
ABSTRACT
Chimeric antigen receptor T cells are T cells genetically engineered with CAR constructs which mainly contain scFV and TCR zeta chain. With promising development in blood cancers, CAR T trials are also applied in solid cancers. However, the treatment effect in solid cancers is lower than expected. This review summarizes difference of CAR T applications in solid and blood cancers. Future challenges of CAR T cell treatment in solid cancer are also discussed using ovarian cancer as an example.
Cancer immunotherapy which harnesses human immune system to attack tumor has become popular in recent years as a treatment modality. This is mainly due to 2 remarkable breakthroughs in the field: PD1 antibody as a representative checkpoint inhibitor in solid cancers, and chimeric antigen receptor T cell (CAR T) treatment in blood cancers. To date, numerous clinical trials, 355 PD1 blockade and 168 CAR T respectively have been registered around the world. (www.clinicaltrials.gov). CAR T treatment have been tried extensively in solid cancers, but the results were disappointing. In this review, we focus on our understanding of CAR T basics and address its challenges in solid cancers using ovarian cancer as an example.
In CAR T system, the key component is CAR construct. The main structure of CAR contains a single chain fraction variable (scFv) and a signaling pathway molecule, TCR zeta chain. The scFv contains an antigen binding site of heavy and light chain of antibody through a linker. The expression of scFv on T cells recognizes antigens on target cells, and TCR zeta chain transfers signal to activate T cells.
The “puzzle” consisting of antibody and TCR components makes it unique. It recognizes the specific antigen through scFv, and the recognition signal passes through TCR zeta chain to activate T cells. Since the recognition is antibody dependent, MHC-based restriction is bypassed for T cell activation. This dominant feature renders non-relevant CD8 T cells to kill cancer cells. This overcomes the barriers to expand very few tumor specific T cells for immunotherapy. When CAR T is activated, it secretes cytokines and expresses FASL and TRAIL on the cell membrane. CAR T also kills cancer cells through secretion of perforin and granzyme B which is similar to T cells through TCR activation.
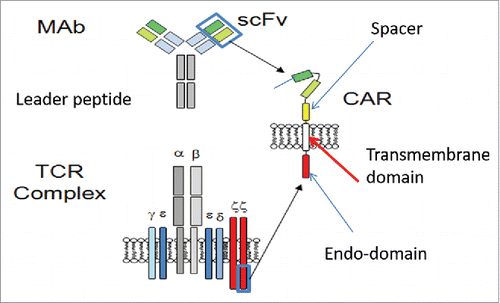
CAR T architecture consists of 5 parts ().Citation1 Leader peptide is the first part which causes CAR protein expression on T cell membrane after secretion from Golgi complex. The second part, scFv, is made mainly from a variable region of heavy and light chain. As mentioned above, its main function is to recognize antigen. Next to scFv is the spacer which gives the heavy and light chain enough space to fold, a prerequisite for antigen binding. Transmembrane segment (fourth part) links the spacer and endo-domain, fifth part, which is usually TCR zeta chain. The function of zeta chain is to transfer stimulating signal to activate T cells. The details of these can be found elsewhere.Citation1,2
Figure 1. Basic structure of CAR. The main parts of CAR are from monoclonal antibody scFv and endo-domain, TCR zeta chain. ScFv is the antigen binding site, and TCR zeta chain is responsible to transfer signal to activate T cell. Other important parts are leader peptide, spacer area, and transmembrane domain.
The seminal feature of CAR T technology (avoids MHC-based restriction) makes it promising in clinical trials. In 2014, scientists reported a CAR T phase I clinical trial at the American Society of Hematology (ASH) meeting. Their product JCAR015, a CD19 chimeric antigen receptor (CAR) T-cell, brought 24 of 27 adults with refractive acute lymphoblastic leukemia (ALL) into remission, and 6 patients remained disease free for more than a year (ASH 2014, Abstract 382). It is worth pointing out this unprecedented response rate was acquired in terminal stage patients with no other treatments. Even though this promising development was observed in blood cancers,Citation3,4,5 clinical trials of CAR T from solid cancer were not that successful,Citation6,7,8 and we list the possible reasons in .
Table 1. Difference of CAR T settings between solid and blood cancers.
Four generations of CAR T: Classification based on the structure
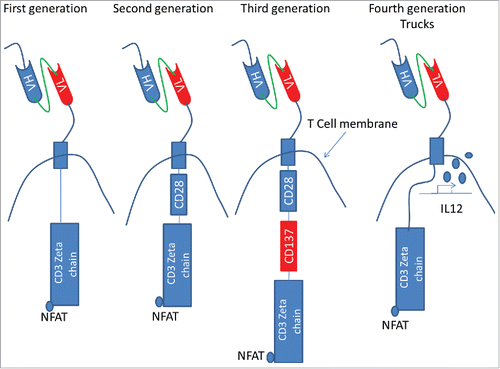
CAR T technology has progressed through 4 generations in the past 30 y. The first generation was developed in 1989 when scientists from a research group in Israel first reported the CAR T experiment. They genetically linked scFv and CD3 zeta chain together and expressed in T cells ().Citation9 Since such CAR T does not respond to antigen stimulation frequently, researchers added a co-stimulatory molecule usually CD28 in the endo-domain (second generation). This design increased CAR T response to antigen, but also increased cell apoptosis.Citation10 To increase survival time after stimulation, researchers developed third generation by incorporating a “survival” molecule such as CD137 (41BB) which is the most frequent molecule to be used. Studies have shown CD137 increases stimulatory signal, generates central memory T cells, and increases survival rate of CAR T cells after antigen stimulation.Citation11,12 Indeed, CD19 CAR T with CD28 and CD137 endo-domains could survive in vivo for 3 years, and proliferate up to 1000 times.Citation13 Other costimulatory markers such as CD27,Citation14 OX40,Citation15 and ICOSCitation16 are also frequently explored with CD28 combination. To further facilitate CAR T activation in tumor microenvironment, researchers transferred cytokine genes into CAR T design which is now called “TRUCK”.Citation17,18,19 It is more complicated and difficult to classify CAR T these days because new forms of CAR T, like bi-specific,Citation20 and Tri-specificCitation21 CAR are developed.
Figure 2. The main difference of four generations of CAR T. The first three generations of CAR T are classified mainly based on the number of co-stimulatory molecules in the intracellular part. CD27, CD28, CD134, CD137, OX-40 are usually the candidates for the molecules. The fourth generation CAR T has cytokine gene together with whatever the endodomain is. The cytokine gene is activated when CAR signal activates T cells. The figure was referred from refs. Citation17 and Citation19.
DNA vs RNA CAR T: Classification based on construct expression
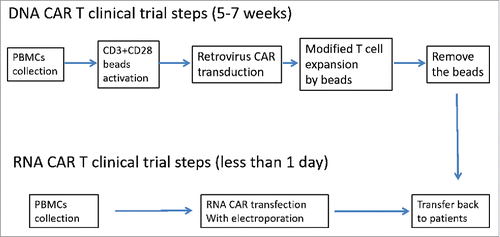
Though not so frequent, there is another CAR T classification as well which is based on DNA vs RNA.Citation22,23 As of today, most CAR T is DNA based. However, RNA based CAR T have increased in recent years. In DNA CAR, the sequence is transferred through retro- or lenti-virus which usually get into nucleus, and integrates into host genome. In contrast, RNA CAR sequence only reaches cytoplasm. Due to enzyme degradation, RNA CAR expression would gradually disappear over time. The advantages and disadvantages of DNA and RNA CAR T are thus decided by this difference ().Citation22,23,24,25
Table 2. Advantage and disadvantage of DNA and RNA CAR Ts.
RNA CAR T has several advantages in clinics in comparison with DNA CAR T. The main advantage of RNA CAR T is less “off tumor” toxicities since the expression of CAR is self-limited. The other advantage of RNA CAR is that it is easier to make in the lab. After integrating a CAR sequence into plasmid, an in vitro transformation system to generate capped/tailed IVT RNA is needed.Citation22,23 It is simpler and cheaper to apply RNA CAR T in clinical trials. The main step is electroporation after mixing the plasmid with blood cells,Citation25 and it takes less than a day to complete the whole treatment (). Furthermore, we have tried freezing CAR T cells after transfection. These cells were used directly for the repeated injection. In line with the other study,Citation26 the frozen cells are fully functional. This simplifies the clinical use of RNA CAR T and further decreases the cost. In contrast, DNA CAR plasmid needs to be packed into lentivirus which is very laborious and unsure. In addition, it usually takes 5–7 weeks to expand DNA CAR T cells in vitro for treatments. This in vitro process is very expensive in terms of contamination prevention. Even worse, all the steps in vitro are empiric, and there is no guarantee that these steps will produce high quality CAR T cells.Citation2,17
Figure 3. Clinical process of DNA and RNA CAR T. DNA CAR T needs more steps to prepare before transferring back to patients. The whole process takes 5–7 weeks. RNA CAR T preparation is much simpler with electroporation; and expansion step can be skipped to avoid in vitro contamination. The whole process can be done in one day.
CAR T in solid tumors—Advantages for ovarian tumors
Despite the disappointment of CAR-T treatment in numerous solid cancers, CAR T still provides a new perspective to treat ovarian cancer. Ovarian cancer is one of the most important cancers in female population. In terms of incidence, it ranks 6th among all cancers in the world. It is the primary reason for morbidity and mortality in female related cancers. Majority of patients (75%) are in the mid or late stage during diagnosis. The main therapy is tumor reduction surgery and chemotherapy. In the past 20 y, despite improvement of surgical techniques and chemotherapy protocols, 5 y survival rate hovers at 30% with little improvement.Citation27 There is a need to find an alternative way to treat the cancer.
To our knowledge, the first CAR T study in ovarian cancer was by Hwu and Rosenberg.Citation28 At the time they genetically expressed a scFV against folate binding protein (MOV18) on tumor infiltrating cells (TILs) from ovarian cancer tissue.Citation28 This modified TILs lysed ovarian cancer cells and opened a new window of opportunity to explore CAR T immunotherapy against cancers. With much effort of several other groups, the achievement of ovarian cancer CAR T studies is at the forefront in CAR T of solid cancers.
There are 2 advantages of studying CAR T to treat ovarian cancers. First, ovarian cancer cell expresses large number of different tumor associated antigens.Citation29 Except CA125, the cancer cell expresses over 60 tumor associated antigens.Citation30 This provides number of available targets for CAR T design. To date, there are 4 clinical trials of CAR T against antigens such as MUC16,Citation31 NY-ESO-1 (NCT01967823), MAGE-A3 (NCT02111850) , and Mesothelin (NCT01583686). Other antigens such as folate receptors,Citation6,32 BH6,Citation33 Her2,Citation34 mesothelinCitation35,36 and follicle-stimulating hormone receptorCitation37 are ongoing in preclinical studies. The second advantage is that CAR T delivery can be applied through intraperitoneal injection (IP) instead of intravenous injection (IV). This is because peritoneal cavity is the main metastasis region of ovarian cancer. It has been proved that chemotherapy reagents through IP has better therapeutic effect than traditional IV.Citation38 The reason could be the direct contact of chemo-drug and cancer cells. In the same principle, IP delivery might also make CAR T cells contact tumor cells directly,Citation39 and avoids homing process through IV delivery. This unique feature provides chance to study effective CAR T delivery and its mechanisms.
CAR T challenges in ovarian tumors
The first challenge of CAR T is side effects. CAR T therapy has been reported to associate with cytokine release syndrome, neurologic toxicity, and anaphylaxis.Citation40 It was not expected that a cancer patient had anaphylaxis to mesothelin RNA CAR T, which is the most likely IgE mediated response to murine sequence of the CAR because of repeated infusions.Citation26 This work indicates that protocols related to the prevention of such anaphylaxis should be scheduled in clinical trials.
The second challenge of CAR T in solid tumor is on target/off tumor toxicities. This side effect is related to how antigen is expressed in normal tissues in vivo. We have been involved in study of 3rd generation CAR T against mesothelin on ovarian cancer cells. Mesothelin is a glycoprotein expressed on ovarian cancer with 67–100%.Citation41 In normal tissues, mesothelin is only expressed in pleura, pericardium and peritoneum. Such distribution makes mesothelin a very specific tumor target, and the side effect could be related to cardiopulmonary membrane cytotoxicity only. As mentioned above, RNA CAR T is believed to have an advantage on target/off tumor toxicities since CAR expression disappears over time.
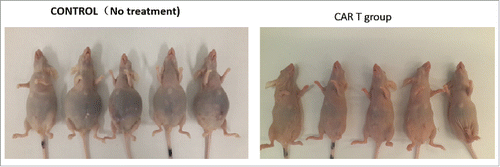
The central challenge is that CAR T is unable to penetrate into solid tumor tissues. This might result from either lacking cytokine or chemokine receptors that CAR T cannot handle homing issue or tumor tissue is too solid to penetrate. To tackle the first issue, we delivered mesothelin RNA CAR T through IP injection in peritoneal metastasis ovarian cancer model. By this way, we believe CAR –T cells was able to “see” the tumor cells directly so homing issue was bypassed. The local injection had dramatic anti-tumor effect in mouse model. showed that CAR T treatment group mice were still healthy after 2 wks, while non-treatment group had to be sacrificed due to high level of ascites. In contrast, IV injection seemed not to work at all (result not shown). Since these mice only had peritoneal tumor, the result clearly indicates CAR T cells injected through vessel could not reach the tumor tissue in peritoneal cavity. This was consistent with a literature report that, in retrovirus transduced α-folate receptor antibody CAR clinical trial, CAR T cells could be tracked in 2 d after injection in 14 ovarian cancer patients.Citation6 However, CAR T cells could not be “seen” in vivo after a month of injection, and no anti-tumor response was observed in these patients.Citation6 So homing issue must be considered when designing CAR-T for solid tumors. Combination of chemotherapy or radiotherapy could be a way to solve homing issues for the time being which we have addressed under the “perspective on CAR T clinical trial of solid tumors” later on. Except this, we still need creative ways to solve this problem in solid tumor therapies.
Figure 4. CAR T treatment in ovarian cancer model (unpublished data). Ovarian cancer cell line ID8 cells (5 × 104) were injected to nude mice through IP injection. A single intra-peritoneal (IP) injection of CAR T (1 × 107) 3 d after cancer cells injection. On day 7, CAR T group mice looked normal and no treatment group showed significant ascites.
The challenge of IP injection is that we need to know how frequent the injection should be in the case of multiple injections. As per our study, one single injection could induce 3–4 weeks durable anti-tumor response. Repeated injections on 3 continuous days induced longer durable anti-tumor response. Since the CAR expression disappeared a week after transfection, we believe that injection once a week might be the optimal condition. Indeed, the injection once a week for 3 weeks also gives better tumor control than the single injection group.
Cell number is also a challenge for treatment. In our mouse study, we needed large number (1 × 108) of CAR T cells for a single treatment which meant more cells will be needed in humans. Indeed, 1 × 1010 CAR T cells were applied in a clinical trial.Citation6 Such many cells could not be easy to get in clinic since cells must be from autologous and cancer patients usually have lower number of white blood cells in their blood. To solve the problem, we did an in vitro activation to increase T cell function so that we could decrease the injection of cell number. It is known that CD3 plus CD28 stimulation on T cells could activate T cells efficiently. By using CD3 plus CD28 dynabead stimulation overnight, we showed that using 10 times less CAR T number can have the same effect to control ovarian tumors in mice. However, the tumors in such situation quickly relapsed (1–2 weeks). Our limited data indicated this might be the phenomenon of activation induced cell death (AICD).Citation42,43
There was no systematic data from clinical trials and the reason as to how exactly AICD affects treatment. So overcoming CAR T AICD could be a big challenge. First, CAR T AICD is from its intrinsic design and repetitive activation. Data have shown that spacer and endo domain area could cause AICD in vitro, and 3rd generation CAR T (better response magnitude) is easier to have AICD than its 2nd generation CAR T cells.Citation42 Repetitive stimulation causes CAR T exhaustion, and studies have shown that PD1 blockade could save AICD of CAR T.Citation43 Second, it has been known CAR T cells had increased expression of FAS/FASL after activation in vitro.Citation42 Such FAS/FASL upregulation and interaction increases AICD.Citation42 Thus, in addition to PD1 blockade,Citation43 blocking FAS/FASL interaction could decrease AICD and increase CAR T function. However, using antibody in this situation is not a good choice. Our experience in blocking PD1 and CTLA4 in vivo showed huge amount of antibody is needed every 2–3 daysCitation44 which could cause significant side effects in practice. Furthermore, blocking FAS could hinder the killing effects since tumor cells also express FAS. On the other hand, FASL blocking by antibody could also decrease the killing effect of CAR-T since FAS L is one of the important weapons for T cells to exert killing.Citation43
There are some ideas about how to solve AICD recently. A group expressed 2 second generation CARs on T cells, which contains CD28 and CD137 respectively. Such arrangement makes the CAR T survive better with less AICD.Citation45 Apart from this, CRISPR/CAS9 technique provides the chance to knock out FAS gene in mammalian cells.Citation46,47 Since siRNA technique already improved the AICD after blocking FAS gene,Citation48 we believe effective delivery of the sgRNA/CAS9 to human CAR T is able to solve the AICD.
Perspective on CAR T clinical trial of solid tumors
CAR T therapy combined with other treatment modalities such as chemotherapy and radiotherapy should be tried in clinical trials. Current clinical trials of CAR T are mainly limited to terminal stage patients with no other treatments. Chemotherapies and radiotherapies are double edged sword for patients as they suppress CAR T function. On the other hand, such treatments might create more antigen exposure on cancer cellsCitation49 and thus provides more available targets for CAR T to attack. So, it is valuable to look into CAR T treatment against cancers at earlier stage with chemo-and radiotherapies.
CAR T therapy combined with PD1 blockade should also be investigated in clinical trials. It is known that CAR T increased PD1 expression after activation.Citation43 PD1 blockade could increase survival time of T cells.Citation43 We believe CAR T combined PD1 blockade would give more positive treatment effect on cancers.
In terms of RNA CAR T clinical trials, low efficiency of transfection by electroporation on large scale of T cells needs to be solved and this process needs to be cGMP-compliant. We have used closed flow microfluidic electroporation for 100 million cells. The transfection process can be done in half an hour, and the efficiency of RNA CAR transfection could reach 95% in 24 hours (manuscripts submitted). Such kind of system is very promising in clinical trials.
Conclusions and future directions
CAR T has emerged as a potential cancer therapy method and opens a very imaginative window for researchers to find a way to cure cancer. Adoptive transfer of CD19-targeted CAR-T has resulted in remarkable responses in blood cancer patients. Some recent studies showed IV delivery of CAR T was also effective to control solid tumors.Citation24,50 However, many other studies and trialsCitation51,52,53,54,55 in solid cancers including ours showed that inability of CAR T to penetrate into tumor tissues is the main problem. IP delivery of CAR T is only available in cancers which metastasize to peritoneal cavity. For most other cancers, including ovarian and cervical cancers, local delivery (intra-tumor) might be straightforward though this is not the best way. Modification of CAR T with cytokine and chemokine receptor expression is a foreseeable solution. In general, we still need to find more imaginative ways to solve the problem.
Except the central challenge, we believe FAS/FASL induced AICD is an important factor to affect CAR T therapies. Solving this problem could significantly decrease T cell number used in clinics.
All these mentioned above are not only challenges in ovarian cancers, but also the challenges for all CAR T studies in solid cancers.
Disclosure of potential conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev 2014; 257:107-26. ISSN 1600-065X. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/24329793; PMID:24329793; https://doi.org/10.1111/imr.12131
- Geyer MB, Brentjens RJ. Review: Current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy 2016; 18(11):1393-1409. ISSN 1477-2566. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27592405; PMID:27592405
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507-17. ISSN 1533-4406. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25317870; PMID:25317870; https://doi.org/10.1056/NEJMoa1407222
- Chen R, Song XT, Chen B. CD19 chimeric antigen receptor T cell therapy for the treatment of B cell lineage acute lymphoblastic leukemia. Discov Med 2015; 20:185-90. ISSN 1944-7930. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26562471; PMID:26562471
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509-18. ISSN 1533-4406. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/23527958; PMID:23527958; https://doi.org/10.1056/NEJMoa1215134
- Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006; 12:6106-15. ISSN 1078-0432. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/17062687; PMID:17062687; https://doi.org/10.1158/1078-0432.CCR-06-1183
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011; 118:6050-6. ISSN 1528-0020. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/21984804; PMID:21984804; https://doi.org/10.1182/blood-2011-05-354449
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013; 21:904-12. ISSN 1525-0024. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/23423337; PMID:23423337; https://doi.org/10.1038/mt.2013.17
- Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989; 86:10024-8. ISSN 0027-8424. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/2513569 >
- Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J 2014; 20:127-33, ISSN 1540-336X. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/24667958; PMID:24667958; https://doi.org/10.1097/PPO.0000000000000034
- Makkouk A, Chester C, Kohrt HE. Rationale for anti-CD137 cancer immunotherapy. Eur J Cancer 2016; 54:112-9. ISSN 1879-0852. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26751393; PMID:26751393; https://doi.org/10.1016/j.ejca.2015.09.026
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21:581-90. ISSN 1546-170X. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25939063; PMID:25939063; https://doi.org/10.1038/nm.3838
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3:95ra73. ISSN 1946-6242. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/21832238; PMID:21832238; https://doi.org/10.1126/scitranslmed.3002842
- Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ, Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012; 119:696-706. ISSN 1528-0020. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/22117050; PMID:22117050; https://doi.org/10.1182/blood-2011-03-344275
- Hombach AA, Abken H. Of chimeric antigen receptors and antibodies: OX40 and 41BB costimulation sharpen up T cell-based immunotherapy of cancer. Immunotherapy 2013; 5:677-81. ISSN 1750-7448. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/23829616; PMID:23829616; https://doi.org/10.2217/imt.13.54
- Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, Lee J, Posey AD, Jr, Scholler J, Scholler N, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014; 124:1070-80. ISSN 1528-0020. Disponível em: <https://www.ncbi.nlm.nih.gov/pubmed/24986688; PMID:24986688; https://doi.org/10.1182/blood-2013-10-535245
- Cheadle EJ, Gornall H, Baldan V, Hanson V, Hawkins RE, Gilham DE. CAR T cells: driving the road from the laboratory to the clinic. Immunol Rev 2014; 257:91-106. ISSN 1600-065X. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/24329792; PMID:24329792; https://doi.org/10.1111/imr.12126
- Chmielewski M, Abken H. Trucks: the fourth generation of CARs. Expert Opin Biol Ther 2015; 15:1145-54. ISSN 1744-7682. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25985798; PMID:25985798; https://doi.org/10.1517/14712598.2015.1046430
- Smith AJ, Oertle J, Warren D, Prato D. Chimeric antigen receptor (CAR) T cell therapy for malignant cancers: Summary and perspective. Journal of Cellular Immunotherapy 2016; 2(2):59-68; https://doi.org/10.1016/j.jocit.2016.08.001
- Kobold S, Steffen J, Chaloupka M, Grassmann S, Henkel J, Castoldi R, Zeng Y, Chmielewski M, Schmollinger JC, Schnurr M, et al. Selective bispecific T cell recruiting antibody and antitumor activity of adoptive T cell transfer. J Natl Cancer Inst 2015; 107:364. ISSN 1460-2105. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25424197; PMID:25424197; https://doi.org/10.1093/jnci/dju364
- Zhen A, Kamata M, Rezek V, Rick J, Levin B, Kasparian S, Chen IS, Yang OO, Zack JA, Kitchen SG. HIV-specific immunity derived from chimeric antigen receptor-engineered stem cells. Mol Ther 2015; 23:1358-67. ISSN 1525-0024. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26050990; PMID:26050990; https://doi.org/10.1038/mt.2015.102
- Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 2013; 24:717-27. ISSN 1557-7422. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/23883116; PMID:23883116; https://doi.org/10.1089/hum.2013.075
- Schutsky K, Song DG, Lynn R, Smith JB, Poussin M, Figini M, Zhao Y, Powell DJ, Jr. Rigorous optimization and validation of potent RNA CAR T cell therapy for the treatment of common epithelial cancers expressing folate receptor. Oncotarget 2015; 6:28911-28. ISSN 1949-2553. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26359629; PMID:26359629
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014; 2:112-20. ISSN 2326-6074. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/24579088; PMID:24579088; https://doi.org/10.1158/2326-6066.CIR-13-0170
- Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 2010; 70:9053-61. ISSN 1538-7445. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/20926399; PMID:20926399; https://doi.org/10.1158/0008-5472.CAN-10-2880
- Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 2013; 1:26-31. ISSN 2326-6074. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/24777247; https://doi.org/10.1158/2326-6066.CIR-13-0006
- Baldwin LA, Huang B, Miller RW, Tucker T, Goodrich ST, Podzielinski I, DeSimone CP, Ueland FR, van Nagell JR, Seamon LG. Ten-year relative survival for epithelial ovarian cancer. Obstet Gynecol 2012; 120:612-8. ISSN 1873-233X. Disponível em: http://www.ncbi.nlm.nih.gov/pubmed/22914471; PMID:22914471; https://doi.org/10.1097/AOG.0b013e318264f794
- Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, Rosenberg SA, Eshhar Z. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med 1993; 178:361-6. ISSN 0022-1007. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/8315392; PMID:8315392; https://doi.org/10.1084/jem.178.1.361
- Rein BJ, Gupta S, Dada R, Safi J, Michener C, Agarwal A. Potential markers for detection and monitoring of ovarian cancer. J Oncol 2011; 2011:475983. ISSN 1687-8469. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/21577260; PMID:21577260; https://doi.org/10.1155/2011/475983
- Chatterjee M, Mohapatra S, Ionan A, Bawa G, Ali-Fehmi R, Wang X, Nowak J, Ye B, Nahhas FA, Lu K, et al. Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res 2006; 66:1181-90. ISSN 0008-5472. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/16424057; PMID:16424057; https://doi.org/10.1158/0008-5472.CAN-04-2962
- Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 2015; 13:102. ISSN 1479-5876. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25890361; PMID:25890361; https://doi.org/10.1186/s12967-015-0460-x
- Kandalaft LE, Powell DJ, Coukos G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J Transl Med 2012; 10:157. ISSN 1479-5876. Disponível em: http://www.ncbi.nlm.nih.gov/pubmed/22863016; PMID:22863016; https://doi.org/10.1186/1479-5876-10-157
- Wu MR, Zhang T, DeMars LR, Sentman CL. B7H6-specific chimeric antigen receptors lead to tumor elimination and host antitumor immunity. Gene Ther 2015; 22:675-84. ISSN 1476-5462. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25830550; PMID:25830550; https://doi.org/10.1038/gt.2015.29
- Lanitis E, Dangaj D, Hagemann IS, Song DG, Best A, Sandaltzopoulos R, Coukos G, Powell DJ, Jr. Primary human ovarian epithelial cancer cells broadly express HER2 at immunologically-detectable levels. PLoS One 2012; 7:e49829. ISSN 1932-6203. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/23189165; PMID:23189165; https://doi.org/10.1371/journal.pone.0049829
- Mayor M, Zeltsman M, McGee E, Adusumilli PS. A regional approach for CAR T-cell therapy for mesothelioma: from mouse models to clinical trial. Immunotherapy 2016; 8:491-4. ISSN 1750-7448. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27140404; PMID:27140404; https://doi.org/10.2217/imt-2016-0008
- O'Hara M, Stashwick C, Haas AR, Tanyi JL. Mesothelin as a target for chimeric antigen receptor-modified T cells as anticancer therapy. Immunotherapy 2016; 8:449-60. ISSN 1750-7448. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26973126; PMID:26973126; https://doi.org/10.2217/imt.16.4
- Urbanska K, Stashwick C, Poussin M, Powell DJ, Jr. Follicle-Stimulating Hormone Receptor as a Target in the Redirected T-cell Therapy for Cancer. Cancer Immunol Res 2015; 3:1130-7. ISSN 2326-6074. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26112923; PMID:26112923; https://doi.org/10.1158/2326-6066.CIR-15-0047
- Mackay HJ, Provencheur D, Heywood M, Tu D, Eisenhauer EA, Oza AM, Meyer R. Phase ii/iii study of intraperitoneal chemotherapy after neoadjuvant chemotherapy for ovarian cancer: ncic ctg ov.21. Curr Oncol 2011; 18:84-90. ISSN 1718-7729. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/21505599; PMID:21505599; https://doi.org/10.3747/co.v18i2.725
- Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med 2014; 6:261ra151. ISSN 1946-6242. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25378643; PMID:25378643; https://doi.org/10.1126/scitranslmed.3010162
- Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 2016; 3:16011. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27626062; PMID:27626062; https://doi.org/10.1038/mto.2016.11
- Hassan R, Laszik ZG, Lerner M, Raffeld M, Postier R, Brackett D. Mesothelin is overexpressed in pancreaticobiliary adenocarcinomas but not in normal pancreas and chronic pancreatitis. Am J Clin Pathol 2005; 124:838-45. ISSN 0002-9173. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/16416732; PMID:16416732; https://doi.org/10.1309/F1B64CL7H8VJKEAF
- Künkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, Jensen MC. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated Due to Cell Fas-FasL-Dependent AICD. Cancer Immunol Res 2015; 3:368-79. ISSN 2326-6074. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25576337; PMID:25576337; https://doi.org/10.1158/2326-6066.CIR-14-0200
- Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID, Brenner MK, Brown MP. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 Blockade. Mol Ther 2016; 24:1135-49. ISSN 1525-0024. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27019998; PMID:27019998; https://doi.org/10.1038/mt.2016.63
- Ma SD, Xu X, Jones R, Delecluse HJ, Zumwalde NA, Sharma A, Gumperz JE, Kenney SC. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog 2016; 12:e1005642. ISSN 1553-7374. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27186886; PMID:27186886; https://doi.org/10.1371/journal.ppat.1005642
- Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol 2009; 183:5563-74. ISSN 1550-6606. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/19843940; PMID:19843940; https://doi.org/10.4049/jimmunol.0900447
- Liang WC, Liang PP, Wong CW, Ng TB, Huang JJ, Zhang JF, Waye MM, Fu WM. Crispr/Cas9 technology targeting FAS gene protects mice from Concanavalin-A induced fulminant hepatic failure. J Cell Biochem 2016; https://www.ncbi.nlm.nih.gov/pubmed/27585307
- Chu VT, Graf R, Wirtz T, Weber T, Favret J, Li X, Petsch K, Tran NT, Sieweke MH, Berek C, et al. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc Natl Acad Sci U S A 2016; 113(44):12514-12519. ISSN 1091-6490. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27729526; PMID:27729526; https://doi.org/10.1073/pnas.1613884113
- Dotti G, Savoldo B, Pule M, Straathof KC, Biagi E, Yvon E, Vigouroux S, Brenner MK, Rooney CM. Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood 2005; 105:4677-84. ISSN 0006-4971. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/15713795; PMID:15713795; https://doi.org/10.1182/blood-2004-08-3337
- Song L, Yang MC, Knoff J, Wu TC, Hung CF. Cancer immunotherapy employing an innovative strategy to enhance CD4+ T cell help in the tumor microenvironment. PLoS One 2014; 9:e115711. ISSN 1932-6203. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25531529; PMID:25531529; https://doi.org/10.1371/journal.pone.0115711
- Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016; 44:1444-54. ISSN 1097-4180. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27332733; PMID:27332733; https://doi.org/10.1016/j.immuni.2016.05.014
- Singh N, Liu X, Hulitt J, Jiang S, June CH, Grupp SA, Barrett DM, Zhao Y. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol Res 2014; 2:1059-70. ISSN 2326-6074. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25104548; PMID:25104548; https://doi.org/10.1158/2326-6066.CIR-14-0051
- Singh N, Barrett DM, Grupp SA. Roadblocks to success for RNA CARs in solid tumors. Oncoimmunology 2014; 3:e962974. ISSN 2162-4011. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25964863; PMID:25964863; https://doi.org/10.4161/21624011.2014.962974
- Beatty GL, O'Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol Ther 2016; 166:30-9. ISSN 1879-016X. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27373504; PMID:27373504; https://doi.org/10.1016/j.pharmthera.2016.06.010
- Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology 2014; 3:e970027. ISSN 2162-4011. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/25941599; PMID:25941599; https://doi.org/10.4161/21624011.2014.970027
- Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, Boutros C, Hanna N, Junghans RP. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther 2016; 23:142-8. ISSN 1476-5500. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/27080226; PMID:27080226; https://doi.org/10.1038/cgt.2016.14