antonio Military Medical Center (San Antonio, TX, US) and Naval Medical Center (Portsmouth, VA, US) between 2009 and 2010. Eligible subjects met the following inclusion criteria: healthy adults aged 18 to 55 years; DoD beneficiary (including active duty members); a negative pregnancy test prior to injection for women of childbearing age; and willingness to provide written informed consent. Exclusion criteria are provided in the Supplementary File. This study was approved by the Uniformed Services University Infectious Disease Institutional Review Board, and all subjects provided written informed consent.
Clinical supplies of vaccine were provided by Nabi Biopharmaceuticals in single-dose vials and stored at 2-8°C. Two placebos were utilized: sterile 0.9% normal saline (provided by each site) and aluminum hydroxide (alum) in phosphate buffered saline (provided by Nabi Biopharmaceuticals in single-dose vials - 1 mL of phosphate buffered saline [0.002 M sodium phosphate, 0.15 M sodium chloride, 0.01% polysorbate 80], and 800 µg/mL aluminum hydroxide adjuvant). All vaccine doses containing rAT or rLukS-PV contained an alum adjuvant. Each study injection was prepared by an independent, unblinded pharmacist. Masked syringes were provided to members of the study team for injection.
Subjects were enrolled successively into one of eleven cohorts. Within each cohort, 16 subjects were randomized (using a web-based randomization system) at a 6:1:1 ratio to receive one dose of monovalent or bivalent toxoid vaccine, alum-placebo, or normal saline placebo, respectively (Supplementary Material and Table S1). The dose levels for the 11 cohorts were monovalent rAT or rLukS-PV at 10, 25, 50 or 100 μg and pre-formulated bivalent rAT:rLukS-PV in a 1:1 ratio at 10, 25 or 50 μg of each antigen. Initial cohorts of enrolled subjects received the lowest dosages of monovalent antigens, with escalation to the respective next dose after review of the 7-day safety data for each cohort by study investigators, independent medical monitors, and the chair of the data safety and monitoring board. Because of the dose-escalation study design, study personnel and subjects were double-blinded to active vaccine vs. placebo within each treatment cohort, but not to volume of vaccine/placebo injected. To evaluate the utility of booster doses, subjects enrolled in the 50 μg bivalent cohort received a second dose of vaccine or placebo on Day 84. Subjects experiencing any grade 3 or higher event following initial dose of vaccine, regardless of treatment relationship, did not receive the second dose.
Follow-up and safety evaluations
Following study injection on Day 0, subjects returned on Days 7, 14, 28, and 84. Those in the bivalent cohort received a second injection on Day 84 and returned on Days 98 and 112. Safety was evaluated by assessment of vital signs and laboratory values, along with any local and systemic reactogenicity events from Days 0 through 7, and adverse events (AEs) throughout the study (definitions included in the Supplementary Material). Additionally, a follow-up safety evaluation phone call was completed either on Day 168 for subjects who received a single injection or Day 252 for subjects that received the second injection.
Assessment of immunogenicity
Geometric mean concentrations (GMCs) of anti-rAT and anti-rLukS-PV IgG were calculated using data from quantitative enzyme-linked immunosorbent assay testing on Days 0, 14, 28 and 84 for the monovalent cohort and Days 98 and 112 for the bivalent cohort. Neutralizing activity was determined using serum samples obtained on Day 0, 28, and 84 for subjects in the monovalent and bivalent 25 and 50 μg cohorts, and Day 112 for subjects in the 50 μg bivalent cohort. Neutralizing activity of rAT IgG was measured by its ability to inhibit hemolysis of rabbit erythrocytes by wild-type α-toxin. Rabbit red blood cells were washed with phosphate buffered saline (PBS) and then aliquoted into 96 well microtiter plates. Following the addition of serial dilutions of either sera from rAT vaccine recipients or rAT-antibody negative human sera, and 50 μL of wild-type α-toxin at a concentration of 2.0 μg/mL to each well, microtiter plates were incubated at 37°C, under humidified conditions for 30 minutes. Plates were then centrifuged for 10 minutes at room temperature. 20 μL of the lysate supernatant was then removed from each well and added to 180 μL PBS in a new microtiter plate, and the optical density of each well was then read at 450 nm. Neutralizing activity of rLukS-PV IgG was measured by inhibition of Panton-Valentine leukocidin (PVL) mediated lysis of neutrophil-like cells, derived from HL-60 cells differentiated with dimethyl sulfoxide (DMSO). Sixty µL of HL-60 cell suspension at 8.5×106 cells/mL (5.0 × 105 cells/well) was placed in 96 well cell culture plates. Following the addition of buffer and 30 μL PVL at a concentration of 1.0µg/mL to each well, PVL-antibody negative human sera, and sera from rLukS-PV vaccine recipients were added to separate wells by serial fold dilution. After incubation at 37°C with 8% CO2 for an additional 24 hours, an XTT tetrazolium salt, 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide, was added. Following incubation for 4 hours allowing cleavage of the yellow XTT tetrazolium salt to a soluble orange formazan dye via the succinatetetrazolium reductase system in the mitochondria of metabolically active cells, cell culture plates were centrifuged for 10 minutes. The supernatant from each cell was then transferred via pipette, and absorbance at 450 nm was measured to determine cell viability.
Neutralizing titers were reported in neutralizing units/mL (NU/mL). A neutralizing unit (NU) was arbitrarily assigned as the amount of anti-rAT antibody resulting in ≥50% inhibition of red blood cell lysis caused by 1 ηg of α-toxin (rAT-IC50). For rLukS-PV antibodies, NU was defined as the lowest serum dilution resulting in ≥50% inhibition of neutrophil lysis by 1 ηg of PVL (rLukS-PV-IC50). All neutralizing titers were log-transformed to calculate geometric mean concentrations titers (GMTs), using a covariate-adjusted mixed model to derive the 95% confidence interval. Neutralizing titers were also reported in term of geometric mean fold increases (GMFIs) and categorical seroresponse to vaccination. GMFI for all initial vaccinations was defined as the fold increase in GMT at day 28 and day 84 compared to day 0. For the booster dose, GMFI was defined as the fold increase at day 112 compared to day 84. Subjects were considered seropositive at baseline (day 0) if the IC50 neutralizing antibody titer was greater than 8. A positive seroresponse to initial vaccination was defined as a ≥2-fold increase in neutralizing antibody titer at Day 28 or Day 84 compared to Day 0. For subjects who received a booster, a positive seroresponse was represented by a ≥2-fold increase in neutralizing antibody titer at Day 112 compared to Day 84.
Statistical analyses
This was an exploratory analysis intended to provide initial estimates for evaluating safety, tolerability and immunogenicity; sample size estimation was not performed. The incidence of reactogenicity events and AEs were compared using chi-square or Fisher's Exact test. Initial analyses found reactogenicity and adverse event rates were similar for saline and alum placebo recipients. Therefore, pooled results from all placebo recipients are presented. For analyses regarding safety, all subjects who received study injections were included.
Immunogenicity endpoints were tested using a null hypothesis that there would be no difference in an immunogenicity endpoint between the different doses of a given formulation (monovalent or bivalent) at a particular time point. All antibody concentrations and neutralizing titers were log-transformed to calculate geometric mean concentrations (GMCs) or titers (GMTs) respectively, using a covariate-adjusted mixed model to derive the 95% confidence interval. A repeated measures analysis approach (i.e., generalized estimating equation or GEE) with the GENMOD procedure was used for the covariate (baseline IgG concentration)-adjusted comparisons among the treatment cohorts at each follow-up visit.
The functionality of rAT and rLukS-PV antibodies was evaluated by plotting the IgG antibody concentration against the neutralizing antibody for day 0, 28, 84 and 112. Subjects who were positive for neutralizing antibody (i.e. IC50 > 8) and IgG concentration by ELISA (i.e., >1 EU/mL) at the specified time-point, were included in the correlation analysis. Correlation between IgG concentration and neutralizing titers at each time point was analyzed by Pearson's correlation coefficient. All p-values were 2-sided with values <0.05 considered statistically significant. There was no adjustment for multiple comparisons. All analyses were conducted using SAS® version 9.1.3 (SAS Institute, Cary, NC).
Disclaimer
Some authors on this paper are military service members and/or employees of the US. Government. As such, this work was prepared as part of official duties. Title 17 USC. 105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 USC. 101 defines a United States Government work as a work prepared by a military service member or employee of the United States Government as part of that person's official duties.
The content of this publication is the sole responsibility of the authors and does not necessarily reflect the views or policies of the National Institute of Health or the Department of Health and Human Services, the DoD or the US. Departments of the Army, Navy or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the US. Government.
GlaxoSmithKline Biologicals SA, who acquired the StaphVax clinical program from Nabi Biopharmaceuticals, was provided the opportunity to review a preliminary version of this manuscript for factual accuracy. But the authors are solely responsible for final content and interpretation. The authors received no financial support or other form of compensation from GSK related to this study or the development of the manuscript.
Abbreviations
| AE | = | Adverse event |
| CA-MRSA | = | Community-associated methicillin-resistant S. aureus |
| CO2 | = | Carbon Dioxide |
| CPS | = | Capsular polysaccharides |
| DMSO | = | Dimethyl sulfoxide |
| DoD | = | Department of Defense |
| ELISA | = | Enzyme-linked immunosorbent assay |
| GENMOD | = | General linear model procedure |
| GEE | = | Generalized estimating equation |
| GMC | = | Geometric mean serum concentrations |
| GMFI | = | Geometric mean fold increases |
| GMT | = | Geometric mean titers |
| GSK | = | GlaxoSmithKline |
| IDCRP | = | Infectious Disease Clinical Research Program |
| IgG | = | Immunoglobulin G |
| IL-17A | = | Interleukin-17A |
| MRSA | = | Methicillin-resistant S. aureus |
| NU/mL | = | Neutralizing units/mL |
| PVL | = | Panton-Valentine leukocidin |
| rAT | = | Recombinant α-toxoid |
| rClf | = | Recombinant clumping factor |
| rLukS-PV | = | Subunit of Panton-Valentine leukocidin |
| SAE | = | Serious adverse event |
| SSTI | = | Skin and soft-tissue infection |
Disclosure of potential conflicts of interest
None- MLL, TL, JDM, DRH, JF, KW, MWE, DRT
Employment by Nabi Biopharmaceuticals- MN, AF, KT, PK, RF.
Supplementary Materials
Download MS Word (485.2 KB)Acknowledgments
The authors wish to thank Dr. Matthew Hohenboken for his many contributions to the design of this study during its development, Steve Fuller for providing expertise in performing the immunogenicity assays for both rAT and rLukS-PV and Luca Illinik for her editorial assistance. Lastly, the authors wish to thank the many members of the study team at each of the clinical sites for the countless hours and exceptional efforts which they devoted to the successful execution of this study.
Funding
Support for this work (IDCRP-035/Nabi-6801) was provided by a grant (DR081318) from the Department of Defense Deployment-Related Medical Research Program (to M.L.L. and partnering principal investigator, P. K.). Additional support was provided by the Infectious Disease Clinical Research Program (IDCRP, www.idcrp.org), a Department of Defense (DoD) program executed through Uniformed Services University of the Health Sciences and from Nabi Biopharmaceuticals, Rockville, MD. This project has been funded in part, with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, under Inter-Agency Agreement Y1-AI-5072.
Related Research Data
References
- CDC. Methicillin-resistant Staphylococcus aureus infections in correctional facilities—Georgia, California, and Texas, 2001-2003. MMWR Morb Mortal Wkly Rep 2003; 52:992-6
- Kazakova SV, Hageman JC, Matava M, Srinivasan A, Phelan L, Garfinkel B, Boo T, McAllister S, Anderson J, Jensen B, et al. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N Engl J Med 2005; 352:468-75; PMID:15689585; https://doi.org/http://dx.doi.org/10.1056/NEJMoa042859
- Zinderman CE, Conner B, Malakooti MA, LaMar JE, Armstrong A, Bohnker BK. Community-acquired methicillin-resistant Staphylococcus aureus among military recruits. Emerg Infect Dis 2004; 10:941-4; PMID:15200838; https://doi.org/http://dx.doi.org/10.3201/eid1005.030604
- Ellis MW, Hospenthal DR, Dooley DP, Gray PJ, Murray CK. Natural history of community-acquired methicillin-resistant Staphylococcus aureus colonization and infection in soldiers. Clin Infect Dis 2004; 39:971-9; PMID:15472848; https://doi.org/http://dx.doi.org/10.1086/423965
- Ellis MW, Griffith ME, Jorgensen JH, Hospenthal DR, Mende K, Patterson JE. Presence and molecular epidemiology of virulence factors in methicillin-resistant Staphylococcus aureus strains colonizing and infecting soldiers. J Clin Microbiol 2009; 47:940-5; PMID:19213694; https://doi.org/http://dx.doi.org/10.1128/JCM.02352-08
- Co E, Keen EF, Aldous WK. Prevalence of methicillin-resistant Staphylococcus aureus in a combat support hospital in Iraq. Military Medicine 2011; 176:89-93; PMID:21305966; https://doi.org/http://dx.doi.org/10.7205/MILMED-D-09-00126
- Huang X, Cash DM, Chahine MA, van Horn GT, Erwin DP, McKay JT, Hamilton LR, Jerke KH, Co EA, Aldous WK, et al. Methicillin-resistant Staphylococcus aureus infection in combat support hospitals in three regions of Iraq. Epidemiol Infect 2011; 139:994-7; PMID:20727246; https://doi.org/http://dx.doi.org/10.1017/S0950268810001950
- Landrum ML, Neumann C, Cook C, Chukwuma U, Ellis MW, Hospenthal DR, Murray CK. Epidemiology of Staphylococcus aureus blood and skin and soft tissue infections in the US military health system, 2005-2010. JAMA 2012; 308:50-9; PMID:22760291; https://doi.org/http://dx.doi.org/10.1001/jama.2012.7139
- Ellis MW, Griffith ME, Dooley DP, McLean JC, Jorgensen JH, Patterson JE, Davis KA, Hawley JS, Regules JA, Rivard RG, et al. Targeted intranasal mupirocin to prevent colonization and infection by community-associated methicillin-resistant Staphylococcus aureus strains in soldiers: a cluster randomized controlled trial. Antimicrob Agents Chemother 2007; 51:3591-8; PMID:17682105; https://doi.org/http://dx.doi.org/10.1128/AAC.01086-06
- Whitman TJ, Herlihy RK, Schlett CD, Murray PR, Grandits GA, Ganesan A, Brown M, Mancuso JD, Adams WB, Tribble DR. Chlorhexidine-impregnated cloths to prevent skin and soft-tissue infection in Marine recruits: a cluster-randomized, double-blind, controlled effectiveness trial. Infect Control Hosp Epidemiol 2010; 31:1207-15; PMID:21028984; https://doi.org/http://dx.doi.org/10.1086/657136
- Army Medical Surveillance Activity. Hospitalizations among members of the active component, US. Armed Forces, 2014. Medical Surveillance Monthly Report 2015; 22:11-17 Medical Surveillance Monthly Report:11-7
- Army Medical Surveillance Activity. Ambulatory Visits Among Members of the Active Component, U.S Armed Forces, 2014. Medical Surveillance Monthly Report 2015; 22:19-24
- Deresinski S. Antistaphylococcal vaccines and immunoglobulins: current status and future prospects. Drugs 2006; 66:1797-806; PMID:17040111; https://doi.org/http://dx.doi.org/10.2165/00003495-200666140-00002
- Holtfreter S, Kolata J, Broker BM. Towards the immune proteome of Staphylococcus aureus- the anti-S. aureus antibody response. Int J Med Microbiol 2010; 300:176-92; PMID:19889576; https://doi.org/http://dx.doi.org/10.1016/j.ijmm.2009.10.002
- Otto M. Targeted immunotherapy for staphylococcal infections: focus on anti-MSCRAMM antibodies. BioDrugs 2008; 22:27-36; PMID:18215088; https://doi.org/http://dx.doi.org/10.2165/00063030-200822010-00003
- Schaffer AC, Lee JC. Vaccination and passive immunisation against Staphylococcus aureus. Int J Antimicrob Agents 2008; 32 Suppl 1:S71-8; PMID:18757184; https://doi.org/http://dx.doi.org/10.1016/j.ijantimicag.2008.06.009
- Botelho-Nevers E, Verhoeven P, Paul S, Grattard F, Pozzetto B, Berthelot P, Lucht F. Staphylococcal vaccine development: review of past failures and plea for a future evaluation of vaccine efficacy not only on staphylococcal infections but also on mucosal carriage. Expert Rev Vaccines 2013; 12:1249-59; PMID:24111513; https://doi.org/http://dx.doi.org/10.1586/14760584.2013.840091
- Levy J, Licini L, Haelterman E, Moris P, Lestrate P, Damaso S, Van Belle P, Boutriau D. Safety and immunogenicity of an investigational 4-component Staphylococcus aureus vaccine with or without AS03B adjuvant: Results of a randomized phase I trial. Hum Vaccin Immunother 2015; 11:620-31; PMID:25715157; https://doi.org/http://dx.doi.org/10.1080/21645515.2015.1011021
- Giersing BK, Dastgheyb SS, Modjarrad K, Moorthy V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine 2016; 34(26):2962-6
- Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. Targeting of α-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis 2010; 202:1050-8; PMID:20726702; https://doi.org/http://dx.doi.org/10.1086/656043
- Karauzum H, Adhikari RP, Sarwar J, Devi VS, Abaandou L, Haudenschild C, Mahmoudieh M, Boroun AR, Vu H, Nguyen T, et al. Structurally designed attenuated subunit vaccines for S. aureus LukS-PV and LukF-PV confer protection in a mouse bacteremia model. PLoS One 2013; 8:e65384; PMID:23762356; https://doi.org/http://dx.doi.org/10.1371/journal.pone.0065384
- Brown EL, Dumitrescu O, Thomas D, Badiou C, Koers EM, Choudhury P, Vazquez V, Etienne J, Lina G, Vandenesch F, et al. The Panton-Valentine leukocidin vaccine protects mice against lung and skin infections caused by Staphylococcus aureus USA300. Clin Microbiol Infect 2009; 15:156-64; https://doi.org/http://dx.doi.org/10.1111/j.1469-0691.2008.02648.x
- Berube BJ, Bubeck Wardenburg J. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins 2013; 5:1140-66; PMID:23888516; https://doi.org/http://dx.doi.org/10.3390/toxins5061140
- Kennedy AD, Wardenburg JB, Gardner DJ, Long RD, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. Targeting of α-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis 2010; 202:1050-8; PMID:20726702; https://doi.org/http://dx.doi.org/10.1086/656043
- Adhikari RP, Ajao AO, Aman MJ, Karauzum H, Sarwar J, Lydecker AD, Johnson JK, Nguyen C, Chen WH, Roghmann MC. Lower antibody levels to Staphylococcus aureus exotoxins are associated with sepsis in hospitalized adults with invasive S. aureus infections. J Infect Dis 2012; 206:915-23; PMID:22807524; https://doi.org/http://dx.doi.org/10.1093/infdis/jis462
- Fritz SA, Tiemann KM, Hogan PG, Epplin EK, Rodriguez M, Al-Zubeidi DN, Bubeck Wardenburg J, Hunstad DA. A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin Infect Dis 2013; 56:1554-61; PMID:23446627; https://doi.org/http://dx.doi.org/10.1093/cid/cit123
- Genestier AL, Michallet MC, Prevost G, Bellot G, Chalabreysse L, Peyrol S, Thivolet F, Etienne J, Lina G, Vallette FM, et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J Clin Invest 2005; 115:3117-27; PMID:16276417; https://doi.org/http://dx.doi.org/10.1172/JCI22684
- Lina G, Piemont Y, Godail-Gamot F, Bes M, Peter MO, Gauduchon V, Vandenesch F, Etienne J. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis 1999; 29:1128-32; PMID:10524952; https://doi.org/http://dx.doi.org/10.1086/313461
- Konig B, Prevost G, Piemont Y, Konig W. Effects of Staphylococcus aureus leukocidins on inflammatory mediator release from human granulocytes. J Infect Dis 1995; 171:607-13; PMID:7533198; https://doi.org/http://dx.doi.org/10.1093/infdis/171.3.607
- Konig B, Koller M, Prevost G, Piemont Y, Alouf JE, Schreiner A, Konig W. Activation of human effector cells by different bacterial toxins (leukocidin, alveolysin, and erythrogenic toxin A): generation of interleukin-8. Infect Immunity 1994; 62:4831-7; PMID:7927762
- Hensler T, Konig B, Prevost G, Piemont Y, Koller M, Konig W. Leukotriene B4 generation and DNA fragmentation induced by leukocidin from Staphylococcus aureus: protective role of granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF for human neutrophils. Infect Immunity 1994; 62:2529-35; PMID:7514577
- Yoong P. Enhancement of bacterial virulence by antibody neutralization of immune-activating toxins. Virulence 2010; 1:409-13; PMID:21178480; https://doi.org/http://dx.doi.org/10.4161/viru.1.5.12705
- Croze M, Dauwalder O, Dumitrescu O, Badiou C, Gillet Y, Genestier AL, Vandenesch F, Etienne J, Lina G. Serum antibodies against Panton-Valentine leukocidin in a normal population and during Staphylococcus aureus infection. Clin Microbiol Infect 2009; 15:144-8; PMID:19154491; https://doi.org/http://dx.doi.org/10.1111/j.1469-0691.2008.02650.x
- Brown EL, Dumitrescu O, Thomas D, Badiou C, Koers EM, Choudhury P, Vazquez V, Etienne J, Lina G, Vandenesch F, et al. The Panton-Valentine leukocidin vaccine protects mice against lung and skin infections caused by Staphylococcus aureus USA300. Clin Microbiol Infect 2009; 15:156-64; https://doi.org/http://dx.doi.org/10.1111/j.1469-0691.2008.02648.x
- Nissen M, Marshall H, Richmond P, Shakib S, Jiang Q, Cooper D, Rill D, Baber J, Eiden J, Gruber W, et al. A randomized phase I study of the safety and immunogenicity of three ascending dose levels of a 3-antigen Staphylococcus aureus vaccine (SA3Ag) in healthy adults. Vaccine 2015; 33:1846-54; PMID:25707693; https://doi.org/http://dx.doi.org/10.1016/j.vaccine.2015.02.024
- Fattom A, Fuller S, Propst M, Winston S, Muenz L, He D, Naso R, Horwith G. Safety and immunogenicity of a booster dose of Staphylococcus aureus types 5 and 8 capsular polysaccharide conjugate vaccine (StaphVAX) in hemodialysis patients. Vaccine 2004; 23:656-63; PMID:15542186; https://doi.org/http://dx.doi.org/10.1016/j.vaccine.2004.06.043
- Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 2011; 11:505-18; PMID:21720387; https://doi.org/http://dx.doi.org/10.1038/nri3010
- Niebuhr M, Mamerow D, Heratizadeh A, Satzger I, Werfel T. Staphylococcal α-toxin induces a higher T cell proliferation and interleukin-31 in atopic dermatitis. Int Arch Allergy Immunol 2011; 156:412-5; PMID:21832830; https://doi.org/http://dx.doi.org/10.1159/000323905
- Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest 2010; 120:1762-73; PMID:20364087; https://doi.org/http://dx.doi.org/10.1172/JCI40891
- Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, Long RD, Dorward DW, Gardner DJ, Lina G, et al. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis 2006; 194:1761-70; PMID:17109350; https://doi.org/http://dx.doi.org/10.1086/509506
- Bubeck Wardenburg J, Palazzolo-Ballance AM, Otto M, Schneewind O, DeLeo FR. Panton-Valentine leukocidin is not a virulence determinant in murine models of community-associated methicillin-resistant Staphylococcus aureus disease. J Infect Dis 2008; 198:1166-70; PMID:18729780; https://doi.org/http://dx.doi.org/10.1086/592053
- Labandeira-Rey M, Couzon F, Boisset S, Brown EL, Bes M, Benito Y, Barbu EM, Vazquez V, Höök M, Etienne J, et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science 2007; 315:1130-3; PMID:17234914; https://doi.org/http://dx.doi.org/10.1126/science.1137165
- Zivkovic A, Sharif O, Stich K, Doninger B, Biaggio M, Colinge J, Bilban M, Mesteri I, Hazemi P, Lemmens-Gruber R, et al. TLR 2 and CD14 mediate innate immunity and lung inflammation to staphylococcal Panton-Valentine leukocidin in vivo. J Immunol 2011; 186:1608-17; https://doi.org/http://dx.doi.org/10.4049/jimmunol.1001665
- Loffler B, Hussain M, Grundmeier M, Bruck M, Holzinger D, Varga G, Roth J, Kahl BC, Proctor RA, Peters G. Staphylococcus aureus panton-valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathogens 2010; 6:e1000715; PMID:20072612; https://doi.org/http://dx.doi.org/10.1371/journal.ppat.1000715
- Spaan AN, Schiepers A, de Haas CJ, van Hooijdonk DD, Badiou C, Contamin H, Vandenesch F, Lina G, Gerard NP, Gerard C, et al. Differential interaction of the staphylococcal Toxins Panton-Valentine Leukocidin and gamma-hemolysin CB with human C5a Receptors. J Immunol 2015; 195:1034-43; https://doi.org/http://dx.doi.org/10.4049/jimmunol.1500604
- Sause WE, Buckley PT, Strohl WR, Lynch AS, Torres VJ. Antibody-Based Biologics and Their Promise to Combat Staphylococcus aureus Infections. Trends Pharmacological Sci 2016; 37:231-41; PMID:26719219; https://doi.org/http://dx.doi.org/10.1016/j.tips.2015.11.008
- Rouha H, Badarau A, Visram ZC, Battles MB, Prinz B, Magyarics Z, Nagy G, Mirkina I, Stulik L, Zerbs M, et al. Five birds, one stone: neutralization of α-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. Mabs 2015; 7:243-54; PMID:25523282; https://doi.org/http://dx.doi.org/10.4161/19420862.2014.985132