
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Cancer Immunotherapetics differ fundamentally from most cancer therapies in that they involve manipulation of the immune system to induce an anti-tumor response. This fundamental difference results in differences in the pre-clinical and clinical development of immunotherapeutics. Even the regulatory culture of developing one drug at a time and demonstrating that it, when added to standard therapy, prolongs patient survival, is often not suitable for the development of effective immunotherapy regimens. In this commentary, we explore some of these differences and describe novel clinical trial designs which may be useful in immunotherapeutics regimen development.
Introduction
Clinical trials, and particularly randomized clinical trials, have played a key role in the development of effective therapeutics and for enabling physicians to avoid use of harmful and ineffective treatments. This has represented an important advance in medical practice. Over this period of time a literature has grown up defining the principles of good clinical trial design and analysis.Citation1
Today the field of oncology is focused as never before on the development of immunotherapeutics. Exciting successes have been observed with T-cell checkpoint inhibitorsCitation2,3 and, in selected patients, with adoptive T-cell therapy.Citation4,5 New technology for sequencing DNA from single circulating or tumor-infiltrating T cells has also become available.Citation6 The challenges are to enhance the durability of these responses, to extend them to more patients and to develop biomarkers that identify the patients most likely to benefit from these expensive treatments.
The purpose of this Commentary is to point out some ways that development of immunotherapy is fundamentally different from treatments targeted directly to the tumor and how these differences influence key aspects of clinical trial design.
Pre-clinical models
Immunotherapy development is hindered by limitations in pre-clinical model systems. Because the host immune system cannot be represented in tumor tissue cultures, immunotherapeutic interventions cannot be easily studied. Mouse models must provide good models of human tumors developing in the context of the human immune system and transplantable mouse tumors have limitations in this regard.Citation7 These limitations inhibit the ability to explore immunological mechanisms and places a heavier burden on human clinical trials for elucidating the biological mechanisms involved in immunotherapy.
Clinical discovery
Because of the complexity of immune-oncology and the limitations of pre-clinical models, exploration of mechanisms must often be performed in the context of clinical trials. This makes the planning and conduct of good immunotherapy trials more complex and demanding than for small molecule clinical trials. Even if there is only one test intervention, discovery most likely requires intensive immunological monitoring of the patients. If this is to be useful it requires reproducible and well-calibrated assays, which often must be developed. Planning the collection and analysis of immune monitoring data requires collaboration with immunologists and perhaps translational systems biologists. All of this is expensive and requires financial support. It also requires individuals with a wide variety of expertise, an organization structured for trans-disciplinary collaboration and leadership that recognizes that a successful program of immunotherapy development must be strongly science based.
Many candidates
Because of the limitations in pre-clinical models and because of the many ways that the immune system can be modulated, developing an effective immunotherapeutic regimen is often complex; more complex than just adding a new small molecule drug to a standard regimen. For some types of immunotherapy, the interventions are not drugs but living cells, e.g. adoptive T cell therapy. The T cells can be collected, cultured, engineered, selected and expanded in many ways and these alternatives may have major effects on the effectiveness of the regimen. Consequently, there is a need for clinical trial designs which permit the screening of multiple interventions simultaneously in a manner that is efficient with regard to the number of patients required.
The usual phase I -> II -> III paradigm breaks down
Some immunological interventions, like tumor vaccines, may be non-toxic on their own although the full regimen of the vaccine with adjuvant and other modulators may have dose limiting toxicity. This means that standard phase I trials may not be applicable.Citation8 For non-toxic modulators, dose levels must be established based on immunological or clinical endpoints and the usual small phase I trials may be not adequate for this purpose.Citation9 Dose selection is of major importance for immunomodulants because the drugs can have very different effects on the immune system at different doses. Hence larger phase I studies with extensive measurement of immunological effects is often necessary compared with determining a maximum tolerated dose of a chemotherapeutic agent.
The phase II study may involve multiple modulators and be based on evaluation of multiple immunological parameters. For some phase II and phase III clinical trials there may be difficult questions of how long to treat because effectiveness of the regimens may require prolonged therapy and conventional measures of tumor progression may be inadequate.Citation10
Durable complete remission of metastatic disease
For some immunotherapy regimens, such as T cell checkpoint inhibitors and adoptive T cell therapies, durable complete remissions of heavily pre-treated patients with metastatic disease has been achieved. Extended randomized trials have not been necessary to recognize the value of these treatments. This experience is quite different from that with chemotherapy or molecularly targeted kinase inhibitors.
One drug is not enough
Small molecules and monoclonal antibodies generally get approved for clinical use by demonstrating that when added to a standard regimen they improve the survival of a defined group of patients. For combination regimens, all components of the combination must be demonstrated to be essential. Although this paradigm worked for T cell checkpoint inhibitors it is likely to be more problematic for some types of adoptive T cell therapy which is often administered in conjunction with lymphodepletion and post-treatment IL2. For maximal effectiveness, it may be necessary for the tumor-immune system to be modulated in several ways simultaneously; e.g., upregulation of MHC expression of neo-antigens and downregulation of regulatory T cells. It may not be practical to design phase III studies that establish the essentiality of all components of such regimens. Flexibility of regulators in using phase II data to qualify complete regimens for phase III evaluation as a whole will be needed.
Alternative clinical trial designs
When the biological understanding of a disease is lacking and therefore the likely of success of any intervention is limited, the best strategy may be to ensure that the clinical trials that are performed serve to enhance understanding of the disease. The objectives may be primary exploratory rather than hypothesis testing and power calculation driven. To establish likely benefit from this approach, however, the investigators may need to convince sponsors to run a series of phase II trials with several modulators with emphasis on immunological monitoring so as to build up the scientific basis for conducting a phase III trial. If these phase II trials are to provide interpretable results, they must use assays of immunological response that have been analytically validated as reproducible. There are no Nobel prizes for analytical validation but using assays of unknown reproducibility and accuracy in immunotherapy development is an obstacle to progress.
Factorial designs
The factorial design is useful for screening candidate interventions for their individual average effect and for finding leads to synergism of combinations.Citation11 With the 2K factorial design, K modulators are involved, say A, B, C for K-3. Patients are randomized to receive the base regimen, say a tumor vaccine, without any of the modulators, or to receive the base regime with one of A, B or C, or to receive the base regimen with AB, or AC or BC, or to receive the base regimen with all 3 modulators. Applicability of the factorial design is limited to cases where the modulators are relatively non-toxic and administration of one does not limit the dose at which the other can be delivered. Instead of the factors A, B and C being used to represent the presence or absence of a modulator, the levels of the factors can represent alternative versions of the same type of modulator. In a 2K design each factor has 2 levels. For example, there may be 2 alternative adjuvants A1 and A2 as well as 2 alternative methods of regulatory cell reduction, B1 and B2.
An example of a 2 × 2 factorial design is shown in . In this example there is a core tumor vaccine. Factor A represents either pre-treatment with low dose cyclophosphamide or not for control of tumor suppressive cells. Factor B represents administration of a PD-1 antibody or not.
Figure 1. Schema for factorial design with two interventional factors, immune ablation and anti-PD1 antibody administration.
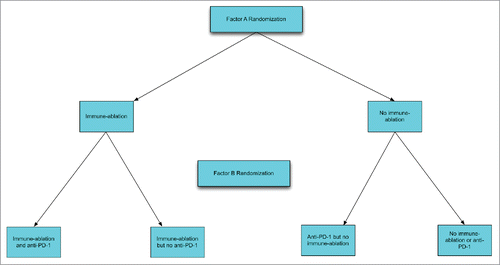
The usual factorial analysis provides for a very efficient evaluation of the average effect of each individual factor. To evaluate whether there the average effect of suppressor depletion is significant, the 2 treatment arms that include use of cyclophosphamide pre-treatment and are pooled and compared with efficacy for the pooled arms not including cyclophosphamide pre treatment. This is equivalent to calculating the effect of cyclophosphamide pre-treatment of the arms involving PD-1 treatment and calculating the effect of cyclophosphamide pre-treatment of the arms not involving PD-1 treatment and then averaging the 2 treatment effects. Similarly, the average effect of PD-1 treatment is estimated by averaging its effect in the arms containing pre-treatment and separately in the arms not involving pre-treatment. Hence the average effect of each factor uses all the data and has the power of a 2-arm trial with the same total number of patients.
Figure 2A. Two-stage phase II/III design; Selection design for selecting among three test regimens followed by phase III design of the selected regimen compared to a standard control regimen.
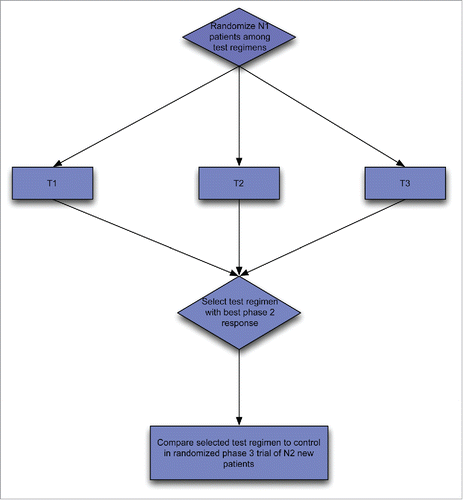
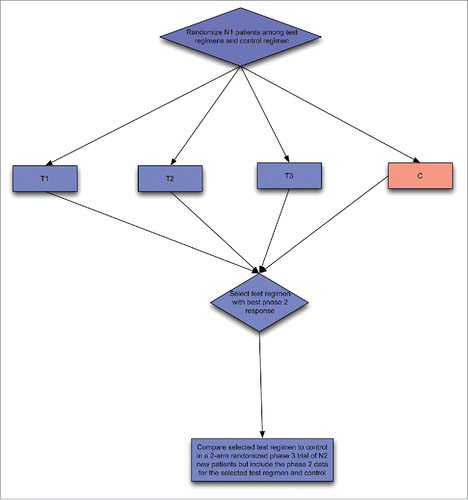
Figure 2B. Two stage phase II/III design; Phase II selection design comparing two test regiment and control followed by phase III design of one of the selected regimens and the control. The phase II data can be used in the final analysis because the control appears in both phase II and phase III portions.
The average effects of the factors are most meaningful when there are not synergistic or antagonistic interactions between the factors. We hope for synergistic interactions however, and these can be identified by analysis using a statistical model. For example, Let x1 = 1 for patients in the arms receiving PD-1 antibody and x1 = 0 for the other patients. Let x2 = 1 when there is pre-treatment with cyclophosphamide and 0 otherwise. A logistic model relating the factors to the probability of response to treatment can be written:(1)
(1)
Y = 1 indicates response and the parameters a,b,c,d are regression coefficients which are determined by fitting the model to the data. The parameter d is the interaction coefficient. One tests whether it is zero. If this hypothesis is not rejected, then there is no significant interaction among the effects of the 2 factors on response. If d is significantly non-zero and is positive then there is a synergistic effect. If d is significantly non-zero and negative then there is an antagonistic effect. In addition to fitting a mode for clinical response, we can fit models for immunological endpoints that were determined based on immune monitoring; for example, CD8+ cells or CD4+ cells in the tumor bed or in the circulation.
The factorial design is particularly useful for screening multiple candidate modulators, each of which has a low prior probability of exhibiting a non-zero average treatment effect and is not dose limiting.
Response-surface designs
A variant of the factorial design is the response-surface design.Citation12 Again there are K factors. Each factor can indicate presence or absence of a modulator or administered concentration of the modulator. There is also a model like (Equation1(1)
(1) ) defining the relationship between the end point (either clinical or immunological) and the factors. The model contains regression coefficients of unknown value. There may be a prior distribution for the value of each parameter of the model. Initially patients are assigned factor levels by randomization and their outcomes are observed. After a training sample is obtained, the probability of clinical or immunological response is computed for subsequent patients as a function of the factor levels. The factor levels are assigned to maximize this probability of response. The response data are continually updated. The objective is to find a combination of factor levels associated with large probability of response with fewer patients than would be required for a standard non-adaptive design.
Selection designs
There are statistical paradigms for clinical trials other than the null hypothesis paradigm commonly used in phase III regulatory clinical trials. One paradigm is that of “selection” of a regimen for further investigation from among several candidate regimens.Citation13 In a selection trial you do not test the null hypothesis of no treatment difference. Instead, one of the treatments is always selected and the trial is sized so that the correct treatment arm is selected if one of the arms is superior by at least a specified difference d. This formulation may require a smaller sample size that a multi-arm clinical trial that compares each experimental arm to an internal control and to each other with a standard hypothesis test. In a selection design, the selected arm may be compared with a control either in a subsequent trial or in an extension phase. Selection designs are not well suited to comparing A + B to A alone. Rather, they are better suited to comparing A + B to A + B’ to A + B” where B, B’ and B” are similar variants. The best is selected and in a subsequent trial one might compare A to the regimen selected.
Phase 2/3 designs
An extension of the selection design is to combine a phase II selection portion with a phase III hypothesis testing portion. Two versions of this are illustrated in . In starts with a selection phase among regimens A + B, A + B’ and A + B.”Citation14 The regimen selected as best is then compared with A alone in the phase III portion. In , a control arm A alone is include in the phase II screening portion of the design.Citation15 In the second portion only the selected combined regimen and the control regimen are continued.
Adaptive designs
The phase 2/3 design is a form of an adaptive design. There are also more complicated types of adaptive designs in which dose levels, treatment arms, and eligibility criteria are modified as the trial proceeds based on interim results.Citation16 The adaption may involve specific biomarker subsets.Citation17 The adaption may also be by dropping arms or eligibility at distinct interim-analysis times or continuously by changing randomization weights.
Discussion
Immuno-oncology treatment development is complex. Some aspects of the standard drug development paradigm do not apply very well. This should be taken into consideration by investigators, sponsors and regulators at the design stage of clinical trials.
Designing clinical trials to support better understanding of biological or immunological mechanism is not what trialists or review committees are used to. Such trials are resource intensive, may require multiple clinical grade reagents, and durable trans-disciplinary collaboration. Industry and cancer centers are generally not organized or committed to such long-term translational research. In many medical centers, translational research too often means making administrative and statistical “services” available to individual investigators to facilitate getting their molecules into patients. It does not really address incentivizing disease focused team research. Industry may be more disease focused, but often prefers settings where the basic research has already laid a firm ground-work for direct translation.
Achieving the promise of immunotherapy may, however, require serious re-organization of immune-oncology centers of excellence to focus on deep and durable trans-disciplinary collaboration among oncologists, immunologists, immuno-statisticians, translational systems biologists and assay developers. This will require support of industry in making clinical reagents available and non-industry sponsors for fostering a new approach to translational research.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Piantadosi S. Clinical trials: A methodologic perspective (2nd edition). New York, NY: Wiley; 2005
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen HB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; https://doi.org/10.1056/NEJMoa1003466
- Topalian SL, Hodi S, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Eng J Med 2012; 366:2443-54; PMID:22658127; https://doi.org/10.1056/NEJMoa1200690
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-Cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550-7; PMID:21498393; https://doi.org/10.1158/1078-0432.CCR-11-0116
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric Antigen Receptor-Modified T cells for Acute Lymphoid Leukemia. N Eng J Med 2013; 368:1509-18; PMID:23527958; https://doi.org/10.1056/NEJMoa1215134
- Kirsch IR, Watanabe R, O'Malley JT, Williamson DW, Scott LL, Elco CP, Teague JE, Gehad A, Lowry EL, LeBoeuf NR, et al. TCR sequencing facilitates diagnosis and identifies mature T cells as the cell of origin in CTCL. Sci Transl Med 2015; 7 Issue 308:308ra158; PMID:26446955; https://doi.org/10.1126/scitranslmed.aaa9122
- Davis MM. Immunology taught by humans. Sci Transl Med 2012; 4:117fs2; PMID:22261029; https://doi.org/10.1126/scitranslmed.3003385
- Rahma OE, Gammoh E, Simon R, Khleif SN. Is the “3+3” dose escalation phase 1 clinical trial design suitable for therapeutic cancer vaccine development? A recommendation for an alternative design. Cancer Res 2014; 20:4758-67.
- Hunsberger S, Rubinstein LV, Dancey J, Korn EL. Dose escalation trial designs based on a molecularly targeted endpoint. Statistics Med 2005; 24:2171-81; PMID:15909289; https://doi.org/10.1002/sim.2102
- Hoos A, Eggermont AMM, Janetzki S, Hodi FS, Ibrahim R, Anderson A, Humphrey R, Blumenstein B, Old L, Wolchok J. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Institute 2010; 18:1388-97; https://doi.org/10.1093/jnci/djq310
- Stampfer MJ, Buring JE, Willett W, Rosner B, EberleinK , Hennekens CH. The 2 × 2 factorial design: Its application to a randomized trial of aspirin and U.S. physicians. Statistics Med 1985; 4:111-6; PMID:4023472; https://doi.org/10.1002/sim.4780040202
- Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Therapeutics 1997; 61:275-91; https://doi.org/10.1016/S0009-9236(97)90160-0
- Simon RM, Steinberg SM, Hamilton M, Hildesheim A, Khleif S, Kwak LW, Mackall CL, Schlom J, Topalian SL, Berzofsky JA. Clinical trial designs for the early clinical development of therapeutic cancer vaccines. J Clin Oncol 2001; 19:1848-54; PMID:11251017; https://doi.org/10.1200/JCO.2001.19.6.1848
- Thall PF, Simon R, Ellenberg SS. Two-stage selection and testing designs for comparative clinical trials. Biometrika 1988; 75:303-10; https://doi.org/10.1093/biomet/75.2.303
- Thall PF, Simon R, Ellenberg SS. A two-stage design for choosing among several experimental treatments and a control in clinical trials. Biometrics 1989; 45:537-48; PMID:2765637; https://doi.org/10.2307/2531495
- Berry DA. Adaptive clinical trials in oncology. Nat Rev Clin Oncol 2012; 9:199-207; https://doi.org/10.1038/nrclinonc.2011.165
- Carey LA, Winer EP. I-SPY 2: Toward more rapid progress in breast cancer treatment. N Eng J Med 2016; 375:83-4; PMID:27406352; https://doi.org/10.1056/NEJMe1603691