
ABSTRACT
While interest in immunotherapies is renewed by the successful introduction of immune checkpoint blocking agent in the clinic, advances in genome sequencing are opening new possibilities in the design of increasingly personalized vaccines. Personalization of medicine can now be realistically contemplated at the single patient level. Unlike the previous generation of cancer vaccines, neoantigen directed vaccines would target truly specific tumor antigens resulting from acquired tumor genome mutations. Immune response induced by this next generation vaccine would not be subject to self-tolerance and will likely result to enhanced efficacy. Nevertheless, this new technologies can hold to their promises only if sponsors manage to meet several scientific, technical, logistical and regulatory challenges. In particular manufacturers will have to design, manufacture, and deliver to the patient a new pharmaceutical grade in a matters of weeks. In this paper, we briefly review current technologies currently tried at the translation of personalized vaccines and explore the possibilities offered by the Modified Vaccinia virus Ankara in this next wave of cancer vaccines.
Introduction
Harnessing the immune system to eradicate tumor cells has become an established therapeutic strategy as demonstrated by FDA approval of sipuleucel-T (Provenge), ipilimumab (Yervoy, anti-CTLA-4), and more recently, the programmed cell death (PD)-1 antibody (pembrolizumab, Keytruda) for the treatment of multiple types of cancer. Despite the great success of checkpoint blockade therapies, more than 50% of cancer patients still fail to respond to immunotherapy. Meanwhile, technological breakthroughs in the field of DNA sequencing, genomics, and bioinformatics have paved the way to the concept of personalized medicine. These advances have hence enhanced our ability to detect new immune targets and tumor-specific neoantigens (mutation-derived antigens), and have accelerated the development of immunotherapies with potentially broader coverage of cancer patients by providing an unprecedented opportunity for the design of mutation targeted immunotherapy. While CAR T-cells constitute an efficient tool to target surface antigens, cancer vaccines provide a mean to target the whole cancer mutanome.Citation1 In this paper, we will discuss the impact of these recent progresses on the role of a well-established cancer vaccine platform: the MVA (Modified Vaccinia virus Ankara).
Designing personalized cancer vaccines would translate into a major paradigm shift. In spite of generating a common vaccine against a known antigen, and then assess if individual patient tumors would express this antigen, the customized approach implies the prior identification of specific antigens for each patient and the subsequent generation of a vaccine targeting these mutations. Although this strategy has been proposed for more than a decade,Citation2 the development of an individualized cancer vaccine faces major challenges in several domains before delivering to its promise.
Personalized cancer vaccines will have to be designed, manufactured and made available quickly after the diagnosis. While needs can vary considerably from one clinical indication to the other, it can be anticipated that the vaccine would need to be available from 4 to 6 weeks after diagnosis to allow a realistic integration in the current workflow of patient care. Within this time frame, vaccine manufacturers will have to identify the most relevant antigens to elicit an efficient T cell response, and manage various issues relating to the generation of a pharmaceutical grade product within acceptable costs of manufacturing. These difficulties may be exacerbated by the absence of appropriate regulatory guidelines. Last, their adoption in routine care will be subordinated to their seamless integration in an ever increasingly complex therapeutic armamentarium.
Several technologies are currently assessed as potential platforms for custom-designed vaccines: nucleic acids, peptides, live bacterial or viral vectors or human cells; all presenting loaded neoantigens to the patient's immune system. In our vision, the combination of technical, operational, and regulatory constraints might represent an opportunity for well-established technologies such as viral-based cancer vaccines. In this respect, the Modified Virus Ankara (MVA), an attenuated vaccinia virus, displays several advantages. It has served as an effective vector in previous attempts to develop cancer vaccines, some of which had demonstrated significant clinical efficacy in phase 2 trials. In this commentary, we will shortly highlight some of the MVA features that may be of added value in the implementation of an individualized vaccination approach.
Finding the most appropriate antigens
One first issue in the design of the cancer vaccine is the selection of relevant target antigens. Current designs have been based on either tumor-associated antigens (TAA), that are highly expressed in tumor cells compared with normal cells (e.g. Her-2, Muc-1), on onco-fetal antigens (e.g., AFP, CEA, 5T4), or on cancer/testis antigens (NY-ESO1, MAGE-A3, CT83). TAAs have been subject to intense effort of research as cancer vaccine targets in the previous generations of cancer vaccines.Citation3 However, all these self antigens may bear the risk of clinical inefficacy due to central immunotolerance or autoimmunity. Hence, non self antigens exhibit lower risk of failure. They are either viral oncogenes (e.g., HPV E6/E7, EBNA-1) or tumor-specific neoantigens (TSNA) resulting from DNA misrepair or chromosomal rearrangement. These non-self antigens are now considered as more efficient than their self counterparts.Citation4-6
Because of high mitotic rates and deficient repair mechanisms, tumor are characterized by the accumulation of somatic mutations from the earliest stage of tumorigenesis.Citation7 The rate of mutation in the tumor exome varies considerably from one cancer to the other from a median of less than one mutation per million of bases in rhabdoid tumors and Ewing sarcoma to more than 10 in a million bases in melanoma or lung cancer.Citation8 The onset of non-synonymous mutations in the coding region of DNA may results in the synthesis of aberrant protein species.
Current, sequencing technologies can reliably identify mutations in tumor material, however, the number of mutations can be relatively high – up to several hundreds – and a selection must be performed to select a panel of vaccine targets. Criteria entering in the selection of most relevant antigens are level of antigen expression, conservation across tumor cells, presentation by the MHC, and potential immunogenicity.Citation9 Noteworthy, neoantigenic landscape of tumors can prove relatively heterogenous and evolve during time, in particular when under immune selective pressure.Citation10,11
Several authors have attempted to identify such neoantigens that are recurrent in patients so that target libraries can be built to develop vaccines.Citation12 These studies did not results in the identification of recurrent somatic mutations but identified mutational hotspotsCitation13 associated with increased probability of mutation. Consequently, development of cancer vaccines requires the identification of neoantigen on a patient by patient basis and consequently, the vaccine design should be tailored and manufactured for individual patient. These considerations require the definition of a robust workflow for the design of the vaccine. Selection of antigens can be assisted by in silico prediction or bioanalytic approaches. In silico predictors are built using machine learning algorithms such as neural networks and trained to identify highly immunogenic sequences.Citation14 The algorithm is then used to classify epitopes for their affinity for HLA class I protein as an index of their potential presentation. Additional filtering can be added by factoring in levels of expression at RNA level using RNAseq. While in silico based approaches have been shown to have some level of predictivity on the immunogenicity of a library of putative epitopes, their current level of validation is unlikely to permit to have a vaccine targeted only to a very restricted number of predicted epitopes as there is a significant risk that these epitopes are not associated with an actual response in vivo.Citation14,15 Also, the reliability of these approaches remains questionable when predicting CD4 epitopes. Last, it should be mentioned that several collaborative consortia, such as TESLA (Tumor Neoantigen Selection Alliance), supported by the Parker Institute,Citation16 or the European APERIM (Advanced bioinformatic platform for personalized cancer immunotherapy),Citation17 are underway to achieve a higher level of validation of these predictive tools. Alternatively to in silico approaches, some authors have proposed the direct identification of presented peptides using LC/MS approaches, potentially after immunoprecipitation of tumor MHC. This approach allows for an unbiased identification of peptides presented by a patient tumor cells. However, it is likely that improvement in analytical sensitivity will be required to allow routine use of this technology.Citation18,19
In practice, the uncertainty around the best method for predicting relevant epitopes and the dynamic nature of the tumor mutational landscape result in the conclusion that, whatever the technology, a personalized vaccine will embark a relatively high number of distinct neoantigens. Thus, the capacity of the vector will a key decision factor.
Manufacturing and regulatory challenges
In complement to antigen discovery and vaccine design, significant efforts are still required to manufacture the product according to pharmaceutical regulations. Quality control and regulatory reviews in the pharmaceutical industry were historically based on extensive chemical and biological characterization of the drug product as well as tight control thereof during routine manufacturing. However, given the time constraints in cancer care, manufacturers of unique personalized vaccines must invent new ways to deliver a product matching FDA/EMA requirements, without engaging into lengthy analytical characterization of product properties and stability. While needs may vary from one indication to the other, a product should be made available within weeks. Necessity for fast manufacturing is certainly the major challenge that this emerging field will face.
When presented with increasingly personalized products, regulators responded with specialized organizations, such as the FDA's Personalized Medicine teams at CDRH and CBER,Citation20 or guidelines, such as the ATMP (advanced therapeutic medicinal product) regulation of the European Medicinal Agency (EMA).Citation21 These guidelines have taken in account some specificities of the next generation of products by ensuring compliance through a “quality by design” and risk-based approach.
The last critical point is cost control. It is of particular relevance in an environment where high drug costs are facing an ever increasing scrutiny. While the final pricing of personalized vaccines will obviously be discussed on the basis of their medical utility, manufacturers should make all possible efforts to control costs of treatment and think of innovative business models. Failure to do so will drastically limit the adoption of this emerging class of therapy. A notorious example is the bankruptcy of Dendreon, the manufacturer of the first personalized CAR-T cell therapy to hit the market.Citation22
Suitable vectors for personalized cancer vaccines
Considering the aforementioned challenges, selecting the best vaccine platform among the multiple possibilities has been a conundrum. A number of technological options are now being considered for the development of a neoantigen-based vaccine (). They can be categorized in 5 major families: peptide-based vaccines, nucleic acids (DNA, mRNA)-based vaccines, human cell-based approaches, live vector vaccines based on engineered viruses or bacteria. Unfortunately, no systematic experiment was performed to compare different formats displaying the same TAA, in terms of intensity or quality of the immune response. Moreover, it is likely that similar antigens displayed by diverse platforms in the same preclinical model resulted in different immunogenicity profiles.Citation23,24
Table 1. List of principal technological platforms for the development of neoantigen-directed vaccines.
Peptide vaccines, based on either whole antigens or short peptides (9-mers), have been largely studied but they generally elicited transient and relatively weak T cell responses, as exemplified by the gp100280-288 peptide in melanomaCitation25 which showed limited clinical efficacy. This disappointing outcome was attributed to poor in vivo stability or to the lack of specificity of peptides for “professional” antigen presenting cells. The use of long peptides (30-mers), adjuvanted with biologically active factors was promoted to circumvent these limitations in several indications (breast,Citation26 pancreatic,Citation27 lungCitation28 and colorectalCitation29 cancers). However, these products did not reach clinical registration and are currently improved in the setting of neoantigen vaccination by different companies.
The demonstration that naked foreign nucleic acids injected in vivo can penetrate somatic cells and can result in the expression of the encoded sequence opened the door to use of DNA or mRNA vaccines.Citation30 Building upon the experience accumulated in the field of gene therapy, DNA plasmid vaccines were designed in conjunction with various technologies intended to increase the rate of transfection such as electroporation or high pressure needless injection devices.Citation31 Despite promising clinical results, these vaccines proved clinically disappointing. Studies administrating up to 2 mg of plasmidic DNA intramuscularly or intranodally in advanced melanoma or colorectal cancer did not provide anti-tumor activity nor clinical benefit. Furthermore, possible integration of exogenous DNA remains a concern. So further improvement of vectors and administration routes are still needed for a DNA based approach to be used routinely; one promising approach is developed by Vaccibody (Oslo, Norway): a DNA plasmid encoding for a multidomain protein comprising a targeting factor, a stimulating cytokine complex and strings of antigens, the plasmid is being delivered using a needless high pressure device.Citation32 Similarly, RNA vaccines utilization has been hampered by difficulties to address sufficient amount of RNA to the target cells to achieve a consistent expression of the antigen. Recent developments of advanced formulations targeting RNA to the spleen, and compatible with repeated administrations, have renewed the interest in RNA vaccines and opened up opportunities to target neoantigens using these approaches.Citation33
One alternative modality of use of peptides and nucleic acids is the use of dendritic cells as vector after in vitro/ex vivo exposure to the antigen. Autologous dendritic cells are pulsed with peptide, sequences of RNA encoding for the antigenic sequence or with tumor lysates prior to administration to the patient. However, most initiatives in the field did not reach late stage clinical development and have been limited by logistical hurdles and excessively high costs. Current efforts aim at tackling these limitations using off the shelf dendritic cells.Citation34
Last, live vectors such as bacteria or viruses constitute an attractive option to overcome limitations of the technologies described above. Live vector vaccines may be based on viral or bacterial platforms. The immune environment elicited by infection of cells by such live vectors has been shown to promote an enhanced immune response in terms of frequency and avidity of cytotoxic T cells.Citation35 Additionally, live vectors constitute an attractive mean to target professional antigen presenting cells with a relatively easier manufacturing and logistical process that dendritic cell vaccines.Citation36 Attenuated forms of Listeria monocytogenes have been used for the development of cancer vaccines.Citation37 This technology is currently applied by Advaxis (Princeton, NJ, US) as a platform for neoantigen vaccine delivery. Besides, the large majority of live vaccine vector are viruses. αviruses, Adenoviruses, viruses from the pox family or lentiviral systems such as ImmuneDesign's ZVex® (Seattle, WA, US) are used as cancer vaccines.Citation36 Each platform has its advantages and limitations, but generally may be limited by safety concerns originating from risks of genome integration and uncontrolled replication in immunocompromised patients. To circumvent these limitations, the Modified Virus Ankara (MVA) has served as a safe and effective delivery platform for cancer antigens in numerous cancer trials.
Advantages of the MVA platform
The Modified Virus Ankara (MVA) has been generated through serial passages in chicken embryo fibroblasts.Citation38 It has been used safely and effectively for small pox vaccination in more than a hundred thousand individuals.Citation39 The MVA has lost the pathogenicity of its parental virus, the Chorioallantois Vaccinia virus Ankara, through alterations of its genome, i.e. 15% of genome loss. The virus is replication defective in human cells, ensuring its safety and tolerability in clinical use. Furthermore, it provides an inexpensive cellular system to produce large quantities of the virus in egg-based manufacturing processes.
Subsequently to its use as a small pox vaccine, MVA was largely used as a genetic vaccine vector in several trials, targeting several cancer antigens in various indications including melanoma, prostate, breast, kidney and lung cancer.Citation40 MVA-based vaccines have achieved good clinical results when given either as a monotherapy or in combination with chemotherapy, radiotherapy or immune checkpoint inhibitor.Citation41
The large variety of antigens used in the MVA platform confirms the robustness and flexibility of the vector. Inserts of up to 11 kilobases can reliably be cloned in the vector, as demonstrated by our experience with the full length cDNA of dystrophin, one of the largest human protein. This high cloning capacity enables the insertion of a large number of protein antigens and also leaves room for additional material intended to increase the immunogenicity of the vaccine. Practically, one single vector can be used to generate a response against different antigens and poly-epitopes.
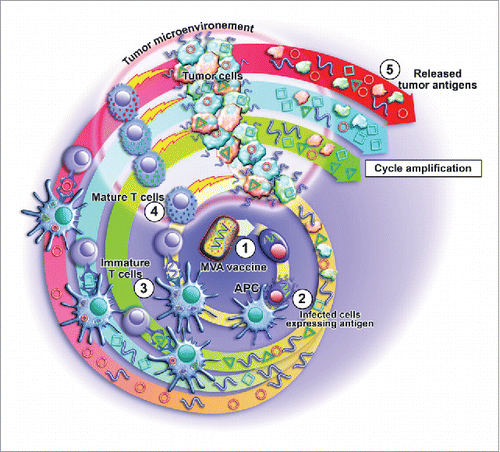
MVA vaccines act on several key points in the tumor immunity cycle (). After targeted expression of the cancer antigen at the site of injection, the viral infection triggers the release of immune alarm signals, then stimulating the intervention of professional APC that will present both encoded antigens and viral components. Noteworthy, all these steps take place outside of the immunosuppressive environment of the tumor. MVA vaccination has the potential to elicit both CD4 and CD8 responses.Citation42 The diversity of these responses leverages an efficient anti-tumor response, and decreases the risks of immune escape by counterselection of negative tumor cell clones. The large cloning capacity can also be used to select mutations targeting respectively CD4 or CD8 responses as both response types are demonstrated to play a role in the development of a durable anti-tumor response.Citation43
Figure 1. Mechanism of action of MVA vaccines at cellular levels. Infection of cell (1), followed by intervention of professional APC (2) and presentation of viral components including antigenic transgene (3). After priming and maturation, effectors T cells home into the site of the tumor and (exert their cytolytic action on tumor cells presenting the targeted antigen (4). As a consequence, tumor cell death might release other cancer antigens that then lead to spreading of the response (5). We could demonstrate this phenomenon in the case of TG4010, a MVA-based vaccine used in patients with NSCLC.
Two recent examples from our portfolio confirm the clinical efficacy of MVA-based vaccines. The first one is TG4001 (Tipapkinogene sovacivec), targeting E6 and E7 antigens from human papilloma virus, was used to treat high grade cervical intraepithelial neoplasia. In a phase I study, 9 out of 10 patients displayed eradication of HPV DNA 6 months after initiation of treatment and lesions regressed in 7 out of 10 patients.Citation44 These results were confirmed in a randomized, controlled phase II study (unpublished data). The second one is TG4010 (Mesmulogene ancovacivec, MVA-Muc1-IL2). It demonstrated a remarkable efficacy in non-small cell lung cancer patients; with doubling of both progression free survival (12 to 30% at 12 months) and overall survival (19 to 40% at 24 months),compared with placebo.Citation45 Immunomonitoring of patient samples from this TIME study demonstrated that clinical benefit was associated to epitope spreading to other cancer associated antigens.Citation46 Similarly, Prostvac® (Rilimogene galvacirepvec), an MVA based vaccine expressing PSA (Prostate specific antigen) and stimulating cytokines (LFA-3, ICAM-1, and B7.1) has shown encouraging results in prostate cancer.Citation47
It is frequently argued that the activity of viral vaccine might be impeded by neutralizing antibodies. In our experience, neither prior vaccination against smallpox, nor repeated administration negatively affected immunogenicity of the product. Hence the platform would be fit for repeated treatment of patients in case of evolution of the tumor mutational landscape.
In addition to these mechanistic advantages, MVA also deserves consideration from a pharmacokinetic et pharmacodynamic point of view. DNA, RNA or peptide vaccines rely on the use of different sequences, which different biophysical behaviors which might potentially lead to product heterogeneity, affect in vivo stability, and thus impair biodistribution properties. In the case of live vectors, one can reasonably assume that biodistribution will be independent of the nature of the neoantigenic cassette, just as it remained constant across the different existing vaccines directed at various TAA.
There is a strong track record for design and manufacturing of MVA-based vaccines. The recent integration of novel genome editing technologies has greatly improved the cloning yields, and thus allowed significant reduction of manufacturing times from 2–3 months to a few weeks. This disruption should favorably impact the economics of the global manufacturing process. In our analysis, the recent progress on MVA manufacturing makes it compatible with the following cost and time targets for a pharmaceutical-grade personalized vaccine: total treatment cost ≤ $15 000 with a lead time from tumor sequencing to vaccine delivery of 6 to 8 weeks.
The future of personalized vaccines and their integration into therapeutic schemes
Neoantigens vaccination is a promising treatment modality, in particular with respect to its capacity to embrace the entire cancer mutanome. Implementation of this approach is however contingent to the successful resolution of technical challenges associated with the one-patient one-product paradigm and integration of this new therapeutic class into the standards of care.
Among them, one should obviously consider the arrival of immune checkpoint inhibitors (ICI) in various oncology indications, either as standalone therapy or in combination with other immunotherapy products, leading to a paradigm shift in the treatment of cancer. We assume neoantigenic vaccine will be used in combination with ICI, taking advantage of potential synergy between these 2 therapeutic classes. For instance, combination with checkpoint blockade is a particularly appealing approach to attempt to increase response rates.Citation48 Among the several clinical trials in progress in this area, we wish to outline the combination trial between the MVA-based vaccine TG4010 and the anti-PD1 Nivolumab in NSCLC patients (NCT02823990). Nevertheless, heterogeneity of response across patients reflects a large diversity of immune mechanisms and highlights the importance to adapt the therapeutic intervention based on tumor immune profile.Citation49 Personalized vaccination will thus be associated with the development of precision medicine and related bioanalytical methods.
Third, the diversity of vaccine platforms also opens up the possibility to act sequentially on different immune mechanisms. This so called heterologous prime/boost vaccination has been shown to leverage synergy between different adaptive response mechanisms.Citation50,51 Given their unique features, it is a reasonable assumption that MVA based platforms will be central in this upcoming developments.
Disclosure of potential conflicts of interest
KB and EQ are full time employees of Transgene SA.
References
- Mullard A. The cancer vaccine resurgence. Nat Rev Drug Discov 2016; 15:663-5; PMID:27681782; https://doi.org/10.1038/nrd.2016.201
- Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wölfel C, Huber C, Wölfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A 2005; 102:16013-8; PMID:16247014; https://doi.org/10.1073/pnas.0500090102
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 2009; 15:5323-37; PMID:19723653; https://doi.org/10.1158/1078-0432.CCR-09-0737
- Wang RF, Wang HY. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res 2017; 27:11-37; PMID:28025978; https://doi.org/10.1038/cr.2016.155
- Buonaguro L, Petrizzo A, Tornesello ML, Buonaguro FM. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol 2011; 18:23-34; PMID:21048000; https://doi.org/10.1128/CVI.00286-10
- Tagliamonte M, Petrizzo A, Tornesello ML, Buonaguro FM, Buonaguro L. Antigen-specific vaccines for cancer treatment. Hum Vaccin Immunother 2014; 10:3332-46; PMID:25483639; https://doi.org/10.4161/21645515.2014.973317
- Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci U S A 1996; 93:14800-3; PMID:8962135; https://doi.org/10.1073/pnas.93.25.14800
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499:214-8; PMID:23770567; https://doi.org/10.1038/nature12213
- Savage PA. Tumor antigenicity revealed. Trends Immunol 2014; 35:47-8; PMID:24439426; https://doi.org/10.1016/j.it.2014.01.001
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016; 375:819-29; PMID:27433843; https://doi.org/10.1056/NEJMoa1604958
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov 2017; 7(3):264–276; PMID:28031159; https://doi.org/10.1158/2159-8290.CD-16-0828
- Tokheim C, Bhattacharya R, Niknafs N, Gygax DM, Kim R, Ryan M, Masica DL, Karchin R. Exome-Scale discovery of hotspot mutation regions in human cancer using 3D protein structure. Cancer Res 2016; 76:3719-31; PMID:27197156; https://doi.org/10.1158/0008-5472.CAN-15-3190
- Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao J, Socci ND, Solit DB, Olshen AB, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016; 34:155-63; PMID:26619011; https://doi.org/10.1038/nbt.3391
- Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, Griffith M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med 2016; 8:11; PMID:26825632; https://doi.org/10.1186/s13073-016-0264-5
- Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J Clin Invest 2015; 125:3413-21; PMID:26258412; https://doi.org/10.1172/JCI80008
- http://www.parkerici.org/media/2016/parker-institute-for-cancer-immunotherapy-cri-neoantigen-alliance
- http://aperim.eu/downloads/publications
- Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun 2016; 7:13404; PMID:27869121; https://doi.org/10.1038/ncomms13404
- Gfeller D, Bassani-Sternberg M, Schmidt J, Luescher IF. Current tools for predicting cancer-specific T cell immunity. Oncoimmunology 2016; 5:e1177691; PMID:27622028; https://doi.org/10.1080/2162402X.2016.1177691
- Hamburg MA. Paving the way for personalized medicine: FDA's role in a new era of medical product development. US Food Drug Admin 2013
- European Medicine Agency. Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells: EMA Committee for Advanced Therapies. London: European Medicine Agency; 2012.
- Ward A, Crow D. Race to control costs of cancer therapy revolution financial times. London: The Financial Times 2015
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016; 534:396-401; PMID:27281205; https://doi.org/10.1038/nature18300
- Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72:1081-91; PMID:22237626; https://doi.org/10.1158/0008-5472.CAN-11-3722
- Slingluff CL Jr., Yamshchikov G, Neese P, Galavotti H, Eastham S, Engelhard VH, Kittlesen D, Deacon D, Hibbitts S, Grosh WW, et al. Phase I trial of a melanoma vaccine with gp100(280–288) peptide and tetanus helper peptide in adjuvant: Immunologic and clinical outcomes. Clin Cancer Res 2001; 7:3012-24. PMID:11595689
- Mittendorf EA, Clifton GT, Holmes JP, Schneble E, van Echo D, Ponniah S, Peoples GE. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol 2014; 25:1735-42; PMID:24907636; https://doi.org/10.1093/annonc/mdu211
- Nishida S, Koido S, Takeda Y, Homma S, Komita H, Takahara A, Morita S, Ito T, Morimoto S, Hara K, et al. Wilms tumor gene (WT1) peptide-based cancer vaccine combined with gemcitabine for patients with advanced pancreatic cancer. J Immunother 2014; 37:105-14; PMID:24509173; https://doi.org/10.1097/CJI.0000000000000020
- Kotsakis A, Papadimitraki E, Vetsika EK, Aggouraki D, Dermitzaki EK, Hatzidaki D, Kentepozidis N, Mavroudis D, Georgoulias V. A phase II trial evaluating the clinical and immunologic response of HLA-A2(+) non-small cell lung cancer patients vaccinated with an hTERT cryptic peptide. Lung Cancer 2014; 86:59-66; PMID:25130084; https://doi.org/10.1016/j.lungcan.2014.07.018
- Okuno K, Sugiura F, Inoue K, Sukegawa Y. Clinical trial of a 7-peptide cocktail vaccine with oral chemotherapy for patients with metastatic colorectal cancer. Anticancer Res 2014; 34:3045-52. PMID:24922671
- Weide B, Garbe C, Rammensee HG, Pascolo S. Plasmid DNA- and messenger RNA-based anti-cancer vaccination. Immunol Lett 2008; 115:33-42; PMID:18006079; https://doi.org/10.1016/j.imlet.2007.09.012
- Graham BS, Enama ME, Nason MC, Gordon IJ, Peel SA, Ledgerwood JE, Plummer SA, Mascola JR, Bailer RT, Roederer M, et al. DNA vaccine delivered by a needle-free injection device improves potency of priming for antibody and CD8+ T-cell responses after rAd5 boost in a randomized clinical trial. PLoS One 2013; 8:e59340; PMID:23577062; https://doi.org/10.1371/journal.pone.0059340
- Grodeland G, Fredriksen AB, Loset GA, Vikse E, Fugger L, Bogen B. Antigen targeting to human HLA class II molecules increases efficacy of DNA vaccination. J Immunol 2016; 197:3575-85; PMID:27671110; https://doi.org/10.4049/jimmunol.1600893
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520:692-6; PMID:25901682; https://doi.org/10.1038/nature14426
- de Gruijl T, Santegoeds S, van Wetering S, Singh SK, Hall A, van de Loosdrecht AA, et al. Allogeneic dendritic cell (DC) vaccination as an “off the shelf” treatment to prevent or delay relapse in elderly acute myeloid leukemia patients: Results of Phase I/IIa safety and feasibility study. J Immunother Cancer 2013; 1:P205-P; ; https://doi.org/10.1186/2051-1426-1-S1-P205
- Kantor J, Irvine K, Abrams S, Kaufman H, DiPietro J, Schlom J. Antitumor activity and immune responses induced by a recombinant carcinoembryonic antigen-vaccinia virus vaccine. J Natl Cancer Inst 1992; 84:1084-91; PMID:1619682; https://doi.org/10.1093/jnci/84.14.1084
- Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J 2011; 17:359-71; PMID:21952287; https://doi.org/10.1097/PPO.0b013e3182325e63
- Toussaint B, Chauchet X, Wang Y, Polack B, Le Gouellec A. Live-attenuated bacteria as a cancer vaccine vector. Expert Rev Vaccines 2013; 12:1139-54; PMID:24124876; https://doi.org/10.1586/14760584.2013.836914
- Mayr A, Munz E. [Changes in the vaccinia virus through continuing passages in chick embryo fibroblast cultures]. Zentralbl Bakteriol Orig 1964; 195:24-35. PMID:5890664
- Stickl H, Hochstein-Mintzel V, Mayr A, Huber HC, Schafer H, Holzner A. [MVA vaccination against smallpox: clinical tests with an attenuated live vaccinia virus strain (MVA) (author's transl)]. Dtsch Med Wochenschr 1974; 99:2386-92; PMID:4426258; https://doi.org/10.1055/s-0028-1108143
- Acres B, Bonnefoy JY. Clinical development of MVA-based therapeutic cancer vaccines. Expert Rev Vaccines 2008; 7:889-93; PMID:18767940; https://doi.org/10.1586/14760584.7.7.889
- Farsaci B, Kwilas A, Hodge J. Design, development, and translation of poxvirus-based vaccines for cancer. In A Bot, M Obrocea, & F Marincola (Eds.), Cancer Vaccines: From Research to Clinical Practice (Vol. 1, pp. 56-77). London: Informa Healthcare; 2011.
- Mooij P, Balla-Jhagjhoorsingh SS, Koopman G, Beenhakker N, van Haaften P, Baak I, Nieuwenhuis IG, Kondova I, Wagner R, Wolf H, et al. Differential CD4+ versus CD8+ T-cell responses elicited by different poxvirus-based human immunodeficiency virus type 1 vaccine candidates provide comparable efficacies in primates. J Virol 2008; 82:2975-88; PMID:18184713; https://doi.org/10.1128/JVI.02216-07
- Galaine J, Borg C, Godet Y, Adotevi O. Interest of tumor-specific CD4 T helper 1 Cells for therapeutic anticancer vaccine. Vaccines (Basel) 2015; 3:490-502; PMID:26350591; https://doi.org/10.3390/vaccines3030490
- Brun JL, Dalstein V, Leveque J, Mathevet P, Raulic P, Baldauf JJ, Scholl S, Huynh B, Douvier S, Riethmuller D, et al. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am J Obstet Gynecol 2011; 204:169 e1-8; PMID:21284968; https://doi.org/10.1016/j.ajog.2010.09.020
- Quoix E, Lena H, Losonczy G, Forget F, Chouaid C, Papai Z, Gervais R, Ottensmeier C, Szczesna A, Kazarnowicz A, et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): Results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol 2016; 17:212-23; PMID:26727163; https://doi.org/10.1016/S1470-2045(15)00483-0
- Tosch C, Bastien B, Barraud L, Grellier B, Nourtier V, Gantzer M, et al. Viral based vaccine TG4010 induces broadening of specific immune response and improves outcome in advanced NSCLC. Submitted for publication; 2017
- Singh P, Pal SK, Alex A, Agarwal N. Development of PROSTVAC immunotherapy in prostate cancer. Future Oncol 2015; 11:2137-48; PMID:26235179; https://doi.org/10.2217/fon.15.120
- Hardwick N, Chung V, Cristea M, Ellenhorn JD, Diamond DJ. Overcoming immunosuppression to enhance a p53MVA vaccine. Oncoimmunology 2014; 3:e958949; PMID:25941580; https://doi.org/10.4161/21624011.2014.958949
- Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013; 39:1-10; PMID:23890059; https://doi.org/10.1016/j.immuni.2013.07.012
- Breitbach CJ, Lichty BD, Bell JC. Oncolytic viruses: Therapeutics with an identity crisis. EBioMedicine 2016; 9:31-6; PMID:27407036; https://doi.org/10.1016/j.ebiom.2016.06.046
- Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol 2009; 21:346-51; PMID:19500964; https://doi.org/10.1016/j.coi.2009.05.016