
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Ebola virus (EBOV) infection is highly lethal and results in severe febrile bleeding disorders that affect humans and non-human primates. One of the therapeutic approaches for treating EBOV infection focus largely on cocktails of monoclonal antibodies (mAbs) that bind to specific regions of the EBOV glycoprotein (GP) and neutralize the virus. Recent structural studies using cryo-electron microscopy have identified key epitopes for several EBOV mAbs. While such information has yielded deep insights into antibody binding, limitations on resolution of these structures often preclude a residue-level analysis of EBOV epitopes. In this study, we performed combinatorial peptide-based epitope mapping of EBOV GP against a broad panel of mAbs and polyclonal sera derived from several animal species vaccinated with EBOV DNA and replicon vaccines and/or exposed to EBOV infection to identify residue-level determinants of antibody binding. The peptide-based epitope mapping obtained from a wide range of serum and mAb samples, combined with available cryo-EM structure reconstructions revealed fine details of antibody-virus interactions, allowing for a more precise and comprehensive mapping of antibody epitopes on EBOV GP. We show how these residue-level epitope definitions can be used to characterize antigenic variation across different filoviruses, and provide a theoretical basis for predicting immunity and cross-neutralization in potential future outbreaks.
Introduction
The genus Ebolavirus includes 5 known virus species (Bundibugyo, Ebola, Reston, Sudan, and Taï Forest viruses) that are endemic to Africa. These viruses affect humans, nonhuman primates (NHPs), or both, often lethally, by causing severe febrile bleeding disorders and organ failure.Citation1 According to the World Health Organization (WHO), the average fatality rate of Ebola virus (EBOV) is approximately 50%, with case fatality rates ranging from 25% to 90% in past outbreaks.Citation2 The most recent outbreak affecting human populations spread to various countries and led to more than 28,000 cases and over 11,000 fatalities. This unprecedented situation prompted the WHO to adopt emergency measures to contain the spreading of the disease for which there is no approved vaccine or antibody (Ab) therapy. The measures included testing of unapproved medical treatments with post-exposure therapeutics. Among these treatments, the use of monoclonal Ab (mAb) cocktails has demonstrated certain degree of success as a passive immunotherapy in NHPs and mice. Two of the most successful cocktails –MB-003, which contains 3 plant-derived mAbs c13C6, h-13F6, and c6D8,Citation3 and ZmAb, which contains mAbs c1H3, c2G4, and c4G74 – have now been combined into a third cocktail, ZMapp,Citation5 which is under development for mass production and continued human use.
The target for all Abs in these cocktails is the EBOV glycoprotein (GP), a class I fusion protein, constituted by 2 disulfide-linked subunits identified as GP1 and GP2. GP molecules assemble into trimer to form a chalice-shaped quaternary structure.Citation6 Three distinct regions can be identified in GP1: a core domain, a glycan cap, and a glycosylated mucin-like domain (MLD). The GP1 subunit also contains a recognition site that binds to the Niemann-Pick C1 receptor. On the other hand, GP2 contains a series of residues that constitute the main fusogenic element of GP, known as the internal fusion loop (FL). GP2 also contains 2 heptad repeat regions identified as HR1and HR2 which come together during fusion to form antiparallel α-helices. The HR2 region folds-back onto HR1 in a process that distorts the virus and host-cell bilayers and brings the 2 membranes into contact to form a hemifusion stalk.. Subsequent opening of the hemifusion stalk results in transfer of the viral genetic material into the host cell.
The mechanisms by which mAbs interact with epitopes on the EBOV surface and neutralize the virus are poorly understood. X-ray crystallography and cryo-electron microscopy (cryo-EM) methods have been used to determine the complexes of that the trimeric EBOV GP forms with mAbs, particulary, with those mAbs in the therapeutic cocktails ZMapp, MB-003, and ZmAbCitation7-9; those derived from human sera of survivors of the 2014 outbreakCitation10; a 1995 Kikwit EBOVCitation11 outbreak; and from EBOV immunized Chinese rhesus macaques.Citation12 However, in many of these cases, the resolution of the cryo-EM structures (> 20Å) is insufficient to provide residue-level information on the antibody epitopes. While these structures provide insight into the overall epitope regions of EBOV GP, residue-level information is needed to help determine how antigenic variation due to amino acid mismatches in GP1 between ebolaviruses can result in these mAbs and sera to lose their ability to neutralize other EBOV species.
The epitopes on a protein surface recognized by Abs are commonly associated with secondary structure elements such as loop regions or α-helices. In many cases, peptides derived from regions of the protein sequence containing such loops can retain secondary structural features, allowing Abs to recognize their epitope and bind both, peptide and protein with similar affinities. These linear B cell epitopes can be mapped by using overlapping peptide libraries that cover the entire protein of interest. In other cases, an Ab recognizes a conformational epitope produced by 2 or more fragments of the polypeptide chain which are far apart in the linear sequence, but come spatially close due to folding of the protein. Although linear epitopes have been associated with protection against infection and in vaccination studies, many protective and neutralizing epitopes tend to be conformational in nature.Citation13-19
One method for peptide-based epitope mapping involves the use of Chemically Linked Peptides on Scaffolds (CLIPS) peptides, where libraries of oligopeptides composed of up to 4 individual peptide fragments bound to a common scaffold can be synthesized.Citation20 Theoretically, within a CLIPS library, some of the peptides will present the appropriate order and configuration required to reconstitute conformational epitopes found on the protein antigen. However, the combinatorial complexity of the CLIPS library is immense because hundreds of epitopes are presented in combination with each other across thousands of peptide constructs. Here we present a novel method for analyzing this combinatorial peptide-based epitope mapping data to identify linear and conformational epitopes in the EBOV GP in a wide range of mAbs and polyclonal sera from animals infected with EBOV and/or immunized EBOV DNA or replicon vaccines. Many of the serum samples from DNA-based vaccines, and additional mutagenesis data used in the present study are described in an accompanying article by Mitchell et al.Citation21 in this special issue. By combining epitope mapping information across large combinatorial peptide libraries, and across a wide range of antigen conditions, we aimed to 1) develop a comprehensive list of EBOV GP epitopes and 2) identify residue-level determinants of binding for EBOV Abs. This information can provide additional insight into the molecular determinants of epitope regions identified in existing cryo-EM studies and provide a basis for analyzing and predicting the effect of antigenic variation in future EBOV disease outbreaks.
Results
CLIPS screening
Peptide libraries covering the entire EBOV GP amino acid sequence were used to map immunologically important regions on the protein. In an attempt to mimic linear and conformational epitopes on the GP surface, a series of CLIPS were synthesized and screened using mAbs and sera from vaccinated and/or convalescent animals. These CLIPS were constructed using various scaffold combinations to produce 2 peptide libraries, LIB1 and LIB2 containing 7286 and 6518 oligopeptides, respectively. These complex CLIPS libraries were expected to produce different arrangements of the component peptide fragments, leading to conformationally-constrained epitopes similar to those present on the surface of EBOV GP trimers. For screening purposes, these CLIPS were bound to solid supports where they could fold, or become constrained in higher order structures such as β-sheets, α-helices and double and triple loops. The type and number of CLIPS included in both libraries are presented in . LIB1 was used for an initial screening intended to identify the most reactive regions of the EBOV GP sequence, whereas LIB2 contained CLIPS synthesized for fine mapping of the most reactive regions previously identified with LIB1. It is important to note that LIB1 and LIB2 represent combinatorial libraries, and even though LIB2 peptides were derived from the epitopes identified in LIB1 screening, they are arranged in different combinations and configurations from LIB1 to further identify possible conformational epitopes. As such, LIB2 presents a different, but related, library of epitopes from LIB1, and while we can expect a general agreement between the 2 libraries, one cannot serve as a validation for epitopes identified in the other.
Table 1. CLIPS peptides.
To reveal both continuous and discontinuous epitopes on GP, the CLIPS libraries were probed with mouse mAbs chosen for their ability to passively protect animals, as well as immune sera from mice, guinea pigs, and NHPs that were immunized with EBOV DNA or replicon vaccines and/or infected with EBOV. As negative controls, we screened all CLIPS using sera from naïve or mock-vaccinated animals.
The details of each serum or mAb sample used in this study are listed in . Polyclonal serum samples were obtained from mice or guinea pigs experimentally vaccinated with DNA constructs expressing EBOV GP, and from rhesus and cynomolgus macaques that survived EBOV infection. Samples also consisted of mouse mAbs 13F6, 13C6, 6D8, 6D3 and 6E3.Citation22 The remaining samples contained pooled sera from mice, guinea pigs, and NHPs vaccinated with EBOV DNA or replicon vaccines, or from convalescent NHPs exposed to EBOV. Additional details on the origin of the samples are provided in an accompanying article by Mitchell et al in this special issue.Citation21
Table 2. Samples tested for epitope mapping.
One major limitation to the present study involves serum sample availability. Because of the wide range of serum samples and host organisms used in this study, from mouse to guinea pig to Rhesus macaques and crab-eating macaques, we were limited to serum samples that were available from many previously conducted experiments. As such, subject-matched ‘pre-immune’ samples were typically not available. We had negative controls matched to each host species to distinguish antigen-specific binding from non-specific binding for both LIB1 and LIB2 screening, except in the LIB1 screening for guinea pig samples, where no negative control sample was available. Furthermore, because different serum samples were available at different stages of the study, we were not able to directly compare the LIB1 and LIB2 epitope mapping results with the same polyclonal serum samples. However, since one of the primary goals is to comprehensively survey the epitope landscape of Ebola GP, the presence of an epitope in the mapping of a given serum sample is more important than the lack of a given epitope in a particular sample. In the subsequent analyses, we combined all the epitopes identified in LIB1 and LIB2 to assemble a comprehensive map of Ebola GP epitopes. As the number of samples, and the breadth of samples (with respect to host organism and antigen condition), increases, there is a higher likelihood that our combined epitope map will capture all possible Ebola GP epitopes.
Epitope mapping
To determine the most antigenic fragments on the EBOV GP sequence, we developed a statistical technique to analyze the OD binding signals produced by the complete set of CLIPS interacting with sera from each specific sample. This procedure combines the OD measurements with sequence information to generate a set of scores representing the propensity of residues from the EBOV GP sequence to bind Abs from a particular sample. In other words, this method assigns each residue p on the GP sequence, scores and
which measure the likelihood of that residue being part of an epitope recognized by Abs from a given sample s.
is designed to detect residues associated with linear epitopes and Rs(p) is designed to indicate the presence of potential conformational epitopes. These scores are expected to provide a measure of specific binding, i.e., binding due to Abs elicited by EBOV infection or the application of Ebola vaccines. To such end, we use information derived from the negative controls samples, as described in “Methods: Detection of specific binding.”
The initial screening by LIB1 was performed using the 15 samples described in , which included mAb 13F6 as an additional control. The epitope of mAb 13F6 is linearCitation22 and we used this sample to evaluate the screening capabilities of our experimental techniques to test our computational analysis tools. We used the scores to identify highly antigenic regions of the sequence. We classified residues as showing strong, medium and weak binding based on
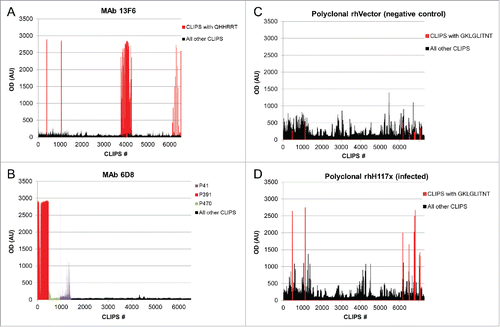
value thresholds of greater than 50, 20 and 10, respectively. shows the OD binding signals for mAb 13F6 and 6D8 when screening against LIB1 and LIB2, respectively, and for the negative control and Ebola-infected polyclonal Rhesus macaque samples screened against LIB1. Peptides containing epitopes identified to have high
scores are colored correspondingly, while all other peptides are shown in black. Overall, we found that in many cases, peptides containing epitopes were clearly distinguishable from other peptides, especially in the case of mAbs with linear epitopes. By contrast, polyclonal sera from negative controls, such as either naïve animals or animals vaccinated with a vector-only control, showed higher background signals across the peptide library. Still, for most cases in the polyclonal sera from infected or immunized animals, our analysis was able to identify epitope regions that consistently showed higher binding signals compared with the background signal found in the corresponding negative controls.
Figure 1. Combinatorial peptide-based epitope mapping of monoclonal and polyclonal sera. Representative examples of epitope mapping of mAbs and polyclonal sera are shown. OD signals, in arbitrary units (AU) are shown for screens of mAb 13F6 against the LIB1 CLIPS library (A), mAb 6D8 against the LIB2 CLIPS library (B), polyclonal sera from a negative control rhesus (rhVector) and polyclonal sera from a Ebola-infected rhesus (rhH117X) against the LIB1 CLIPS library (C and D, respectively). Peptides containing epitopes identified through the combinatorial analysis are colored as described in the respective legends for each sample; all other peptides are shown in black
A summary of the regions in the GP sequence exhibiting high probability scores according to our analysis of 15 serum and mAb samples (see ) screened against LIB1 is shown in . Likewise, a summary of the antigenic regions of GP identified from the analysis of 19 serum and mAb samples screened against LIB2 is shown in . Unfortunately, due to differences in sample availability between the 2 phases of the study, only 2 samples (mAb 13F6 and 13C6) were included in both LIB1 and LIB2 screens.
Figure 2. Initial epitope mapping of EBOV GP: The non-weighted (R) and weighted (Q) scores derived from the OD signals produced during screening of CLIPS from LIB1 are shown.
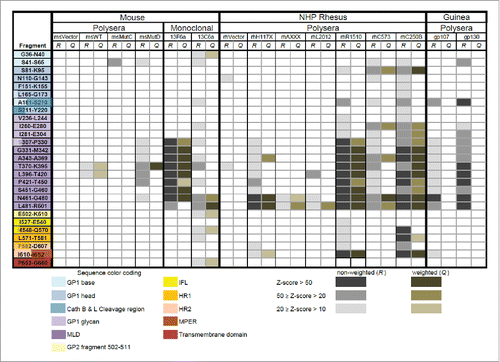
Figure 3. Fine epitope mapping of EBOV GP: The non-weighted (R) and weighted (Q) scores derived from the OD signals produced during screening of CLIPS from LIB2 are shown.
Polyclonal sera
Polyclonal serum samples that served as negative controls showed little or no binding to oligopeptides in the LIB1 or LIB2 libraries as assessed by our Z-score analysis ( and ). This shows that background signals due to non-specific binding are relatively rare in this analysis and suggests that observed binding signals likely indicate specific binding, regardless of whether a negative control is available for comparison. For some polyclonal serum samples from vaccinated animals such as msMutC and msVRP from mouse, gpVRP from guinea pig and rhCVRP from rhesus, we found binding that was relatively weak (just above threshold) or non-existent. The reason for the weak binding in these samples is unclear – it may be due to poor sample quality or low vaccine-induced antibody responses in these animals.
Overall, across all the polyclonal serum samples tested, we were able to identify 13 highly antigenic fragments, hereafter referred to as epitopes, on the sequence of EBOV GP based on the score computed with a cutoff value of 20 (). For convenience, these peptide fragments are identified by using the letter “P” followed by a number that corresponds to the global position in the EBOV GP sequence of the first residue of the fragment under consideration.
Table 3. Epitope regions in Ebola GP.
There was significant overlap in the epitopes identified by the different polyclonal sera in the LIB1 screen (). A stretch of the sequence spanning residues S307 to R501 that include peptides P391 and P470 listed in , showed significant responses (i.e., with R and/or Q scores greater than 50) in 3 out of 6 mouse samples (msMutD, 13F6a and 13C6a), both guinea pig serum samples, and 4 of 7 NHP serum samples (rhH117X, rhR1510, rhC573 and rhC250B), as shown in . A portion of that region, from residues 461 to 501, was identified in 8 out of 13 polyclonal samples with R and/or Q scores greater than 20. As expected, the negative control samples (msVector and rhVector) did not show binding except for low R scores for the fragments N110-G143 and T370-K395 on rhVector ().Citation23 The LIB2 screen also identified several common epitope regions across serum and mAb samples (). Fragments 391–415 and 470–494 were both recognized in 2 out of 5 mouse polyclonal samples. In addition, a stretch of 5 peptides spanning 189 to 308 was recognized in 3 out of 5 mouse polyclonal samples. Overall, fragments 391–415 and 470–494 were found to be highly reactive in samples from mice. In the guinea pig, and NHP serum samples the overall level of specific binding was low, despite the fact that some (non-specific) binding was observed in the samples (see Supplemental Fig. S1). Only one of the NHP samples (i.e., rhVRP) showed specific binding within fragments 391–415 and 470–494.
Monoclonal antibodies
We performed the screening of the peptide libraries using 5 mAbs: 13F6, 13C6, 6D3, 6E3, and 6D8. Binding signals detected for all 5 mAbs and the corresponding epitope regions are summarized in . To reduce the chances of errors in the epitope assignment that may originated from non-specific or spurious binding, we do not report in fragments associated with weak binding (i.e., R and Q scores below 20). Screening of both LIB1 and LIB2 with mAbs 13F6 and 6D8 produced the strongest binding signals for CLIPS that included fragment 391–415, consistent with previously identified linear epitopes for both mAbs ( and ).Citation22 Screening of LIB2 with mAb 6E3 produced high R and Q residue-scores for a fragment 391–415, which also agrees with a previously identified epitope for this mAb.Citation22
Table 4. Monoclonal Ab epitopes.
Probable binding for other fragments was found during screening of LIB2 for mAbs 6D3, 13F6, and 6D8. We found that the R and Q scores produced by our method point unequivocally to residues in the actual linear epitopes of mAbs 13F6, 6D8 (see and ), and 6E3. The assignment of conformational epitopes in mAbs 6D3 and 13C6 was more challenging. For mAb 6D3, our method identified fragments 201–206, 240–270, 286–291 and 470–494 as probable epitopes. Of these fragments, 201–206 and 240–270 are consistent with the data from Shedlock et al.Citation24 In our binding assays, we found that a single CLIPS, containing fragment Y261-TSGKRSNTTGKLI-W276, generated a signal that far exceeded all others (see Figure S2). For mAb 13C6, binding to 2 fragments corresponding to a stretch of residues from 461 to 501 was detected in the LIB1 screen, and a similar region, represented by fragment 470–494 was identified in the LIB2 screen. Because this epitope definition for 13C6 is at odds with previous studies that pointed to residues in the glycan cap as epitopes,Citation7,25,26 we repeated the experiment with a new aliquot of 13C6 and found the same result. We also examined the probability scores for fragments corresponding to the previously identified epitope for 13C6, including residues 270–27225,26, but found no evidence of specific binding. CLIPS-based screens of both LIB1 and LIB2 definitively identified residues 470–494 to be associated with 13C6 binding.
Overall we found good agreement between epitopes identified in this study with both the mutational analysis performed in an accompanying article in this special issue by Mitchell et al.,Citation21 and with previous experiments characterizing both residue- and fragment-level epitope definitions (). In particular, mutational analysis identified key residues in the epitope definitions for mAbs 13F6, 6D8, 6E3 and 13C6, providing an independent validation of our combinatorial peptide-based epitope mapping approach.
Mapping epitopes to GP structure
We modified the initial model of EBOV GP structure derived from X-ray crystallographic dataCitation6 by using homology-based modeling. With the exception of those residues belonging to the MLD, we added all other missing residues in the GP monomer, and subsequently built a 3D model of the chalice-shaped GP trimer. We used the models to visualize the approximate expected location of the Ab binding regions identified during CLIPS screening. shows 9 of the 13 epitopes mapped onto the structural model of the GP trimer based on whether they were found in vaccinated (left) or infected (right) animals. The 2 other epitopes detected by CLIPS screening are part of the unstructured MLD. Overall, the 13 epitopes associated with Ab binding can be ascribed to 5 regions on the surface of the GP trimer: 1) the MLD, 2) the glycan cap subdomain, 3) the central region of the chalice, 4) the base of the GP chalice, and 5) the membrane-proximal external region (MPER).
Figure 4. Antigenic regions of GP derived from CLIPS screening. A surface model of the GP trimer is shown in white. Smooth regions of the model correspond to regions of the structure determined experimentally.Citation6 Regions that were modeled “de novo” are shown as collections of dots are used to visualize the approximate location where missing epitopes are located. The 3D representation omits the MLD, which is considered unstructured. Nine of the 13 epitopes identified as the most antigenic regions of the GP sequence, based on our analysis, are indicated in various colors. The remaining 2 epitopes identified with CLIPS fall within the MLD, and are not shown in the present structural model. A separate model is shown from epitopes from (A) EBOV DNA and replicon vaccines and (B) for EBOV infections.
It is possible that the epitopes presented during immunization and vaccination may differ in a systematic manner. To address this, we compared the composite epitope maps from vaccinated and infected animals with the understanding that a direct within-host comparison may be confounded by the differences in the host organisms and antigen conditions between the infections and vaccinations used in this study. Overall, we found substantial similarity in the areas of the chalice that were recognized in infection and vaccination, with 7 fragments in common (P189, P256, P281, P391, P470, P544 and P610). In particular, CLIPS derived from the MLD region, primarily P391 and P470 (not shown in ), were well recognized in samples from infected animals, and also detected in samples derived from vaccination.
We did observe several differences between the infection and vaccination epitope maps. We found that antibody responses from vaccinated animals bound strongly to fragments P36 and P41 associated with the base region of GP1, while recognition of these fragments was poor in samples from infected animals. On the contrary, fragment P81, associated with the GP1 head region, was recognized in samples from infected animals, but was found to bind poorly in samples obtained from vaccinated animals. Finally, CLIPS mapping to fragment P582 in the HR2 and MPER regions of GP, elicited strong binding signals with samples from infected animals, but binding went mostly undetected in samples from vaccinated animals.
A substantial amount of data on mAbs complex with EBOV-GP derived from cryo-EM and X-ray crystallography have recently become available. Such data are providing important information on the regions of the GP trimer surface targeted by mAbs, primarily of human origin.Citation7-12 We conducted an analysis of the available structural information with the results from our CLIPS-based epitope detection approach. For this, we retrieved 19 cryo-EM maps of complexes of mAbs with EBOV GP trimers from the EM Data Bank.Citation27 A list of the maps used for this analysis is provided in . A total of 14 out of 19 EM maps have been obtained at resolutions exceeding 20Å, and consequently, only provide a coarse definition of these epitopes. We superimposed all the electron density maps onto our 3D homology model of the GP trimer. The fragments identified as binding strongly according to our CLIPS analysis showed good agreement with regions of the GP surface where mAbs are found to bind experimentally.
Table 5. X-ray crystallography and cryo-EM structures used for epitope mapping.
In , for each epitope fragment identified by our peptide-based epitope mapping, we list Ebola GP-specific mAbs that have previously been shown to bind to those epitope regions. It is important to note that since the epitope list in came from mapping of polyclonal sera, it represents the aggregate binding of many monoclonal antibodies that each bind to distinct, and possibly overlapping epitopes. As such, it is unlikely that any single monoclonal antibody will precisely reproduce the polyclonal epitopes identified here.
As examples, EM density maps of the EBOV GP trimer bound with mAbs c4G7 and ADI-15731, superimposed onto a structural model of the GP trimer in and , respectively. In , a cross-section of the electron density corresponding to the bound mAb c4G7 exposes the side of the GP chalice, revealing fragments P36, P41, P502 and P544, which were identified with CLIPS. Similarly, shows the binding site of the mAb ADI-15731 located in the glycan cap subdomain, on top of the GP chalice. A slice through the EM density corresponding to the bound ADI-15731 reveals a surface region largely made up of fragments P256 and P281 from CLIPS. Supplemental Figs S3 through S5 show similar superposition of density maps of complexes involving representative examples from mAbs described in . Strikingly, in every case except in one (mAb100), the epitope regions identified in the cryo-EM density map corresponded to one or more of the epitope fragments shown in . This suggests that the epitope fragment list in may capture all major epitope regions of EBOV GP.
Figure 5. (A) Electron density map (colored green and red) of the complex of mAbs c4G7 and c13C6 with the GP trimer. The c4G7 mAbs bind to the middle of the chalice viewed here from a side view. Two c4G7 mAbs are modeled by a smooth surface representation colored in gray, and fitted within the electron density map. The third c4G7 model has been omitted, and the corresponding regions of the electron density map were sliced to show the antibody binding site formed by epitopes P41, P506, and P544. (B) Electron density map (in blue) of the complex of mAb ADI-15731 with the GP trimer viewed from the top. Two ADI-15731 mAbs, modeled as smooth surfaces in gray, are fitted within the electron density map on top of the glycan cap of GP. A third mAb has been omitted and the electron density map sliced to show the antibody-binding site formed primarily by epitopes P256 and P281.
For mAb100, Misasi et al. reported binding to the FL of GP.Citation11 Our CLIPS analysis shows little or no binding to peptides containing residues from the FL region by Abs in the polyclonal sera. However, most of the remaining key contacts of the antibody with GP are mapped to the interactions with residues in fragments P41, P506 and P544. The quaternary epitope recognized by mAb100 appears to include residues from the β13-β14 loop in GP included in the fragment P189. P189 was identified in our work as a strong antigenic region but we have found no report based on structural data to confirm our findings. We note, however, that the cryo-EM structural study of mAb100 shows weak electron density that the authors ascribed to residues from the β13-β14 loop interacting with Fab100. Binding of mAb100 to GP reduces cathepsin L cleavage of the β13-β14 loop, and removal of the loop affects slightly Ab binding.Citation25 Interestingly, Misasi et al. found that removal β13-β14 loop has a stronger effect in mAb KZ52 binding than in mAb100. Specifically, at pH 5.3, Misasi et al. detected a reduction in the affinity of KZ52 for thermolysin-cleaved GP by a factor of more than 1000 as compared with uncleaved GP. The conformational epitope of KZ52 was reported to involved EBOV GP: residues 42–43 at the N terminus of GP1, and 505–514 and 549–556 at the N terminus of GP2. (Lee et al). In an attempt to understand how KZ52 binding is affected by thermolysin cleavage, we superimposed the electron density maps of a KZ52 – GP-trimer complex (emd 6153) to that of Fab100-GP complex (emd 3310). Such exercise revealed that a portion of the electron density in the Fab100-GP complex, broadly associated with the β13-β14 loop, overlaps slightly with the electron density for a KZ52 Fab in the other complex. Since this is the only portion of the fragment removed by thermolysin in close proximity to the KZ52 binding site, the large change in affinity due to cleavage suggests that P189 may also be part of the conformational epitope of KZ52.
Antigenic variation in EBOV GP epitopes
Single mutations in an epitope can have large effects in the binding affinity of an Ab. Conversely, Abs binding to epitopes that are associated with highly conserved residues may have the potential to show broad cross-reactivity across various strains or even species. Using combinatorial peptide-based epitope mapping we identified 13 epitopes of EBOV GP1. Here we sought to measure the degree of sequence conservation within these epitope regions to determine how antibody binding to these epitopes may be sensitive to antigenic variation across EBOV species.
We retrieved all complete GP sequences for the 5 Ebolavirus species, Zaire, Bundibugyo, Sudan, Reston and Tai Forest from the NCBI sequence database,Citation28 and performed a multiple sequence analysis for each species using unique sequences only. The results from this analysis are shown in where residue conservation is plotted as a function of residue position in the EBOV GP sequence. Residue conservation associated with the 13 epitopes identified from the CLIPS analysis for each 5 species is highlighted. The graph shows that epitope fragments within the central portion of the GP sequence, residues 280 to 450, tend to be less conserved (< 90%) than those located toward the N- and C-termini of this region. Additional details on the sequence variability affecting the 13 epitope fragments, summarizing the specific positions where residue variability occurs in Zaire ebolavirus, and the substitutions and residue-variability found in the other 4 species are shown in . Overall, the N and C-terminal peptides P36, P41, P506, P81, P544, P582 and P610, showed high sequence conservation across ebolaviruses, whereas peptides P189, P211, P256, P281, P391, and P470 showed substantial sequence variation.
Figure 6. Plot of residue-type conservation as a function of position in the EBOV GP sequence derived from a multiple sequence alignment of the Ebolavirus genus. The 13 regions from the EBOV GP sequence identified as highly antigenic are indicated in red over the horizontal axis. The residue conservation on these epitopes is color-coded based on the EBOV species.
Figure 7. Summary of the multiple sequence alignment analysis of the Ebolavirus genus. Sequences of the 13 epitopes identified in the CLIPS study are listed using a single-letter residue code. Letters inside the black boxes indicate the non-conserved residues in Zaire ebolavirus. Below the sequence of each fragment, we indicate the observed residue substitutions in the Bundibugyo, Tai Forest, Reston, and Sudan species. Dashes indicate positions with residue variability in species other than Zaire ebolavirus.
P391 and P470, which show the highest antigenic variation are located on the MLD, whereas P211, P256, and P281, which show more moderate variation are found on the glycan-cap region (). The highly conserved epitope fragments in the N and C-terminal regions are largely found in the base of the chalice and the MPER. These results suggest that, while the MLD may be highly immunogenic, it may also largely elicit a strain or type-specific response, whereas antibodies targeting the GP1 chalice, and in particular, its base, may be more cross-reactive across EBOV species.
Discussion
By screening CLIPS libraries with polyclonal sera from 4 different animal species, and mAb samples, we were able to identify 13 highly antigenic regions within the EBOV GP sequence. Mapping of these 13 regions to the EBOV GP1 structure showed that primarily 5 areas of the protein were targeted: the MLD, the glycan-cap region, the central region of the chalice, the base of the chalice, and the MPER. A comparison with the existing structural data on over 20 mAbs, suggested that these fragments constitute the major epitope regions of EBOV GP. Additionally, multiple sequence analyses showed that fragments P36, P41, P81, P189, 211, P256, P506, P544, P582, and P610 belong to regions of the GP sequence having low variability, particularly in Zaire ebolavirus. These epitopes are primarily located in the glycan-cap region and the base of the chalice in the GP1 structure. Therefore, vaccines that focus the antibody response to these regions of EBOV GP may have the potential to protect across a broader range of EBOV species.
We also identified several host-specific differences in the fine-specificity of the polyclonal antibody response to EBOV GP, although further research is warranted due to the small number of samples for each animal species studied here. The MLD, particularly P391 and P470, was the most antigenic region for samples from mice in screens with LIB1 and LIB2. Similar results were obtained for samples from guinea pigs and NHPs during screens with LIB1. However, screens with LIB2 showed specific binding to the MLD region only in one of the rhesus samples. Antibody binding to the glycan cap region was detected in rhesus macaques and guinea pigs, but not in mice during screens with LIB1. On the other hand, binding to such region was detected during screen of LIB2 with 3 polyclonal mouse samples (msZ19, msZ16, and msCVRP), but no specific binding was found during the screens with LIB2 for samples from guinea pigs or NHP vaccinated with the VEEV virus-replicon particle-based vaccine. Binding to the central chalice region was detected in NHPs and mice, while binding to the base of the chalice was found in all 3 species. Finally, binding to the MPER of GP was found primarily in samples from rhesus macaques in LIB1, and one sample of guinea pigs in screens with LIB2. Differences between the LIB1 and LIB2 results may be, in part, due to the fact that different serum sample were used for each screen.
In addition to epitope mapping of polyclonal sera, we also identified epitope regions for several EBOV mAbs, some of which have been extensively characterized. We performed the screening of the peptide libraries on 5 mAbs: 13F6, 13C6, 6D3, 6E3, and 6D8. Monoclonal Abs 13F6, 6E3, and 6D8 recognized linear epitopes.Citation22 The epitopes recognized by mAbs 13C6 and 6D3 have also been identified as conformational. According to Shedlock et al., both of these mAbs bind GP mutants lacking fragments 302–479 or 494–635 but not to mutants where fragment 49–277 is removed,Citation24 indicating that epitopes of these mAbs should be within the later fragment of GP. A low-resolution cryo-EM structure of c13C6 bound to a GP trimer has also shown that the 13C6 epitope resides on the glycan cap region of GP.Citation7 Furthermore, using alanine scanning techniques, Davison et al. identified residues T270 and K272 as critical for 13C6 binding to GP.Citation25 In addition, a report by Kugelman et al. on the emergence of EBOV escape variants described a series of escape mutations involving residues G271 and K272 which substantially reduced the binding of mAb 13C6.Citation26
Previous studies have shown that mAb 13C6 binds to a conformational epitope consisting of residues 270–272 in the glycan cap region of EBOV GP. However, in this study, CLIPS-based peptide scanning from both LIB1 and LIB2 peptide screens identified a span of residues from 461 to 502 in EBOV GP as being associated with 13C6 binding. Mitchell et al. performed positional mutation scanning, where all possible single-point (non-cysteine) mutations along the peptide P470 were made and binding to 13C6 was measured by ELISA.Citation21 They found that mutations in S475-I482 and G488-T494 significantly abrogated 13C6 binding, which corroborate our findings of residues 461–502 as being involved in a 13C6 epitope. Our results for 13C6 lead us to conclude that the Ab may show some promiscuity in its binding. If 13C6 is capable of binding to 2 different epitope regions of EBOV GP, one corresponding to residues 270–272 and the other, to residues 475–494, a common sequence motif between these epitopes that is recognized by this mAb may exist. We compared the 2 epitope regions and identified a tripeptide motif – GKL –that was common to both regions (residues 271–273 and residues 488–490, respectively). In support of this, the most recent high-resolution structural data of mAb 13C6 bound to GP and sGP shows the G271-K272-L273 tripeptide plays a central role as part of the conformational epitope of the Ab.Citation8 Furthermore, the positional mutation scanning of 13C6 along P470 described by Mitchell et al. resulted in a loss of binding when residues in the GKL tripeptide and neighboring positions were substituted by negatively-charged GLU or ASP residues.Citation21 Mapping these residues onto the structure of Ab-GP and Ab-sGP complexes, revealed that such substitutions in the GKL epitope core (GP residues 269 to 273) would likely generate strong repulsive electrostatic interactions with 3 negatively charged residues nearby, D54, D56, and D100A, in the c13C6 paratope. These results support the hypothesis that 13C6 binds to 2 separate epitopes on EBOV GP through a common GKL tripeptide motif.
Finally, our results agree well with a recent study on the conservancy of mAb epitopes from previous outbreaks.Citation29 Based on a comparison with that work, all functional epitopes reported in the Immune Epitope Database, which includes mAbs for which structural information is unavailable, can be assigned to 7 of the 13 epitope regions identified in this study. This further supports our finding that these epitopes represent a comprehensive description of all the major epitope regions in EBOV GP.
Conclusions
This work shows that CLIPS technology constitutes a useful tool for mapping antigenic regions of a molecule. Screening of serum samples from different animal species and with mAbs resulted in the detection of 11 highly immunogenic fragments in the GP sequence. When these fragments were mapped into a structural model of the GP chalice, they defined 5 distinct areas on the surface of the GP trimer. A comparison with existing structural data from Abs bound to GP showed that the fragments identified by CLIPS coincided well with sites where the Abs are experimentally detected to bind. The information provided by our CLIPS binding assays thus complements that obtained from structural studies. Combining CLIPS analysis, coupled with positional scanning, enabled a fine mapping of the interaction sites of the Abs to detect residues most likely to contribute to binding.
Notably, our study revealed that mAb 13C6 can bind promiscuously to EBOV GP and certain oligopeptides. The common element of recognition appears to be the tripeptide GKL. As shown by X-ray crystallography of complexes with the GP trimer and sGP, 13C6 binds to a conformational epitope that includes residues G271-K272-L273. However, our CLIPS analysis suggests that 13C6 also recognizes linear fragments that include residues surrounding the GKL tripeptide at position 477 to 479 of EBOV GP1.
Materials and methods
Screening of CLIPS
The set of CLIPS was synthesized using the sequence of the Mayinga strain of EBOV virus, (NCBI Accession No. AAN37507.1). For screening purposes, CLIPS are bound to solid supports where they are folded or constrained to mimic higher order structures such as loops, β sheets, and α-helices.Citation20 Both continuous and discontinuous epitopes can be revealed by probing the CLIPS with immune sera or mAbs.
Analysis of epitope mapping data
Sample distribution
We analyzed the Optical Density (OD) data corresponding to each sample used in the Pepscan experiments as follows,
| 1. | We computed the average OD value, | ||||
| 2. | To determine the distribution of the data, we generated a histogram for the magnitude Δx, which is given by: | ||||
Detection of binding
While a strong OD signal is indicative of Ab binding, OD measures are not actual true/false signals that unequivocally distinguish “binding” from “absence of binding;” and the assignment of an intermediate OD value is often difficult or impossible. To detect putative binding regions on EBOV GP, we computed 2 probability scores for each residue in the target protein: and
(where s indicates the sample under consideration and p the position of the residue in the sequence of the target protein). Both probability scores are associated with the overall binding strength at a particular residue in the protein sequence. The procedure we used to obtain them is as follows:
We used a sliding window encompassing m residues (with m ≥ 4) that runs over the entire GP sequence. The size of the window, m, was selected in such a manner that every m-letter word defined by the window is unique. The later requirement guarantees that the contributions to the probability scores come from peptides designed to map the same specific amino acid region in the sequence.
For each word generated, we produced a subset
of OD measurements from sample s. To be included in
, a measure was required to satisfy the following conditions: (a) the associated peptide should contain the word
as part of the sequence, and (b) the measured OD value,
, should represent probable Ab binding, which we defined as a value that exceeded the threshold
, where the parameter k represents an adjustable integer that allowed us to test more restrictive binding assumptions by imposing higher thresholds at higher k values.
For each word a Z-score,Citation30
, was computed as described in Eq Equation1
(1)
(1) .
(1)
(1) where
is the mean OD value of
, and n corresponds to the number of measures included in such subset. This Z-score is expressed in units of the standard deviation of the distribution.
The score for a residue at position p in the GP sequence was computed as a weighted-average of the Z-scores from the set of m words
(i = 1, …, m) that include position p, according to the formula:
(2)
(2) where
corresponds to the number of measures included in the subset
.
To compute the score for residue p in the GP sequence, we use a different weighted-average of the Z-scores:
(3)
(3) where
represents the total number of measures (binding and non-binding) from sample s that contain the word
. The additional factor
rewards words
that appear with high frequency in measures associated with binding, i.e., when ni →Ni. Thus, the
score is expected to be large for residues in words that constitute the core of linear epitopes. On the other hand, residues with large
but low
scores may indicate the presence of a conformational epitope, because the latter requires the presence of more than one peptide fragment. By construction, both
and
scores are positive, and their values are indicative of the strength of the OD binding signal at site p.
Detection of specific binding
To differentiate between antigen-specific and non-specific binding, we adapted the above procedure to include information from negative control (nc) samples. Because the Pepscan experiments included data from nc samples, step 2 of the procedure described above for Detection of Binding was modified by requiring that any measure to be included in
also satisfy the following condition:
where
is the corresponding OD value measured for the same peptide in the nc sample, and
and
represent the mean OD value and corresponding standard deviation, respectively, of the nc sample. Thus, the new condition established that detection of specific binding should only consider data from peptides showing a high OD signal in sample s, and a corresponding low OD (i.e., no binding) signal in the nc sample.
Sequence analysis
We initially obtained 1189 full-length sequences from the National Center for Biotechnology Information (NCBI) (retrieved on 11/30/2016) corresponding to the Ebolavirus genus GP. After elimination of identical sequences, we used the program MUSCLECitation31 to carry out multiple sequence alignments and derived information on residue conservation.
Generation of structural models of GP
We built 3-dimensional (3-D) models of the EBOV GP, using the Protein Structure Prediction Pipeline (PPSP) package.Citation32 Models of the EBOV GP were generated by using the X-ray crystallographic data of the trimeric form of GP in complex with MAb KZ52 (PDB id: 3CSY) as the template.Citation7 Because the MLD is considered unstructured, our GP model omits residues from such domains.
The structural superposition of the best GP1-GP2 monomer model and its template, monomer 3CSY, shows that few distortions were needed to accommodate the new sequence. The optimal superposition between the template and model was obtained by matching 313 Cα atoms with a root mean square difference (rmssd) of 0.47 Å. Superposition of the model and template was performed using the program PYMOL.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Material.pptx
Download MS Power Point (4.1 MB)Acknowledgments
We thank Dr. Ilja Khavrutskii for helping with the multiple sequence alignment analysis. We would also like to thank Dr. Mariano Sanchez-Lockhart and Dr. Gustavo F. Palacios for their help in revising the manuscript.
Funding
The authors were supported by the US. Army Medical Research and Materiel Command (Ft. Detrick, MD), the US Department of Defense High-Performance Computing Modernization Program, and the Defense Threat Reduction Agency. The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the US. Army or the US. Department of Defense.
References
- Kuhn JH. Filoviruses. A compendium of 40 years of epidemiological, clinical, and laboratory studies. Arch Virol Suppl 2008;20:13-360. PMID:18637412
- World Health Organization. 2016 Ebola virus disease Fact Sheet. Retrieved from http://www.who.int/mediacentre/factsheets/fs103/en/.
- Olinger GG, Jr, Pettitt J, Kim D, Working C, Bohorov O, Bratcher B, Hiatt E, Hume SD, Johnson AK, Morton J, et al. Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc Natl Acad Sci U S A. 2012;109(44):18030-5. doi:10.1073/pnas.1213709109. PMID:23071322
- Qiu X, Audet J, Wong G, Pillet S, Bello A, Cabral T, Strong JE, Plummer F, Corbett CR, Alimonti JB, et al. Successful treatment of ebola virus-infected cynomolgus macaques with monoclonal antibodies. Sci Transl Med. 2012;4(138):3003876. doi:10.1126/scitranslmed.3003876
- Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514(7520):47-53. PMID:25171469
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454(7201):177-82. doi:10.1038/nature07082. PMID:18615077
- Murin CD, Fusco ML, Bornholdt ZA, Qiu X, Olinger GG, Zeitlin L, Kobinger GP, Ward AB, Saphire EO. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc Natl Acad Sci U S A. 2014;111(48):17182-7. doi:10.1073/pnas.1414164111. PMID:25404321
- Pallesen J, Murin CD, de Val N, Cottrell CA, Hastie KM, Turner HL, Fusco ML, Flyak AI, Zeitlin L, Crowe JE, Jr, et al. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies. Nat Microbiol. 2016;1(9):16128. doi:10.1038/nmicrobiol.2016.128
- Tran EE, Nelson EA, Bonagiri P, Simmons JA, Shoemaker CJ, Schmaljohn CS, Kobinger GP, Zeitlin L, Subramaniam S, White JM. Mapping of ebolavirus neutralization by monoclonal antibodies in the ZMapp cocktail using cryo-electron tomography and studies of cellular entry. J Virol. 2016;90(17):7618-27. doi:10.1128/JVI.00406-16. PMID:27279622
- Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al.. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science. 2016;351(6277):1078-83. doi:10.1126/science.aad5788. PMID:26912366
- Misasi J, Gilman MS, Kanekiyo M, Gui M, Cagigi A, Mulangu S, Corti D, Ledgerwood JE, Lanzavecchia A, Cunningham J, et al.. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science. 2016;351(6279):1343-6. doi:10.1126/science.aad6117. PMID:26917592
- Zhang Q, Gui M, Niu X, He S, Wang R, Feng Y, Kroeker A, Zuo Y, Wang H, Wang Y, et al.. Potent neutralizing monoclonal antibodies against Ebola virus infection. Sci Rep. 2016;6:25856. doi:10.1038/srep25856. PMID:27181584
- Cerino A, Bremer CM, Glebe D, Mondelli MU. A human monoclonal antibody against Hepatitis B surface antigen with potent neutralizing activity. PLoS One 2015;10:4. doi:10.1371/journal.pone.0125704
- Read AJ, Casey JL, Coley AM, Foley M, Gauci CG, Jackson DC, Lightowlers MW. Isolation of antibodies specific to a single conformation-dependant antigenic determinant on the EG95 hydatid vaccine. Vaccine. 2009;27(7):1024-31. doi:10.1016/j.vaccine.2008.11.096. PMID:19095030
- Kuwata T, Takaki K, Yoshimura K, Enomoto I, Wu F, Ourmanov I, Hirsch VM, Yokoyama M, Sato H, Matsushita S. Conformational epitope consisting of the V3 and V4 loops as a target for potent and broad neutralization of simian immunodeficiency viruses. J Virol. 2013;87(10):5424-36. doi:10.1128/JVI.00201-13. PMID:23468483
- Varshney AK, Wang X, Cook E, Dutta K, Scharff MD, Goger MJ, Fries BC. Generation, characterization, and epitope mapping of neutralizing and protective monoclonal antibodies against staphylococcal enterotoxin B-induced lethal shock. J Biol Chem. 2011;286(11):9737-47. doi:10.1074/jbc.M110.212407. PMID:21233204
- Hou VC, Moe GR, Raad Z, Wuorimaa T, Granoff DM. Conformational epitopes recognized by protective anti-neisserial surface protein A antibodies. Infect Immun. 2003;71(12):6844-9. doi:10.1128/IAI.71.12.6844-6849.2003. PMID:14638771
- Wild MA, Xin H, Maruyama T, Nolan MJ, Calveley PM, Malone JD, Wallace MR, Bowdish KS. Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat Biotechnol. 2003;21(11):1305-6. doi:10.1038/nbt891. PMID:14555959
- Woollard DJ, Gauci CG, Heath DD, Lightowlers MW. Protection against hydatid disease induced with the EG95 vaccine is associated with conformational epitopes. Vaccine. 2000;19(4-5):498-507. doi:10.1016/S0264-410X(00)00192-4. PMID:11027814
- Timmerman P, Beld J, Puijk WC, Meloen RH. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem. 2005;6(5):821-4. doi:10.1002/cbic.200400374. PMID:15812852
- Mitchell DAJ, Dupuy LC, Sanchez-Lockhart M, Palacios G, Back JW, Shimanovskaya K, Chaudhury S, Ripoll DR, Anders Wallqvist A, Schmaljohn CS. Epitope mapping of Ebola virus dominant and subdominant glycoprotein epitopes facilitates construction of an epitope-based DNA vaccine able to focus the antibody response in mice. Hum Vaccin Immunother 2017 Jul 12:0. [Epub ahead of print]; doi:10.1080/21645515.2017.1347740.
- Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, Hart MK. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287(5458):1664-6. doi:10.1126/science.287.5458.1664. PMID:10698744
- Dowling W, Thompson E, Badger C, Mellquist JL, Garrison AR, Smith JM, Paragas J, Hogan RJ, Schmaljohn C. Influences of glycosylation on antigenicity, immunogenicity, and protective efficacy of ebola virus GP DNA vaccines. J Virol. 2007;81(4):1821-37. doi:10.1128/JVI.02098-06. PMID:17151111
- Shedlock DJ, Bailey MA, Popernack PM, Cunningham JM, Burton DR, Sullivan NJ. Antibody-mediated neutralization of Ebola virus can occur by two distinct mechanisms. Virology. 2010;401(2):228-35. doi:10.1016/j.virol.2010.02.029. PMID:20304456
- Davidson E, Bryan C, Fong RH, Barnes T, Pfaff JM, Mabila M, Rucker JB, Doranz BJ. Mechanism of binding to ebola virus Glycoprotein by the ZMapp, ZMAb, and MB-003 cocktail antibodies. J Virol. 2015;89(21):10982-92. doi:10.1128/JVI.01490-15. PMID:26311869
- Kugelman JR, Kugelman-Tonos J, Ladner JT, Pettit J, Keeton CM, Nagle ER, Garcia KY, Froude JW, Kuehne AI, Kuhn JH, et al.. Emergence of ebola virus escape variants in infected nonhuman primates treated with the MB-003 antibody cocktail. Cell Rep. 2015;12(12):2111-20. doi:10.1016/j.celrep.2015.08.038. PMID:26365189
- Lawson CL, Baker ML, Best C, Bi C, Dougherty M, Feng P, van Ginkel G, Devkota B, Lagerstedt I, Ludtke SJ, et al.. EMDataBank.org: unified data resource for CryoEM. Nucleic Acids Res. 2011;39(Database issue):D456-64
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733-45. doi:10.1093/nar/gkv1189
- Ponomarenko J, Vaughan K, Sette A, Maurer-Stroh S. Conservancy of mAb Epitopes in Ebolavirus Glycoproteins of Previous and 2014 Outbreaks. PLoS Curr. 2014;6. pii: ecurrents.outbreaks.f1a7028a13ce1c5f0bdbb4b0cc0b919b. doi:10.1371/currents.outbreaks.f1a7028a13ce1c5f0bdbb4b0cc0b919b
- Larsen RJ, Marx ML. An Introduction to mathematical statistics and Its applications. 3rd ed. Upper Saddle River, NJ: Prentice Hall. 2002:p. 282. ISBN 0-13-922303-7.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792-7. doi:10.1093/nar/gkh340. PMID:15034147
- Lee MS, Bondugula R, Desai V, Zavaljevski N, Yeh IC, Wallqvist A, Reifman J. PSPP: a protein structure prediction pipeline for computing clusters. PLoS One. 2009;4:e6254. doi:10.1371/journal.pone.0006254. PMID:19606223
- Warfield KL, Swenson DL, Olinger GG, Nichols DK, Pratt WD, Blouch R, Stein DA, Aman MJ, Iversen PL, Bavari S. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog. 2006;2(1):e1. doi:10.1371/journal.ppat.0020001. PMID:16415982
- Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, Young HA, Fredeking TM, Rote WE, Vlasuk GP. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet. 2003;362(9400):1953-8. doi:10.1016/S0140-6736(03)15012-X. PMID:14683653
- Grant-Klein RJ, Van Deusen NM, Badger CV, Hannaman D, Dupuy LC, Schmaljohn CS. A multiagent filovirus DNA vaccine delivered by intramuscular electroporation completely protects mice from ebola and Marburg virus challenge. Hum Vaccin Immunother. 2012;8(11):1703-6. doi:10.4161/hv.21873. PMID:22922764
- Pushko P, Bray M, Ludwig GV, Parker M, Schmaljohn A, Sanchez A, Jahrling PB, Smith JF. Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine. 2000;19(1):142-53. doi:10.1016/S0264-410X(00)00113-4. PMID:10924796
- Geisbert TW, Pushko P, Anderson K, Smith J, Davis KJ, Jahrling PB. Evaluation in nonhuman primates of vaccines against Ebola virus. Emerg Infect Dis. 2002;8(5):503-7. doi:10.3201/eid0805.010284. PMID:11996686
- Lee JE, Kuehne A, Abelson DM, Fusco ML, Hart MK, Saphire EO. Complex of a protective antibody with its Ebola virus GP peptide epitope: unusual features of a V lambda x light chain. J Mol Biol. 2008;375(1):202-16. doi:10.1016/j.jmb.2007.10.017. PMID:18005986
- Olal D, Kuehne AI, Bale S, Halfmann P, Hashiguchi T, Fusco ML, Lee JE, King LB, Kawaoka Y, Dye JM, Jr, et al.. Structure of an antibody in complex with its mucin domain linear epitope that is protective against Ebola virus. J Virol. 2012;86(5):2809-16. doi:10.1128/JVI.05549-11. PMID:22171276
- Bale S, Dias JM, Fusco ML, Hashiguchi T, Wong AC, Liu T, Keuhne AI, Li S, Woods VL, Jr, Chandran K, et al.. Structural basis for differential neutralization of ebolaviruses. Viruses. 2012;4(4):447-70. doi:10.3390/v4040447. PMID:22590681