
ABSTRACT
The recurrent nasopharyngeal carcinoma of head-and-neck cancers pathology showed unique symptoms and clinical characteristics. The complexity of pathology poses challenges for developing therapeutic interventional approaches against nasopharyngeal carcinoma (NPC). The conventional treatment regimens offer limited local control and survival, which, leads to adverse delayed complications. Our study present a generic monocyte derived dendritic cell (MoDC) vaccine strategy for NPC in which RNA is used as a source of tumor-associated antigens (TAAgs). The RNA extracted from well-characterized highly immunogenic NPC cells (C666–1) was transfected into MoDCs. The formulated and characterized cationic liposomes were used to achieving efficient RNA transfection of immature DCs. Further, DCs were forcibly matured with a cytokine cocktail to achieve greater expression of MHC and co-stimulatory molecules. Moreover, our results did not see any effect of RNA or lipids on MoDCs phenotype or cytokine expression. RNA loaded DCs derived from HLA-A2-positive donors were shown to activate effector memory cytotoxic T lymphocytes (CTLs) specific for TAAg ligand expressed by C666–1 cells. Our results show the comparison of cytotoxic response mounted against RNA-loaded DCs with those directly stimulated by C666–1 tumor cells. Our findings suggest that DCs expressing tumor cell RNA primed naïve T cells show T cells priming with lesser cytotoxicity and cytokine secretion when exposed with with C666–1 tumor cells. These results surface the potential of DCs to deliver RNA in NPCs, sufficient presentation of RNA to provoke perdurable immune responses against nasopharyngeal carcinoma. Our results implies that DC based vaccine approach may be useful to develop therapeutic interventional approach in the form of vaccine to address NPCs.
Introduction
Nasopharyngeal carcinoma (NPC) is well-documented head and neck cancer pathology in South-East Asia and Southern China.Citation1,2 NPC constitutes 90% of malignant nasopharyngeal tumors and is associated with higher morbidity and mortality. The nasopharyngeal carcinoma is divided in 2 clinically distinct pathologies: epidermoid carcinoma, and undifferentiated carcinoma of nasopharyngeal type (UCNT). The later showed greater prevalence rate,Citation3 and Epstein-Barr virus (EBV) is the primary reason for UCNT pathology. The crucial role played by Epstein–Barr virus infection in NPC carcinogenesis is well-documented. The environmental factors and genetic susceptibility may also be crucial players in NPC condition.Citation4
Cancer immunotherapy, an approach to use immune system of cancer patients to diagnose and treat tumors, has been recently established as a potential cancer treatment strategy.Citation5 The results obtained from clinical trials have provided experimental evidence that immune effectors and mediators such as B, T, natural killer (NK), natural killer T (NKT) cells, and inflammatory cytokines help controlling pre-malignancy.Citation6 Cancer immunotherapy involves amenable and effective methods to activate immune system and immune effecters to destroy cancerous cells. Therefore, immune effecters of cancers patients may be targeted by using 1) antigen specific antibodies for cancer therapy, 2) specific proteins of cancer patients for immunizing cancer patients, and 3) passive transfer of cytotoxic T lymphocytes or dendritic cells to cancer patient.Citation7
Dendritic cells (DCs), highly efficient and professional antigen-presenting cells (APC), can stimulate naive T cell and activate antigen-specific cytotoxic T Lymphocytes (CTLs).Citation8,9 The antigen activated DCs are reported to render anti-tumor effect both in vitro as well as in vivo.Citation10,11 Also, DCs loaded with various tumor antigens have shown to produce specific anti-tumor effects in cancer cells and cancer patients.Citation12,13 The RNA transfection, transduction, peptide and protein pulsing, exosomes plus peptides, tumor lysates and whole cell proteins (oragalactosylceramide) targeting NK-T cells have been widely used as antigenic formulations for pulsing of tumor associated antigens into DCs.Citation14,15 Therefore, tumor derived RNA loaded DCs arrangement has been shown to be a promising approachCitation16 to address nasopharyngeal carcinoma.
Nasopharyngeal carcinoma (NPC) has been reported to be a rare cancer in the Western hemisphere with low incidence whereas regions such as southern China, southeast Asia, Mediterranean basin and Alaska show the higher prevalence (80 per 100,000 individuals). The tumor in the western hemisphere presents the different histology to the endemic form [12]. This demographic information suggests that genetic and environmental factors play a very crucial role in the pathogenesis of NPC. The radiotherapy is effective and NPC is a sensitive therapy, and deemed as gold standard treatment for early-stage NPC (T1–2a; N0; M0). The patients with local disease (restricted to specific areas) are reportedly treated using radiation and showed 5 y control ranging from 80–95% cases [22–25].
The development of an effective vaccine using autologous DCs loaded with tumor-derived RNA presents a plausible and promising approach to overcome the limitations. It also allows patients expressing various MHC allo-types to be licensed and to enter clinical trials.Citation16 Our findings show that RNA derived from well-characterized C666–1 tumor cells could be used not only as a source of TAAgs, but also could serve as a tool for vaccination monitoring. Present study is designed to formulate and characterize RNA loaded liposomes to achieve significant transfection in human monocyte derived DCs and in vitro assessment and detection of the mediators of systemic inflammation rendering lesser toxicity. Liposomes are made of phospholipid molecules capable of encapsulating various types of oligonucleotides, drugs, or vaccine candidates within their aqueous core. Liposomes have been engineered to protect carrier molecules (DNA, RNA, protein and drug) from harsh gastric environment when administered orally or degradation because of ribonucleases when administered intramuscularly in an animal model, and to enhance intracellular cytosolic delivery.Citation17,18 In conclusion, we report this strategy for the development of a generic mRNA pulsed DC vaccine which may be deployed as therapeutic vaccine for NPC.
Results
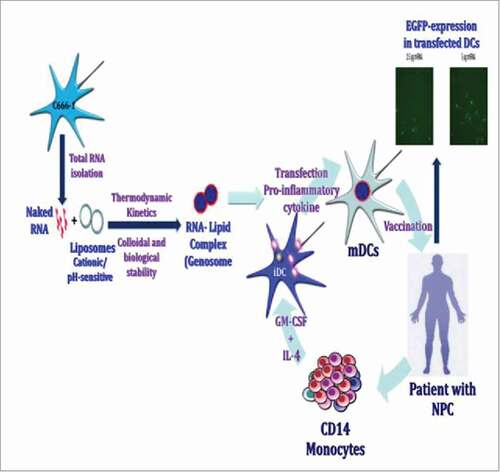
Strategy for a generic RNA-pulsed DC vaccine for NPC patients
This schema of proposed strategy for development and application of RNA-pulsed DC vaccine approach for NPC (CitationSchematic 1). The CD14+ monocytes were selected by positive selection through RosetteSep (Life Technologies, USA) from peripheral blood of NPC patient, and EBV transformed LCLs were generated for long-term therapeutic studies. CD14+ monocytes are differentiated into DC-SIGN and CD1c expressing immature dendritic cells (iMoDCs) in the presence of stimulators (GM-CSF & IL-4). The iMoDCs were transfected with total RNA extracted from C666–1 cells, and forced maturation was induced using a cocktail of inflammatory mediators for 48 hr. RNA extracted from a well characterized nasopharyngeal carcinoma cells drove us to bypass the requirement of RNA preparation from tumor tissue of individual patient. We deployed RNA pulsed matured DCs as vaccine arrangement to develop vaccination interventional approach for NP carcinoma. The autologous LCLs are known to activate, expand the long-lasting perseverance of specific T-cell responses post vaccine immunization. The transfected autologous LCLs with NPC-derived RNA generate specific MHC expressing APCs with various tumor associated antigen (TAAg) epitopes. The culture of RNA pulsed DCs with autologous post-treatment PBMCs allowed to assess the development of MHC class I and class II-restricted T-cells known to provoke anti-tumor immunity. Therefore, we determined specific T-cell responses with NP carcinoma cells.Citation19
Schematics 1. Schematics of a strategy for a generic RNA-pulsed DC vaccine for nasopharyngeal carcinoma.
Transcriptome analysis of ex-vivo generated DCs liposome mediated transfection of transcribed RNA
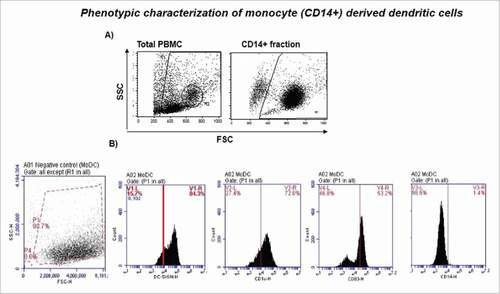
We ex-vivo generated significant population of monocyte-derived iDCs in the presence of GM-CSF and IL-4 yielding considerable MoDC cell numbers (50% of total CD14 monocytes) (). The iMoDCs were activated and forcibly matured by the stimulation with pro-inflammatory cytokine cocktail (TNF-α, IL-1β, PGE2 and IL-6) for 48 hr. The ex-vivo generated MoDCs were confirmed for their immature DC phenotype (CD14lowCD83−CD1c+DC-SIGN+) on day 6 of cultivation by flow cytometer.
Figure 1. Phenotypic characterization of monocyte (CD14+) derived dendritic cells. A) Monocytes were isolated using human monocyte enrichment cocktail (RosetteSep), and cultured in presence of growth factors (GM-CSF and IL-4) for 5–6 d to generate monocyte derived DCs (MoDCs) ex-vivo. The phenotype of immature MoDC (iMoDC) was characterized by flow cytometer. B) 200K-400K monocytes/well were distributed in 6-well plate. MoDCs were confirmed for their immature DC phenotype (CD14lowCD83−CD1c+DC-SIGN+) on day 6.
Transcriptome analysis
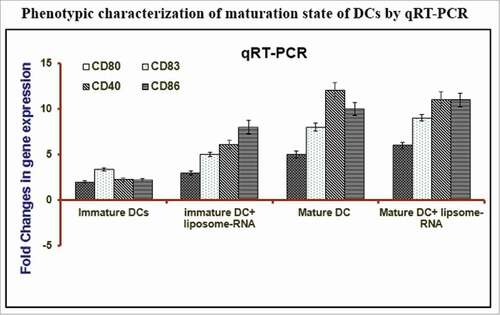
Our group has developed RNA loaded liposomal nanoparticles and validated the utility of this carrier for the transfection of DCs with RNA extracted from nasopharyngeal carcinoma cells. We obliged to characterize different gene expression profile by customized PCR micro-array (). The fundamental cytokines, chemokines, and transcription factors playing a pivotal role in inflammation were analyzed on cultured C666–1 cells. RNA loaded-liposomes and free RNA formulations were normalized to baseline untreated control. The pro-inflammatory cytokines led activation resulted in fully mature DCs (mDCs) showing greater expression of maturation and activation markers (CD86, CD80, CD83, CD40), transcription factors (FOXO1), apoptosis regulators (Bcl2l1, Bcl-2), and inflammatory mediators (IL-6, IL-1β, IL-12) (). The CD14 markers for monocyte were seen the downregulation (). The loading of DCs with RNA has been achieved using various techniques,Citation20-25 but optimal and standardized procedure for generating fully immunocompetent RNA expressing DCs is yet to be optimized and established. Here, we utilized the versatile delivery vehicle, liposome, formulated from cationic lipids (DOTAP) for loading DCs with tumor-derived RNA for evoking tumor-specific immune response.
Table 1. Relative expression of monocyte-derived DCs (MoDCs) regulate expression of various markers relative to monocytes. Monocytes were isolated using human monocyte enrichment cocktail (RosetteSep), and were cultured in presence of growth factors (GM-CSF and IL-4) for 6 d to generated MoDCs, and 200K-400K monocytes/well distributed in 6-well plate. MoDCs were confirmed for their immature DC phenotype (CD14lowCD83−CD1c+DC-SIGN+) on day 6. All biomarkers were designed in triplicates on array plates. The fold change (in gene expression) value greater than one indicates a positive value, or an upregulation of gene expression, and less than one indicates a negative value, or downregulation of gene expression
Formulation and characterization of mRNA-liposomes
Liposomes were used to efficiently transfect RNA into monocyte derived dendritic cells, and these liposomes loaded with RNA was used to provoke humoral and cell mediated immune response against NPC. The negatively charged RNA molecules were encapsulated in conventional or engineered liposome formulation. The liposomes of defined and required size range (100–400 nm) were obtained by sonication and extrusion. The prepared liposomes were characterized for their spherical shape with smooth surfaces through scanning electron microscope (SEM) (). Additionally, spherical shape without agglomeration was confirmed by transmission electron microscopy (TEM) (). The SEM and TEM image analysis confirmed the size (∼210 nm) of prepared liposomes. SEM and TEM results were complemented by liposomal particles (167.7 ± 2.0 nm) determined by size analyzer (Delsa Nano particle analyzer, USA) (). The zeta potential of liposomes was determined by determining electrophoretic movement of charged particles. Particle size for plain liposomes showing no complexation with mRNA was calculated to be 161.3 ± 6.0 nm. However, significant changes in particle size (213 nm) were observed upon complexation with mRNA (p < 0.001). The observed zeta potential of mRNA liposomes was positive 6.87 ± 1.23 mV () against plain liposomes which was calculated to be 36.385 ± 2.8 mV. The decrease in zeta potential of mRNA liposomes in comparison to plain formulation is suggestive of cationic liposome and anionic RNA complex formation. Moreover, both liposome formulations (with and without mRNA) were seen uniform confirmed by lower (≤ 2) polydispersity index PDI ().
Figure 2. Characterization of mRNA loaded liposomes with respect to shape and size A) mRNA loaded liposomes were characterized with respect to shape and size by Scanning Electron Microscopy (SEM) technique, arrows indicate the formulated (mRNA loaded) liposomes, and B) High resolution negative staining Transmission electron Microscopy (TEM) technique was used to investigate the morphology of liposomes produced by thin film method. At the same magnification of micrograph results, the lamelarity of prepared liposomes is seen different.
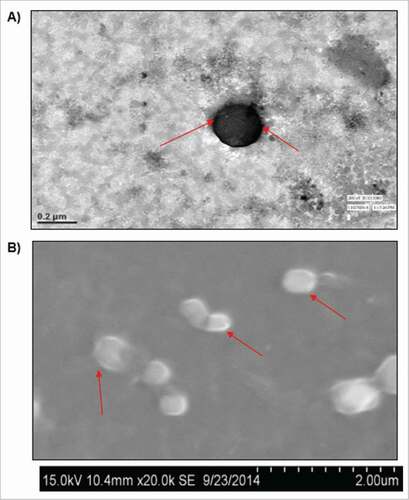
Table 2. The preparation and characterization of cationic liposomes for achieving an efficient mRNA transfection. The liposomes formulation was analysis for particle size, zeta potential and polydispersity index of plain and mRNA-loaded cationic liposomes. The in-process stability/shelf-life of mRNA-loaded liposomes was evaluated for 3 months
Next, we assessed the stability of mRNA-loaded liposomes with respect to particle size and zeta potential over the period of 3 months at 4°C (). This temperature was chosen based on available commercial liposome formulations. We did not observe any significant changes in particle size and zeta potential during this storage period (). These results suggest the stability of optimized liposome formulations with no aggregation and advocates for intact complexation.
Liposome mediated RNA transfection of immature DCs
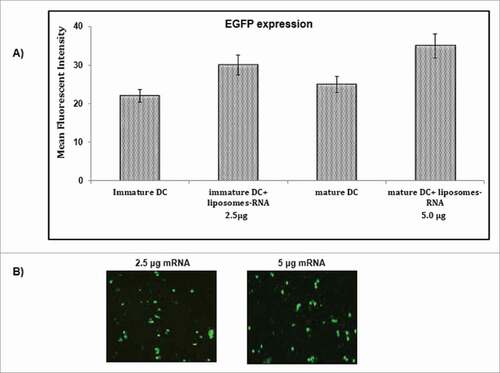
RNA transfection efficiency was determined by the transfection of iDCs with in vitro-transcribed EGFP labeled mRNA loaded liposomes. Liposomes were used due to their biocompatibility and relatively less toxicity and better transfection efficiency than other transfection reagents. Flow cytometry and fold regulation of RNA transcript analysis of EGFP expression after 48 hr of transfection showed an average transfection rate of 20–40% (). Matured DCs showed greater protein expression as increase in EGFP-positive cells was seen when transfected DCs (2.5 μg mRNA/0.5 million cells) were treated with maturation cocktail for 48 hr (). The 5.0 μg mRNA/0.5 million cells led to an increase in percentage of positive cells by 4–5% than that seen with 2.5 μg mRNA (). Liposome mediated mRNA transfection showed broad range of protein density, specificity of DCs with varying degree of EGF protein expression ( & ).
Figure 3. Transfection of immature and mature DCs with EGFP mRNA when delivered through cationic liposomes. The immature and mature DCs were transfected through cationic liposomes using DMRIE-C and in vitro-transcribed EGFP labeled mRNA. A) 2.5 μg EGFP mRNA/0.5–1 million iDCs were transfected (iDC + Liposome-RNA, 2.5 µg) and mature DCs (matured by using a pro-inflammatory cytokine cocktail) were transfected (iDC + Liposome-RNA, 5.0µg). 0.5–1 million DCs were transfected and were matured. EGFP expression was analyzed by flow cytometry 48 hr after transfection. B) EGFP-expression in transfected DCs was visualized using a fluorescent microscope. This experiment was the mean of 3 independent experiments (n = 3).
Liposome mediated RNA transfection of DCs
The interference of DCs maturation after RNA transfection was evaluated by the incubation of transfected iDCs with RNA loaded liposomes formulation and with or without maturation cytokine cocktail. Phenotypic characterization of DCs was performed by flow cytometer after 48 hr treatment. We did not see any effect of liposome-RNA on the maturation of DCs (). The fully mature DC phenotype could be induced by the treatment of pro-inflammatory cytokine cocktail which resulted in the greater expression of co-stimulatory molecule (CD40) as well as higher expression of maturation markers (CD86 & CD83) expressionCitation26 (). Next, we decided to analyze whether loading of iDCs with RNA altered the cytokine production capacity by determining the expression of cytokine secreted by DCs. We tested supernatants of differentially treated DCs on day 7 of culture, and iDCs were incubated with liposomes loaded with RNA (liposome alone, RNA alone or RNA-liposome). DCs were either matured (by inflammatory mediators) or kept in immature stage for 48 hr before harvesting the supernatants, and analyzed.
Figure 4. Characterization of phenotype of mature DCs by estimating the transcript level expression of various maturation markers (CD80, CD83, CD86, CD40). The incubation of monocyte-derived iDCs with a pro-inflammatory cytokine cocktail (IL-1β, IL-6, TNF-α & PGE2) for 48 hr induced a fully mature DC phenotype. The maturation of DCs was assessed by customized microarray plates designed for specific biomarkers. The qPCR was performed with following formulations; mature DCs, mature DCs incubated with liposome-RNA, immature DCs, immature DC incubated with RNA.
Stimulation of C666–1 specific cytotoxic-T lymphocyte lines using RNA-loaded DCs
The de novo priming of antigen-specific T-cell responses for RNA-pulsed DCs was performed with DCs generated from MHC positive healthy donors. We used them as APCs after liposome mediated RNA (total RNA derived from either C666–1 or C15 cells: 5.0 μg RNA/0.5 million DCs) was transfected to DCs and cells were allowed to mature for 48 hr. These mature DCs were cultured with tumor infiltrating lymphocytes. A11-restricted EBNA3B-specific CTL clones (CMc23), a tumor antigen-specific CTL, was expanded and propagated from tumor-infiltrating lymphocyte population of patient. The C666–1 associated determinant was recognized and presented by HLA-encoded molecules, and A11-restricted EBNA3B-specific CTL clones (CMc23)Citation27 was used in the experiments. The supernatants of CMc23 cultured with DCs were harvested and assessed for IFN-γ secretion by ELISA after 24 hr. The EBNA3B-specific CTL clones (CMc23) was cultivated and analyzed with different cell lines (). The TIL-26 (GG) clone was co-cultivated with different cell lines to demonstrate immunogenic specificity. The co-culture with C666–1 (nasopharyngeal carcinoma) cells, but not with autologous LCL-26, the normal kidney cell line (NKC-26), or the HLA-A#0201-matched tumor cell line human Daudi lymphoblastoid cells with IFN-α in order to identify the biochemical pathways of transmembrane signalingCitation28 (). K562 cells are of the erythroleukemia typeCitation29 and are non-adherent and rounded. These cells are positive for the bcr:abl fusion gene, and bear some proteomic resemblance to both undifferentiated granulocytes and erythrocytes.Citation30,31 C666–1 induced IFN-γ-release, indicating that only C666–1 cells provided specific ligand for TIL activation.
Figure 5. RNA pulsed DC mounts Ag-specific response. Reconstitution of the TIL epitope in HLA-A-positive DCs after liposome mediated transfection with C666–1 and C15-derived total RNA. A) The specificity of TIL-CMc23 for an epitope provided by C666–1, different tumor cell lines were cultivated with T cell clone in a ratio of 4:1 and supernatants were analyzed after 24 hr, B) HLA-A-positive iDCs were transfected using DMRIE-C with total RNA derived from nasopharyngeal carcinoma cells. 48 hr after transfection and maturation mDCs were cultivated with TIL-CMc23 T cell clone at a ratio of 4:1 for 24 hr. All experiments were performed in triplicates and activation of T cells was estimated IFN-γ secretion (n = 3).
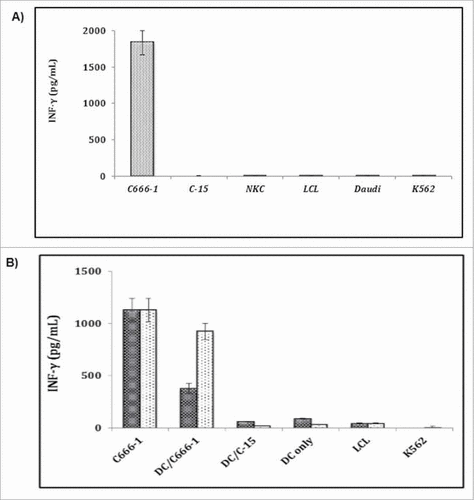
The culture with C666–1 cells with normal kidney cells (NKC), or C15 induced IFN-γ release and indicates that only C666–1 cells provided specific ligand for TIL activation (), but no cytokine secretion was seen with autologous LCL. Moreover, LAK or NK activity against Daudi or K562 cells by using CMc23 clone was not observed. DCs transfected with C666–1 derived RNA were seen to induce IFN-γ secretion in TIL (CMc23) cells, but could not induce IFN-γ production by DCs alone. These results suggest that C666–1 derived RNA provides TIL-CMc23 specific epitope to DCs. However, DCs loaded with RNA derived from C15 or LCL-15 (HLAA2-positive cells) which does not express TIL-CMc23 epitope () were not recognized by T cell clone, and confirmed the specificity of T cell activation by RNA-pulsed DCs (). The responses stimulated by DCs reached levels (20%–80%) seen by the stimulation of C666–1 cells. These variations in the stimulation capacity of RNA-loaded DCs may be reasoned because of the variations in donors and their genetic constitutions, which in turn is due to the use of biocompatible cationic liposomes for the transfection of RNA into DCs. However, this variability was not readily apparent from phenotypic characterization of DCs. The TIL activation capacity of C666–1 transfected DCs and stimulation potential of C666–1 cells were seen same (). The RNA-loaded DC stimulation was estimated about 30% of the level seen with C666–1 cells ().
Antigen processing and presentation of transfected NPC cell lines
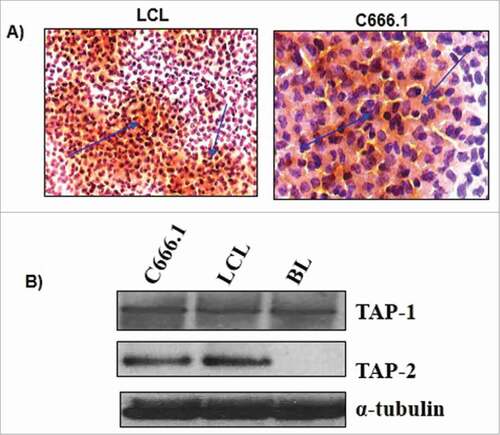
Next, we decided to test upon the functioning of C666.1 NPC cell line after transfection with RNA derived from NPC tumor of southern Chinese originCitation32(). We could see that the integrity and expression of TAP1 and TAP2 remained intact even after the transfection with RNA isolated from NPC cells. The outcome of HLA/MHC typing of C666.1 cell line showed that it carries specific HLA B58, but no other non-specific HLA-A or B alleles. The immunocytochemistry analysis through immunofluorescence assay using HLA class I-specific mAb W632 show that HLA class I molecules expression on the surface of the c666.1 line was lesser compare with than seen with LCL (). We examined the expression of TAP1 and TAP2 proteins by the immunoblot analysis of C666.1 and compared with Burkitt tumor and an LCL cell line. Consistent to the earlier results, we saw little or no expression of TAP1 & TAP2 on Burkitt line, whereas C666.1 showed similar level of expression of TAP1 and TAP2 with LCL ().
Figure 6. Viable cells from c666.1 and an LCL were analyzed through A) immunocytochemistry for the surface expression of HLA class I using the anti-W6/32 antibody, B) immunoblot analysis was done to analyze the expression of TAP1 and TAP2 in c666.1, BL cell line and LCL to assess the in-process integrity of transfected RNA. α-tubulin was taken as loading control.
Discussion
Present study was designed to develop a generic RNA-pulsed DC vaccine for NPC. The poor rate of prognosis and availability of limited alternatives to study NPC in early symptomatic stage. Dendritic cells are potent antigen-presenting cells with heterogeneous population of myeloid origin that express a wide variety of pattern recognition and lectin receptors. These cells are responsible for cross-talk between innate and adaptive immunity.Citation33,34 A DC-based vaccine approach presenting NPC-associated antigens provokes anti-tumor responses and capable of de novo priming of antigen-specific CD4+ and CD8+ effector cells in vivo.Citation35,36 Myeloid DCs are generated ex-vivo from CD14+ monocytes when cultured in presence of stimulators, GM-CSF & IL-4.Citation37 However, meagre evidences are available with TAAgs in context of NPC than melanomas where different peptide-specific immunization approaches have been extensively studied. Therefore, limited clinical studies are reported to evaluate the safety and feasibility of DCs loaded with autologous tumor lysates vaccines for the treatment of NPC.Citation38,39
The limited patient's material (tissue samples to extract RNA and blood to generate MoDC) availability often limits this vaccination approach. The tumor lysates prepared from established C666.1 cell lines was utilized in clinical trial that overcomes the limitations.Citation40 The efficiency of various TAAgs transfected by cell lysates is not clearly understood. Thus, DCs transfected with defined or total tumor-derived RNA can be used as therapeutic vaccine for NPC treatment. Moreover, TAAgs can be quantified using available surrogate marker RNA species expressed by the tumor cells.Citation25 A recent clinical study of RCC patients immunized with DCs transfected with autologous tumor-derived RNA showed the minimum adverse effects post-immunization.Citation41 The iMoDCs were incubated with naked RNA on the day of administration, which showed limited transfection of DCs. The iDCs induced tolerance.Citation42,43 has accomplished MoDCs transfected tumor RNA an amenable strategy. Several methods including electroporation and lipofection to achieving significant RNA delivery to DCs have been reportedCitation20-25,44,45 However, optimized RNA transfection method in which DCs elicit significant T-cell responses in vivo has yet to be established. Although, electroporation has been shown to yield higher percentage of DCs expressing protein of interest, protein expression on per cell basis were seen better in lipofected DCs.Citation25
Here, we transfected DCs with C666–1-derived total RNA via liposomes prepared from cationic lipids (DOTAP/DOTMA). The cationic lipid reagent (DMRIE-C) was chosen due to efficient transfer of RNA and that too with minimal toxicity rendered by transfected DCs. We report to achieve transfection rate of up to 25–40% with 85–90% viability, when iDCs were transfected with EGFP labeled mRNA, and maturation was achieved using a cytokine cocktail. We based on earlier reports of mRNA transfection to DCs analyzed the activation of transfected DCs. We did not observe any negative effect of lipid and RNA on maturation of DC when liposome mediated transfection of DCs was conducted. However, when mature DCs transfected with RNA from immunogenic patient cells C666–1, there would be insufficient transfection as well as lower expression of MHC which in turn leads to the sub-optimal immune response against pathogen. This observation suggests that pro-inflammatory cytokine cocktail is indeed necessary to induce distinct mature DC phenotype. Also, cytokine production by RNA transfected DCs was seen normal. The DC phenotype or cytokine secretion levels were seen unaffected by the liposomes mediated delivery of RNA.
To achieve effective T-cell responses in patients, ex-vivo generated DCs should be competent to re-activate existing effector-memory CTLs, and to prime specific freshly generated T-cells. The anti-tumor CTL responses often detected in NPC patients,Citation46 elicits T-cell responses are not sufficient enough to control disease. It is explained because of the late developing general immune suppression or defective antigen-presenting cells.Citation47-50 The stimulation of memory T cells by antigen-loaded DCs generated ex-vivo might overcome the said limitations, and mount tumor-specific T cell immune responses in patients to address NPC. We used tumor-specific T cell clone (TIL-CMc23) specific for TAAg expressed by C666–1 cells to test the capability of RNA loaded DCs in reactivating specific CTLs.Citation27 Our results suggest the potent reactivation of competent TIL cells by DCs lipofected with C666–1 derived RNA. DCs transfected with RNA derived from C15 cells (not expressing TIL-CMc23 epitope), did not show an activation of TIL-CMc23 cells. These results demonstrates the epitope specificity for the mounted immune response. It is therefore observed that RNA-transfected DCs showed variation in their re-stimulating capacity of TIL-CMc23 cells which is weaker than that seen with C666–1 tumor cells. The short half-life of RNA in DCs might limit MHC presentation of peptide, but variation in ligand reconstitution of DCs may generate newer transcripts. This may provide a continuous supply of peptides for MHC presentation.
Our results showed functional characterization of RNA loaded DCs in their capacity to prime specific T cells. DCs transfected with liposome mediated RNA derived from C666–1 or C15 cells were shown to be able to induce specific T-cell responses against the corresponding tumorous cells. Although in vitro-primed T cells showed some non-MHC-restricted activity directed against NK-sensitive K562 or LAK-sensitive Daudi cells, robust responses were seen against the specific tumor cell lines whose RNA was used for priming of T cells. Therefore, need of sizeable DCs for potent APCs is critical during generation and characterization of specific T-cells. The autologous LCLs transfected with NPC derived RNA were substituted as APCs for re-stimulation during generation of C666–1 specific T cell lines. It showed that RNA-transfected LCLs would efficiently provide pMHC desired ligands and could be an alternative to RNA-loaded DCs. We, however, believe that LCLs might replace DCs only when specific-T cell responses were developed to refrain EBV-specific T cells from being cultivated. The autologous LCL may provide a tool to elucidate T-cell specificity. It is the decisive point in assessing the immune response provoked against a tumor RNA based generic vaccine. Further, RNA loaded DCs may allow epitopes to be presented in both class I and class II MHC molecules, and clinical trials have reportedly shown T-cell responses were detected against onco-fetal antigen,Citation41,51 human telomerase reverse transcriptase (hTERT) & G250/CA-IX antigensCitation41 following DC vaccination. Transfection of LCLs with mRNA encoding the specific TAAgs could be useful to evaluate specific T cell responses.
Therefore, results obtained in this study will be useful for designing a clinical trial based on RNA-loaded DCs. RNA extracted from these GMP-certified C666–1 cells encode an array of TAAgs present in tumor cells. We saw overexpression of both vaccine variants of oncofetal antigen (PRAMlE), adipophilin and hTERT-RNAs.Citation19 Further, these antigens are seen overexpressed in fresh tumors, and serve as targets for evoking T-cell responses.Citation52-55 Further, we analyzed NPC cell lines C15 and C666.1, and showed that TAP-dependent HLA class I processing pathway is seen intact in NPC and that the tumor cells can be lysed by the specific CTL. Our results are consistent to what was reported earlier with C15 and NPC cell lines derived from a North African patient. Our work suggests that C666.1 cell line maintain the intact Ag-processing function in NPC tumor cells of Chinese origin.
Present study is one of the innovative strategies to develop vaccine intervention against NPC based on RNA (extracted from NP cell lines generated from patients) transfected monocyte derived DCs. This vaccine approach presents a plausible solution for NPC. However, scarce RNA extracted from patients and sufficient antigen presentation through MHC I and II will be some of the key limitations. Besides, transfection of RNA to DCs may vary as entrapment efficiency of cationic liposomes will be the variable that has to be optimized to achieving significant transfection efficiency.
Conclusion
Present study describes DC-based vaccine strategy for NPC patients by using a generic source of RNA derived from a well-characterized tumor cells in vitro. The DCs derived from HLA-A2-positive donors were loaded with tumor cell line-derived RNA mediated by liposome, and shown to reactivate effector-memory CTLs. It was capable of priming T cells to mediate cytotoxicity or secrete cytokine upon contact with NPC cells. The availability of GMP-certified cells, expressing a variety of TAAgs sharing many NPC may allow this strategy to be more easily adapted in clinical settings. Furthermore, the ability to load tumor cell line-derived RNA into autologous LCLs might provide a useful tool in immune monitoring.
This study might be deployed to monitor the tools specific for such molecules that prove useful to establish T-cell responses. Further, it can be used to analyze specific T cell responses to generic DC-based vaccine strategy. Moreover, information regarding specific TAAgs can be used to target immune responses against NPC. In brief, it would be possible to develop specific RNA based DCs vaccine against NPC. This study opens new avenues to develop and implement therapeutic approach to address and treat NPC condition.
Materials and methods
Cell lines and cultures
Lymphoblastoid Cell Lines (LCLs) were generated in vitro by transformation of B cells using the standard EBV isolate B95.8Citation56 and cultured in RPMI-1640 mixed with 10% Fetal Calf Serum (FCS), 2 mM L-glutamine, 100 mg/ml streptomycin, and 100 IU/ml penicillin. The cell lines (C15 and c666.1) were derived from NPC biopsies and sequencing of EBV genome was performed as reported elsewhere.Citation32 C15 was sub-inoculated in nude mice, whereas C666.1 was cultured in vitro using growth medium containing only 1% FCS. Both cells (C15 and c666.1) were HLA typed by PCR-based DNA typing method. All cell lines were cultivated in RPMI-1640 medium supplemented with 12% fetal bovine serum (FBS), 2 mM L-glutamate, 1 mM sodium pyruvate and 1-X non-essential amino acids. The tumor inducing leucocytes cell line was obtained from primary patient's tumor, cultured in RPMI-1640 medium mixed with 7.5% FBS, 7.5% heat-de-complemented human serum, 2mM L-glutamine, 1 mM sodium-pyruvate and 1X none essential amino acids. Tumor-infiltrating lymphocytes (TILs) were re-stimulated every 14 d with irradiation attenuated tumor cells.
Ex-vivo generation of monocyte-derived DCs
The peripheral blood mononuclear cells (PBMCs) were isolated by standard Ficoll (PAN Biotech, Aidenbach, Germany) density gradient centrifugation of cancer patient's peripheral blood and washed 2 times with PBS. The CD14+ cells were purified and selectively enriched by human monocyte enrichment cocktail (RosetteSep) purchased from StemCell Technologies, Canada. CD14+cells were cultured in X-Vivo 15 medium (Lonza, USA) supplemented with 800 U/ml GM-CSF and 10 ng/ml IL-4 (Gemini bioproducts, USA) at 1.5 × 106 cells/ml in 6-well culture plates (TPP, Germany) to generate monocytes derived DCs. iMoDCs The cells were harvested on day 6th or 7th with a cytokine cocktail formulation containing 250 U/ml TNF-α, 20 ng/ml IL-1β (Promocell Bioscience), 1μM PGE2 (Sigma, Germany) and 1000 U/ml IL-6 (R&D Systems, USA) for 48 hr to induce maturation of immature DCs. Cell cultures were supplemented with 800 U/ml GM-CSF and 10 ng/ml IL-4.
Quantitative RT-PCR assay
qRT-PCR of inflammatory mediators and transcription factors involved in different molecular events in nasopharyngeal carcinoma was performed with human C666–1 cells. These cells were maintained in RPMI-1640 medium supplemented with 10% inactivated fetal calf serum (FCS) and 50 μg/ml gentamycin (Invitrogen, USA). Cells were seeded in 96-well plates at 4 × 104 cells per well. After culture for 24 hr, cells were added with 100 nM PMA (Wako, Japan) for the differentiation into macrophages. After culture for 48 hr, cells were incubated with plane liposomes and RNA loaded liposomes for 2 hr, and subsequently stimulated with 10 ng/ml ultrapure lipopolysaccharide (LPS) for 24 hr. The analysis of genes expression in C666–1 cells treated with plane and RNA loaded liposome was performed using reverse transcription PCR (RT-PCR). Total RNA was isolated from cells treated with liposomes loaded with RNA by RNeasy kit. The cDNA was synthesized by reverse-transcription reaction (Applied biosystems). The RT-PCR was performed on TaqMan array fast plates using TaqMan fast universal PCR master mix (2X). The fold regulations in the gene expression were calculated by using 2-ΔΔCt method. The fold regulation of gene TNF-α, IL-6, MMP1, CD80, CD83, CD86, FOXO1, IL-1β, IL-10, BCL2 and BCL2l1 () in culture medium treated with RNA-liposome and liposome and/or RNA were determined relative to their untreated control with custom-designed PCR array plates (Life technologies, USA).
Isolation of total tumor RNA
Total tumor RNA was extracted from human C666–1 and C15 cell lines using Tri Reagent® (Sigma, USA) following the manufacturer's recommendations.
In vitro transcribed mRNA
The in vitro-transcribed (IVT) enhanced green fluorescent protein (EGFP) mRNA was prepared using the vector pGEM4Z/GFP/A64Citation44 which already contained the poly (A) template. Further, in vitro transcription under the control ofT7 promoter was performed using the mMESSAGEm MACHINE™ T7 kit (Ambion, USA). IVT mRNA was purified by using RNeasy Mini Kit (Qiagen, Hilden, Germany) after RNA clean up protocol.
Transfection of DCs
Transfection of DCs with RNA loaded liposomes (Lipo-RNA) was performed by the preparation of liposome using the cationic lipid reagent DOTAP. Immature dendritic cells (iMoDCs) were harvested and re-suspended in OptiMEM®I (Invitrogen) at 1.0 × 106 DCs/ml, and cells were distributed immediately in 24-well plates (0.5 ml/well = 0.5 × 106 iDCs/well). The RNA/lipid solution was prepared by adding DC culture 0.5 ml with Opti-MEM®I and was mixed with 5 μl DOTAP in a polystyrene tube followed by the addition of desired quantity of RNA. In general, 2.5–5.0 μg IVT-RNA or 5.0 μg total tumor RNA were used for the transfection of 0.5 million iMoDCs. The RNA/lipid suspension was transferred immediately to iDCs at 37°C.The transfection solution was removed and replaced by X-VIVO 15 medium supplemented with cytokine cocktail after 4 hr incubation period to stimulate the maturation of DC.
Characterization of mRNA loaded liposomes
The mRNA and liposomes morphology was examined using scanning electron microscopy (SEM) and transmission electronic microscopy (TEM). mRNA loaded liposomes were observed by a Philips/FEI CM10 TEM (Hillsboro, OR) instrument, and images were captured using a Gatan Bioscan Model 792 digital camera system. The particle size and zeta potential determination of RNA-liposomes, particle size, and zeta potentials of plain and RNA loaded liposomes were characterized with a Beckman Coulter Delsa Nano C Particle Analyzer (Brea, CA, USA). Photon correlation spectroscopy was used to determine the particle size by measuring the rate of fluctuations of the laser light intensity scattered by particles as they were diffused through the fluid. mRNA-liposomal sample solutions were dispensed into micro-size cells for the measurement with 100 pinhole set to 20 mm and a refractive index of 1.3328 for 70 continuous sample accumulation times. The electrophoretic light scattering was used to measure the zeta potential by determining the electrophoretic movement of charged particles under specified electric field. The measurements were performed in a disposable flow cell position with 20 accumulations. Each analysis was repeated 3 times to achieve average particle size and zeta potential.
The physicochemical stability of liposomal mRNA was assessed by determining the changes in particle size and zeta potential upon storage of formulations at 4°C for nearly 3 months. The particle size and zeta potential of formulations were measured at 0, 1 and 3 months.
Flow cytometry analysis
The phenotypic characterization of DCs was performed by immunofluorescence staining of dendritic-monocyte differentiation and activation markers. Cells were stained with mouse monoclonal antibodies (mAbs) unconjugated or conjugated with fluorescein isothiocyanate (FITC), or phycoerythrin (PE). The FITC conjugated mAbs used were anti-HLA-DR, anti-CD83 (IgG1, clone HB15e), anti-DC-SIGN (CD209), anti-CD14 (BD PharMingen, San Diego, USA). The PE conjugated mAbs used were: anti-CD86, anti-CD80 (BD PharMingen), anti-CD40 (Beckman Coulter, Germany). The indirect immunofluorescence was performed using monoclonal biotin-conjugated rat-anti-mouse IgM in combination with PE-conjugated streptavidin (BD PharMingen). PE/FITC isotype-matched mouse immunoglobulins were used as experimental negative controls (BD PharMingen). The PBS containing 1% FBS washed cells were incubated with appropriate mAb for 30 min on ice in PBS/1% FBS. Cells were washed once again and fixed in 1% paraformaldehyde, and samples were acquired using a FACS Calibur instrument (BD Bioscience, USA). Data was analyzed using Cell Quest Pro-software. DCs were harvested approximately 24 hr after transfection, washed once and directly analyzed by flow cytometry as well as by molecular analysis through RT-PCR.
Immunofluorescence assay
Surface expression of HLA class I molecules on C666.1 cell lines was measured by incubating viable cells with mAb W6/32 for 30 min on ice. After washing with PBS containing 10% (v/v) normal goat serum (NGS), bound Ab was detected by incubation with FITC-labeled goat anti-mouse IgG (Sigma-Aldrich, St. Louis, MO) for 20 min on ice. After 3 washes in PBS and 10% NGS, immunocytochemical study was performed. Cells were cytospin onto slide for 4 minutes at 1,000 rpm, and were fixed with 4% paraformaldehyde in PBS (10 min at room temperature) followed by 3 times washing in distilled water for 5 minutes each. The cells were incubated in 3% hydrogen peroxide for 10 minutes and were washed twice in distilled water for 5 minutes. Cells were then blocked with 100–400 µl blocking solution (TBST/5% normal goat serum) for 1 hr at room temperature. Further, cells were incubated overnight with 100–400 µl mAb W6/32 diluted in Signal Stain antibody diluent at 4°C. The cells were washed 3 times with wash buffer (TBST) for 5 minutes and cells were covered with 1–3 drops of boost detection reagent in a humidified chamber for 30 min at room temperature. Washing of cells as mentioned-above and 100–400 µl DAB plus substrate was applied for 5–15 minutes to have required staining intensity. After washing with water, cells were counter-stained with hematoxylin following manufacturer's recommendations. Slides were mounted with mounting medium and images were taken under a microscope (Nikon instruments).
Analysis of cytokine expression
The release of cytokine by DCs was estimated using highly sensitive Bio-Plex human cytokine reagent kit (Bio-Rad Laboratories Inc. CA, USA) according to the manufacturer's recommendations. The data was analyzed by using BioRad Array Operation System (BioRad).
T-cell stimulation assay
The stimulated cells were harvested and plated in triplicates in 96-well U-bottomed plates at 1.5 ×104 cells/well in 100 μl T-cell medium. T cells were added in a concentration of 3,000 cells/well (TIL clone) or 1.5 × 104 cells/well (de novo primed T cell lines) in 100 μl yielding a total volume 200 μl/well. The culture supernatants were harvested after 24 hr incubation period at 37°C, and release of interferon-gamma (IFN-γ) was measured by ELISA kit (BD PharMingen).
T-cell priming
The generation of anti-tumor T cell lines was performed using autologous cells derived from blood of healthy volunteers. Monocyte-derived iDCs were transfected with total tumor RNA and maturation was induced for 48 hr using cytokine cocktail. 1.25 × 106 mDCs were incubated with 1 × 106 autologous PBMCs in 2 ml/well in RPMI-1640 medium supplemented with 12% heat inactivated human serum, 2 mM L-glutamine, 1 mM sodium pyruvate and 1X non-essential amino acids. Cells were re-stimulated with autologous DCs transfected with total RNA in 4:1 (PBMC: MoDC) ratio. The second round of re-stimulation was performed by by replacing stimulator DCs every 7 d. IL-7 (Promocell Bioscience), 10 ng/ml and 20 U/ml IL-2 (Proleukine®, Chiron, CA, USA) were mixed with the culture 3 times per week starting on days 12 and 18, respectively. The cytolytic activity of induced CTLs was analyzed on day 5 after last round of re-stimulation is over. The release of IFN-γ following T cell stimulation was assessed by ELISA (BD PharMingen) on day 10 following the last round of re-stimulation.
Western blotting
Cells were lysed by sonication in 10 mM Tris buffer (pH 7), concentration of extracted protein was was determined using a protein assay kit (Richmond, CA). The obtained lysate was diluted in electrophoresis sample buffer, and 50–60 µg protein was loaded per well of discontinuous SDS-PAGE with 7.5% acrylamide resolving gel. The electrophoresed proteins were then transferred onto nitrocellulose/PVDF membrane. The TAP1 and TAP2 proteins were detected using anti-TAP1 and anti-TAP 2monoclonal antibodies following the manufacturer's recommendations.
Disclosure of potential conflicts of interest
The author(s) declare that they have no competing interests.
Acknowledgments
Authors offer their thanks to all the working staff who contributed directly or indirectly to the present work.
Funding
This work was supported by grants from Govt of India and NIH, USA.
References
- Perri F, Bosso D, Buonerba C, Lorenzo GD, Scarpati GD. Locally advanced nasopharyngeal carcinoma: Current and emerging treatment strategies. World 2011; 2:377-83; https://doi.org/http://dx.doi.org/10.5306/wjco.v2.i12.377
- Yoshizaki T, Ito M, Murono S, Wakisaka N, Kondo S, Endo K. Current understanding and management of nasopharyngeal carcinoma. Auris Nasus Larynx 2012; 39:137-44; PMID:21592702; https://doi.org/http://dx.doi.org/10.1016/j.anl.2011.02.012
- Bensouda Y, Kaikani W, Ahbeddou N, Rahhali R, Jabri M, Mrabti H, Boussen H, Errihani H. Treatment for metastatic nasopharyngeal carcinoma. Eur Ann Otorhinolaryngol Head Neck Dis 2011; 128:79-85; PMID:21177151; https://doi.org/http://dx.doi.org/10.1016/j.anorl.2010.10.003
- Spano JP, Busson P, Atlan D, Bourhis J, Pignon JP, Esteban C, Armand JP. Nasopharyngeal carcinomas: an update. Eur J Cancer 2003; 39:2121-35; PMID:14522369; https://doi.org/http://dx.doi.org/10.1016/S0959-8049(03)00367-8
- Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013; 342:1432-3; PMID:24357284; https://doi.org/http://dx.doi.org/10.1126/science.342.6165.1432
- Riether C, Schurch C, Ochsenbein AF. From “magic bullets” to specific cancer immunotherapy. Swiss 2013; 143:w13734.:https://doi.org/10.4414/smw.2013.13734
- Karlitepe A, Ozalp O, Avci CB. New approaches for cancer immunotherapy. Tumour Biol 2015; 36:4075-8; PMID:25934338; https://doi.org/http://dx.doi.org/10.1007/s13277-015-3491-2
- Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 1991; 9:271-96; PMID:1910679; https://doi.org/http://dx.doi.org/10.1146/annurev.iy.09.040191.001415
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998; 392:245-52; PMID:9521319; https://doi.org/http://dx.doi.org/10.1038/32588
- Nestle FO, Banchereau J, Hart D. Dendritic cells: On the move from bench to bedside. Nat Med 2001; 7:761-5; PMID:11433329; https://doi.org/http://dx.doi.org/10.1038/89863
- Kawada M, Ikeda H, Takahashi T, Ishizu A, Ishikura H, Katoh H, Yoshiki T. Vaccination of fusion cells of rat dendritic and carcinoma cells prevents tumor growth in vivo. Int J Cancer 2003; 105:520-6; PMID:12712444; https://doi.org/http://dx.doi.org/10.1002/ijc.11120
- Redchenko IV, Rickinson AB. Accessing Epstein-Barr virus-specific T-cell memory with peptide-loaded dendritic cells. J Virol 1999; 73:334-42; PMID:9847337
- Zitvogel L, Mayordomo JI, Tjandrawan T, DeLeo AB, Clarke MR, Lotze MT, Storkus WJ. Therapy of murine tumors with tumor peptide-pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell 1-associated cytokines. J Exp Med 1996; 183:87-97; PMID:8551248; https://doi.org/http://dx.doi.org/10.1084/jem.183.1.87
- de Vries IJ, Lesterhuis WJ, Barentsz JO, Verdijk P, van Krieken JH, Boerman OC, Oyen WJ, Bonenkamp JJ, Boezeman JB, Adema GJ, et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat Biotechnol 2005; 23:1407-13; PMID:16258544; https://doi.org/http://dx.doi.org/10.1038/nbt1154
- O'Neill DW, Adams S, Bhardwaj N. Manipulating dendritic cell biology for the active immunotherapy of cancer. Blood 2004; 104:2235-46; PMID:15231572; https://doi.org/http://dx.doi.org/10.1182/blood-2003-12-4392
- Gilboa E, Vieweg J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev 2004; 199:251-63; PMID:15233739; https://doi.org/http://dx.doi.org/10.1111/j.0105-2896.2004.00139.x
- Garg NK, Dwivedi P, Prabha P, Tyagi RK. RNA pulsed dendritic cells: an approach for cancer immunotherapy. Vaccine 2013; 31:1141-56; PMID:23306369; https://doi.org/http://dx.doi.org/10.1016/j.vaccine.2012.12.027
- Tyagi RK, Garg NK, Jadon R, Sahu T, Katare OP, Dalai SK, Awasthi A, Marepally SK. Elastic liposome-mediated transdermal immunization enhanced the immunogenicity of P. falciparum surface antigen, MSP-119. Vaccine 2015; 33:4630-8.
- Frankenberger B, Pohla H, Noessner E, Willimsky G, Papier B, Pezzutto A, Kopp J, Oberneder R, Blankenstein T, Schendel DJ. Influence of CD80, interleukin-2, and interleukin-7 expression in human renal cell carcinoma on the expansion, function, and survival of tumor-specific CTLs. Clin Cancer Res 2005; 11:1733-42; PMID:15755994; https://doi.org/http://dx.doi.org/10.1158/1078-0432.CCR-04-1883
- Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001; 98:49-56; PMID:11418462; https://doi.org/http://dx.doi.org/10.1182/blood.V98.1.49
- Heiser A, Coleman D, Dannull J, Yancey D, Maurice MA, Lallas CD, Dahm P, Niedzwiecki D, Gilboa E, Vieweg J. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest 2002; 109:409-17; PMID:11828001; https://doi.org/http://dx.doi.org/10.1172/JCI0214364
- Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, Zhang Y, Van Der Bruggen P, Thielemans K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol 2004; 172:6649-57; PMID:15153480; https://doi.org/http://dx.doi.org/10.4049/jimmunol.172.11.6649
- Liao X, Li Y, Bonini C, Nair S, Gilboa E, Greenberg PD, Yee C. Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high-affinity tumor-reactive cytotoxic T lymphocytes. Mol Ther 2004; 9:757-64; PMID:15120337; https://doi.org/http://dx.doi.org/10.1016/j.ymthe.2004.02.011
- Schaft N, Dorrie J, Thumann P, Beck VE, Muller I, Schultz ES, Kämpgen E, Dieckmann D, Schuler G. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfecting defined RNAs after rather than before maturation. J Immunol 2005; 174:3087-97; PMID:15728524; https://doi.org/http://dx.doi.org/10.4049/jimmunol.174.5.3087
- Javorovic M, Pohla H, Frankenberger B, Wolfel T, Schendel DJ. RNA transfer by electroporation into mature dendritic cells leading to reactivation of effector-memory cytotoxic T lymphocytes: a quantitative analysis. Mol Ther 2005; 12:734-43; PMID:15921959; https://doi.org/http://dx.doi.org/10.1016/j.ymthe.2005.03.034
- Luft T, Jefford M, Luetjens P, Toy T, Hochrein H, Masterman KA, Maliszewski C, Shortman K, Cebon J, Maraskovsky E. Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E(2) regulates the migratory capacity of specific DC subsets. Blood 2002; 100:1362-72; PMID:12149219; https://doi.org/http://dx.doi.org/10.1182/blood-2001-12-0360
- Lee SP, Chan AT, Cheung ST, Thomas WA, CroomCarter D, Dawson CW, Tsai CH, Leung SF, Johnson PJ, Huang DP. CTL control of EBV in nasopharyngeal carcinoma (NPC): EBV-specific CTL responses in the blood and tumors of NPC patients and the antigen-processing function of the tumor cells. J Immunol 2000; 165:573-82; PMID:10861098; https://doi.org/http://dx.doi.org/10.4049/jimmunol.165.1.573
- Pfeffer LM, Eisenkraft BL, Reich NC, Improta T, Baxter G, Daniel-Issakani S, Strulovici B. Transmembrane signaling by interferon alpha involves diacylglycerol production and activation of the epsilon isoform of protein kinase C in Daudi cells. Proc Natl Acad Sci U S A 1991; 88:7988-92; PMID:1832772; https://doi.org/http://dx.doi.org/10.1073/pnas.88.18.7988
- Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 1975; 45:321-34; PMID:163658
- Klein E, Ben-Bassat H, Neumann H, Ralph P, Zeuthen J, Polliack A, Vánky F. Properties of the K562 cell line, derived from a patient with chronic myeloid leukemia. Internat J Cancer 1976; 18:421-31; PMID:789258; https://doi.org/http://dx.doi.org/10.1002/ijc.2910180405
- Andersson LC, Nilsson K, Gahmberg CG. K562–a human erythroleukemic cell line. Internat J Cancer 1979; 23:143-7; PMID:367973; https://doi.org/http://dx.doi.org/10.1002/ijc.2910230202
- Cheung ST, Huang DP, Hui AB, Lo KW, Ko CW, Tsang YS, Wong N, Whitney BM, Lee JC. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int J Cancer 1999; 83:121-6; PMID:10449618; https://doi.org/http://dx.doi.org/10.1002/(SICI)1097-0215(19990924)83:1%3c121::AID-IJC21%3e3.0.CO;2-F
- Sela U, Olds P, Park A, Schlesinger SJ, Steinman RM. Dendritic cells induce antigen-specific regulatory T cells that prevent graft versus host disease and persist in mice. J Exp Med 2011; 208:2489-96; PMID:22084406; https://doi.org/http://dx.doi.org/10.1084/jem.20110466
- Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O'Keeffe M, Bahlo M, Papenfuss A. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 2007; 8:1217-26; PMID:17922015; https://doi.org/http://dx.doi.org/10.1038/ni1522
- Steinman RM, Dhodapkar M. Active immunization against cancer with dendritic cells: the near future. Int J Cancer 2001; 94:459-73; PMID:11745430; https://doi.org/http://dx.doi.org/10.1002/ijc.1503
- Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 2005; 5:296-306; PMID:15803149; https://doi.org/http://dx.doi.org/10.1038/nri1592
- Romani N, Gruner S, Brang D, Kampgen E, Lenz A, Trockenbacher B, Konwalinka G, Fritsch PO, Steinman RM, Schuler G. Proliferating dendritic cell progenitors in human blood. J Exp Med 1994; 180:83-93; PMID:8006603; https://doi.org/http://dx.doi.org/10.1084/jem.180.1.83
- Yang Q, Lin H, Wu S, Lei F, Zhu X, Song L, Hong M, Guo L. Prostate Tumor Overexpressed 1 (PTOV1) is a novel prognostic marker for nasopharyngeal carcinoma progression and poor survival outcomes. PLoS One 2015; 10:e0136448; PMID:26305455; https://doi.org/http://dx.doi.org/10.1371/journal.pone.0136448
- Jiang J, Wu H, Huang M, Wu Y, Wang Q, Zhao J, Yang W, Chen W, Feng Q. Variability of gross tumor volume in nasopharyngeal carcinoma using 11C-Choline and 18F-FDG PET/CT. PLoS One 2015; 10:e0131801; PMID:26161910; https://doi.org/http://dx.doi.org/10.1371/journal.pone.0131801
- Moselhy J, Sarkar S, Chia MC, Mocanu JD, Taulier N, Liu FF, Wu XY. Evaluation of copolymers of N-isopropylacrylamide and 2-dimethyl(aminoethyl)methacrylate in nonviral and adenoviral vectors for gene delivery to nasopharyngeal carcinoma. Int J Nanomedicine 2007; 2:461-78; PMID:18019844
- Su Z, Dannull J, Heiser A, Yancey D, Pruitt S, Madden J, Coleman D, Niedzwiecki D, Gilboa E, Vieweg J. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res 2003; 63:2127-33; PMID:12727829
- Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol 2002; 23:445-9; PMID:12200066; https://doi.org/http://dx.doi.org/10.1016/S1471-4906(02)02281-0
- Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A 2002; 99:351-8; PMID:11773639; https://doi.org/http://dx.doi.org/10.1073/pnas.231606698
- Nair SK, Boczkowski D, Morse M, Cumming RI, Lyerly HK, Gilboa E. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat Biotechnol 1998; 16:364-9; PMID:9555728; https://doi.org/http://dx.doi.org/10.1038/nbt0498-364
- Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med 1996; 184:465-72; PMID:8760800; https://doi.org/http://dx.doi.org/10.1084/jem.184.2.465
- Zhang YL, Li J, Mo HY, Qiu F, Zheng LM, Qian CN, Zeng YX. Different subsets of tumor infiltrating lymphocytes correlate with NPC progression in different ways. Mol Cancer 2010; 9:4; PMID:20064222; https://doi.org/http://dx.doi.org/10.1186/1476-4598-9-4
- Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP, Gabrilovich DI. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res 2000; 6:1755-66; PMID:10815894
- Ahmad M, Rees RC, Ali SA. Escape from immunotherapy: possible mechanisms that influence tumor regression/progression. Cancer Immunol Immunother 2004; 53:844-54; PMID:15197495; https://doi.org/http://dx.doi.org/10.1007/s00262-004-0540-x
- Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother 2004; 27:184-90; PMID:15076135; https://doi.org/http://dx.doi.org/10.1097/00002371-200405000-00002
- Menetrier-Caux C, Montmain G, Dieu MC, Bain C, Favrot MC, Caux C, Blay JY. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood 1998; 92:4778-91; PMID:9845545
- Holtl L, Zelle-Rieser C, Gander H, Papesh C, Ramoner R, Bartsch G, Rogatsch H, Barsoum AL, Coggin JH Jr, Thurnher M. Immunotherapy of metastatic renal cell carcinoma with tumor lysate-pulsed autologous dendritic cells. Clin Cancer Res 2002; 8:3369-76; PMID:12429623
- Grunebach F, Muller MR, Nencioni A, Brossart P. Delivery of tumor-derived RNA for the induction of cytotoxic T-lymphocytes. Gene Ther 2003; 10:367-74; PMID:12601391; https://doi.org/http://dx.doi.org/10.1038/sj.gt.3301901
- Weinschenk T, Gouttefangeas C, Schirle M, Obermayr F, Walter S, Schoor O, Kurek R, Loeser W, Bichler KH, Wernet D, et al. Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer Res 2002; 62:5818-27; PMID:12384544
- Schmidt SM, Schag K, Muller MR, Weinschenk T, Appel S, Schoor O, Weck MM, Grünebach F, Kanz L, Stevanovic S. Induction of adipophilin-specific cytotoxic T lymphocytes using a novel HLA-A2-binding peptide that mediates tumor cell lysis. Cancer Res 2004; 64:1164-70; PMID:14871853; https://doi.org/http://dx.doi.org/10.1158/0008-5472.CAN-03-2538
- Sievers E, Albers P, Schmidt-Wolf IG, Marten A. Telomerase pulsed dendritic cells for immunotherapy for renal cell carcinoma. J Urol 2004; 171:114-9; PMID:14665857; https://doi.org/http://dx.doi.org/10.1097/01.ju.0000094803.60928.d7
- Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Séguin C, et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 1984; 310:207-11; PMID:6087149; https://doi.org/http://dx.doi.org/10.1038/310207a0