
ABSTRACT
This study was a phase I double-blind, randomized, placebo-controlled trial to evaluate the safety and immunogenicity of a Serbian-produced seasonal trivalent split, inactivated influenza vaccine in healthy adults. The vaccine was manufactured in eggs by the Torlak Institute of Virology, Vaccines and Sera, Belgrade, Serbia and contained A/H1N1, A/H3N2 and B viruses. The clinical trial took place at the Clinical Center of Serbia in Belgrade. Sixty healthy volunteers, aged 18–45 years, were enrolled in the trial. On the day of immunization, volunteers were randomly assigned to receive either a single dose of the trivalent seasonal influenza vaccine (15 μg of hemagglutinin per strain) or placebo (phosphate-buffered saline). Subjects were monitored for adverse events through a clinical history and physical examination, and blood was taken for testing at screening and on day 8 to assess vaccine safety. Serum samples obtained before and 21 days after immunization were tested for influenza antibody titers using hemagglutination-inhibition (HAI) and microneutralization (MN) tests. No serious adverse events were reported. Pain and tenderness at the injection site were the most commonly reported symptoms in both vaccine and placebo groups. Overall, serum HAI responses of fourfold or greater magnitude were observed to H1, H3, and B antigen in 80%, 75%, and 70% of subjects, respectively. Seroprotection rates as measured by HAI were also high (100%, 100% and 86.67%, respectively, for H1, H3 and B). Thus, Torlak's seasonal trivalent influenza vaccine was not associated with adverse events, was well-tolerated and immunogenic. It should be further evaluated in clinical trials to provide sufficient safety and immunogenicity data for licensing in Serbia.
Introduction
Influenza is a significant cause of respiratory morbidity and mortality, with the highest burden of severe disease occurring in at-risk populations.Citation1,Citation2,Citation3,Citation4 In temperate regions, seasonal influenza epidemics occur each year during the winter months, generally between November and April, causing tens of thousands of deaths in Europe alone.Citation5,Citation6 Because of the ability of the influenza virus to mutate, the strains included in seasonal vaccines need to be updated on a regular basis. Annual vaccination can reduce an individual's risk of getting influenza and the World Health Organization (WHO) recommends vaccination of high-risk groups, including pregnant women, young children, individuals with specific chronic diseases, people aged 65 years or over and health care workers.Citation7
Influenza A viruses also have the ability to cause pandemics when antigenic shift occurs and have caused four pandemics in the last 100 years, the most severe of which occurred in 1918 and is estimated to have caused over 50 million deaths globally.Citation6 Vaccines are one of the best defences against a pandemic; however, in 2009, vaccine availability was limited before the second wave of the pandemic and deliveries of WHO vaccine donations to developing countries were significantly delayed.Citation8 The establishment of local production of influenza vaccines in developing countries has been promoted by WHO's Global Action Plan for Influenza Vaccines (GAP), which was a ten-year strategy to reduce the anticipated shortfall in influenza vaccine supply during a pandemic.Citation9 Since 2009 the Torlak Institute in Serbia has participated in GAP's technology transfer project to establish local production of influenza vaccines, with support from international donors, including the Biomedical Advanced Research and Development Authority (BARDA) of the US Department of Health and Human Services.Citation10 PATH has also been involved in supporting GAP vaccine manufacturers and more precisely in providing technical assistance to the Torlak Institute for process development and design and implementation of phase I clinical trials.Citation11
The Government of Serbia has a policy to immunize people older than 65 years, those with chronic diseases, pregnant women, young children, and health care workers, as well as certain travellers.Citation12 As influenza vaccination is not obligatory, uptake differs from year to year, but the Torlak Institute estimates that between 200 000 and 250 000 doses are used in Serbia each year.
Since joining the GAP programme, the Torlak Institute has established a process for the production of a seasonal, trivalent, split, inactivated influenza vaccine produced on eggs; this vaccine has been shown to be safe and immunogenic in animal studies.Citation13 We evaluated the safety profile and immunogenicity of a single intramuscular dose of the vaccine in healthy adults in a phase I clinical trial.
Challenge/capacity building
Many challenges were faced during the set-up of the Phase 1 clinical trial. It was the first time that a clinical trial for a vaccine was performed in Serbia. PATH and WHO supported initiation of a collaboration between the Serbian Ministry of Health, the national regulatory agency “Medicines and Medical Devices Agency of Serbia (ALIMS)” and Institute Torlak through the WHO Country Office (CO) in Serbia. The goal was to engage in monthly meetings with Torlak and PATH through facilitation by the WHO Serbia office to ensure that the vaccine programme was moving along and any issues were resolved immediately. Capacity building included the strengthening of the ALIMS to assist with vaccines regulation (GMP enforcement of the domestic vaccine manufacturer, approval of clinical trials). Strengthening capacity to conduct clinical trials in Serbia was also provided to ensure that clinical sites assessed would be able to perform the phase 1 clinical trial of Torlak's seasonal influenza vaccine under Good Clinical Practices (GCP). An assessment visit by an independent GCP auditor from Switzerland at the selected clinical site was performed and a Corrective Action Preventive Action (CAPA) plan was shared with the site prior to study initiation. These recommendations were addressed through equipment purchases and specific training activities. WHO/PATH/Torlak developed a crisis communications plan and trained relevant staff for the seasonal vaccine phase 1 trial to deal with any crisis or media enquiries that could have arisen during the trial. Finally, PATH team provided technical assistance to Torlak in all clinical trial documentation preparation.
Results
Demographic and other baseline characteristics
Sixty-six (66) volunteers were screened for enrolment in the study. Six patients did not fulfil required inclusion criteria and finally, 60 healthy male and female adults 18 to 45 years old from Serbia were enrolled in the study; 30 received the vaccine and 30 the placebo ( participant flow diagram). All subjects were of white Caucasian race; 18 (30%) were female and 42 (70%) were male, equally divided between vaccine and placebo groups. The overall mean age was 30.85 years. Basic demographic characteristics are presented in . All subjects completed the study and were included in the per-protocol population analysed for immunogenicity.
Figure 1. Participant Flow Chart.
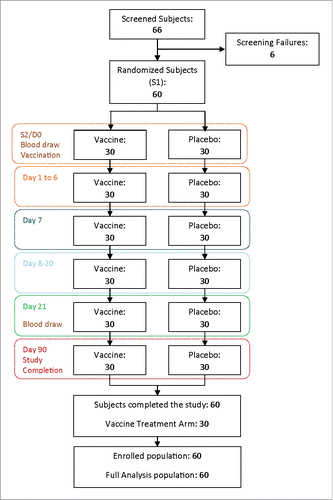
Table 1. Demographic characteristics of study subjects.
Immunogenicity
Baseline HAI and MN seropositivity rates were similar in the two study groups: in the HAI assay, 27% of subjects receiving vaccine and 33% of subjects receiving placebo had antibody titres of 1:40 or more to H1, for H3 this was 63% in both groups and for B antigen this was 10% in the vaccine group and 17% in the placebo group; in the MN assay, 33% of subjects receiving vaccine and 43% of placebo recipients were seropositive to H1, for H3 antigen this was 23% in vaccinated subjects and 27% in placebo recipients and for B antigen this was 77% for vaccinated subjects and 73% for placebo recipients.
Seroconversion was observed in the majority of vaccinated subjects in both the HAI and the MN assays. By HAI, 25 of the 30 subjects (83.33%) had a seroconversion response to H1, 23 (76.67%) to H3 and 21 (70%) to the B antigen. None of the placebo recipients showed a seroconversion response in the HAI assay (). In the MN assay, the seroconversion rates for the three antigens were 83.33% for H1, 70% for H3 and 53.33% for B. Two subjects in the placebo group showed seroconversion to H1 (6.67%), and two to B (6.67%); none showed seroconversion to H3 ().
Table 2. Subjects showing seroconversion in HAI and MN tests, vaccine and placebo recipients.
Post-vaccination seroprotection rates, as measured by HAI (titre ≥ 1/40), were very high among vaccine recipients: 100% for H1 and H3 and 86.67% for B. Among those receiving placebo, the rates were 33.33% for H1, 63.33% for H3 and 16.67% for H3 (). Post-vaccination seroprotection rates measured by MN assay (titer ≥ 40) were detected in 93.33% of vaccine recipients for H1, 86.67% for H3, and 100% for B. Among placebo recipients, post-vaccination seroprotection rates were 53.33% for H1, 33.33% for H3, and 80% for B ().
Table 3. Subjects with seroprotection, by HAI and MN tests, vaccine and placebo recipients.
The geometric mean titers rose significantly among vaccine recipients for all three antigens, in both the HAI and the MN tests. GMTs at baseline were low and there was no rise in antibodies among those given placebo in either test (point estimate for fold rise for all antigens tested by either test was ≤ 1.5). In the HAI assay, the highest GMT was observed for H3 (439.67) and the highest fold rise for H1 (18.49). While the GMT for B antigen was the lowest, it still showed a 7.25-fold increase, from 10.47 (baseline) to 75.95 (post-vaccination). The MN assays showed similar trends with regard to fold changes. However, the strongest GMT response was to B antigen (331.28), followed by H1 (211.12) and H3 (125.53) (). None of the placebo recipients developed a fourfold rise in MN antibodies following vaccination.
Table 4. GMT antibody response to influenza H1, H3 and B antigens, by HAI and MN tests, vaccine and placebo groups.
The number and corresponding percentage of subjects with a fourfold or greater rise from baseline as tested by the MN assay in the vaccine and placebo groups were: for H1, 25 (83.33%) vs 2 (6.67%); for H3, 22 (73.33%) vs 0 (0%); and for B antigen, 14 (46.67%) vs 2 (6.67%).
Adverse events
No allergic reactions were observed during the 30 minutes after administration of either Torlak seasonal influenza vaccine or placebo.
During the seven days after administration, 32 local solicited reactions were reported in the vaccine group and six in the placebo group (). Pain at the injection site was the most frequently reported solicited local symptom, occurring in 46.66% (14/30) of those given vaccine and 13.33% (4/30) of those who received placebo. Other local solicited events were: tenderness (10 (33.33%) subjects in the vaccine group and one subject (3.33%) in the placebo group), and redness at injection site (20% of subjects in the vaccine group and none in the placebo group). Hardness and swelling were reported by less than 10% of the subjects. All local solicited events were assessed as mild in intensity, except for two cases each of pain and tenderness, which were assessed as moderate. Injection-site AEs lasted for 4–5 days in the vaccination group and up to 3 days in the placebo group and resolved without treatment.
Table 5. Solicited symptoms in the 7-day period after immunization with Torlak seasonal influenza vaccine.
Systemic adverse events were less commonly reported and occurred more frequently in the vaccine recipients than placebo recipients. The majority of those were short-lived and mild in intensity. The most commonly reported solicited systemic events were tiredness and headache with each symptom being reported by 16.66% of subjects in vaccine group. None of the subjects in the placebo group reported tiredness and only 6.67% reported headache. Other systemic solicited events, reported by fewer than 10% of subjects, included nausea, fever (axillary temp >37 °C), chills, muscle aches, joint aches and vomiting. Most cases were mild in intensity, with only four episodes (two of tiredness and one each of muscle aches and headache) reported as moderate (). All symptoms resolved within seven days. In all instances, solicited events were assessed as causally related to the vaccination.
During the 21-day follow-up period, one or more unsolicited adverse events were reported for 6.7% (2/30) of subjects in both vaccine and placebo groups. Four systemic AEs were observed in the placebo group: diarrhea, back pain, cough and increased ALAT levels. Three systemic AEs were reported in the vaccine group: influenza-like illness, respiratory tract infection and back pain. All the systemic adverse events were mild in intensity, except for the one case of back pain in the vaccine group, which was considered moderate. None of these events were assessed to be causally related to the treatment administered.
No deaths, and no serious and significant adverse events were reported during the conduct of the study.
Mild elevations in liver enzyme levels were observed in a few subjects in both treatment groups, with no specific pattern or correlation with treatment. The incidence of abnormal laboratory findings was comparable in the two groups. Most episodes were considered as not clinically significant, except for one patient in the placebo group, who had a clinically significant elevation in ALAT (assessed as of mild severity).
Discussion
The remarkable ability of the influenza virus to mutate presents a significant challenge to public health, which is currently addressed by yearly identification of circulating strains and development of matched vaccines.Citation14 Local development of influenza vaccines, as is being done in Serbia, will allow expansion of influenza vaccination and significantly reduce respiratory illnesses and time away from work. Protection from influenza through vaccination can thus contribute to national and regional growth and development.
The high baseline titres in over half of the subjects in this study, particularly to the recently circulating H3 strain, suggest that influenza is widespread in Serbia. The particularly high prevalence of H3N2 among the study participants is consistent with the influenza surveillance report from WHO on seasonal influenza in Serbia.Citation15
The vaccine used in this study was shown to be safe and well tolerated. The reactogenicity profile observed, particularly the frequency and severity of solicited and unsolicited adverse events, was similar to those of already licensed inactivated seasonal influenza vaccines in adults.Citation16,Citation17 Events included local reactions at the injection site, typical of those seen with parenteral vaccination (pain, tenderness, hardness); reactions were mostly mild and of short duration. Systemic adverse events, including laboratory abnormalities, were uncommon, mild, and of no risk to the health and well-being of the individuals.Citation17 There was no difference in the rate of unsolicited systemic AEs between the vaccine and placebo groups, and the AEs observed were considered not to be related to the study vaccine. No SAEs were observed among the vaccine recipients, and no immediate post-vaccination AEs, such as shock and anaphylaxis, were reported.
It should be acknowledged, however, that the sample size in this phase I clinical study was insufficient to detect AEs occurring at low frequency; the full safety profile of the vaccine is yet to be determined in larger-scale studies. Testing also needs to include individuals at higher risk, such as the elderly, individuals with pre-existing conditions and pregnant women. Such testing can be conducted through post marketing surveillance, once the vaccine has been further evaluated in healthy adults and licensed.
The results also indicated that the Torlak vaccine is capable of eliciting robust immune responses, both in naïve subjects and in those previously primed by infection or vaccination. The immunogenicity criteria required for licensure by the European Medicines Agency for seasonal vaccines were met convincingly.Citation18 Seroconversion rates were greater than 80% for H1, 75% for H3, and 70% for B subtypes, as measured by the HAI assay. Slightly lower rates for the H3 and B antigens were observed with the MN test. Seroprotection rates measured by both HAI and MN assays were over 86% for all three subtypes. Similarly, substantial rises in GMTs were observed in the HAI assay in the vaccine group between baseline (pre-immunization) and 21 days after immunization for all three strains, indicating a strong immune response against all three vaccine antigens. GMT ratios (vaccine/placebo) measured by MN assay were also significantly high, the lowest being >4 for the B antigen. Overall, there was a close correlation between HAI and MN results in terms of seroconversion and seroprotection rates and at the individual level. Other studies have demonstrated correlation between HAI and MN results, as well as the higher sensitivity of MN assays, particularly for detection of antibodies to influenza B viruses.Citation19
In conclusion, the Torlak seasonal, trivalent, split, inactivated influenza vaccine was shown to be safe and well tolerated, and induced high levels of seroconversion and seroprotection rates in immunologically naïve individuals as well as those with pre-existing HAI antibodies. These clinical data provide a basis for planning a larger trial, with a view to eventual licensing of the vaccine.
Materials and methods
Study design
This phase I, double-blind, randomized, placebo-controlled study had two objectives: to evaluate the safety profile of a single intramuscular dose of influenza vaccine and to evaluate its immunogenicity. The study was conducted at the Clinic for Infectious and Tropical Diseases, Clinical Centre of Serbia, Belgrade. Screening tests were conducted on volunteers, who gave their informed consent to join the study. Volunteers were randomized to receive either vaccine or placebo on Day 0, prior to receiving the vaccine blood was taken for baseline immunogenicity analyses. During the first week following vaccination, subjects were asked to record local and systemic signs and symptoms using pre-printed Memory Aids, a thermometer, and a small ruler. In addition to solicited signs, subjects were asked to report any other adverse events. The volunteers returned to the clinic seven days after vaccination at which time the study staff reviewed the Memory Aids and transcribed all solicited reactogenicity and other adverse events onto the case report forms. At this visit blood was taken for biochemical and hematological blood tests. Participants returned again for the clinic for their final visit on Day 21 post vaccination where blood was taken again for immunogenicity analyses. Participants were followed up for 90 Days post vaccination with a final phone call to volunteers on Day 90 (± 5 days). The schedule of events are shown in (Supplementary Table 1). The vaccine was used in accordance with Research Protocol of Vaccine Clinical Trial, Protocol Number: TORLAK-100.
Study population
Sixty-six healthy Serbian male and female adult volunteers aged 18–45 years were screened for inclusion in the study. Inclusion criteria were that subjects were literate (by self-report), healthy (as established by the medical history, physical examination, and screening laboratory evaluations), capable and willing to complete a Memory Aid, and willing to return for all follow-up visits. In addition, the women had to have a negative pregnancy test, not be breastfeeding, and be willing to use reliable birth control measures (e.g. intrauterine device, hormonal contraception, condoms) for three weeks from the day of vaccination. Exclusion criteria included: presence of any acute or chronic illness, taking immunoglobulin or medication that could interfere with assessment of safety or immunogenicity, participating in any other clinical trial, hypersensitivity to any vaccine, chicken or egg protein, and drug or alcohol dependence. Subjects who had received influenza vaccine in the previous year were also excluded. Participants needed to be available for vaccination and all follow-up visits.
This study was approved by the ethics review committees of the Clinical Centre of Serbia, the World Health Organization and the National Regulatory Agency (ALIMS). Written informed consent was obtained from all participants before any study-related activities took place.
Treatments
Eligible subjects were randomized 1:1 to receive either vaccine or placebo. The seasonal trivalent influenza vaccine tested was manufactured by the Torlak Institute using embryonated chicken eggs and supplied in prefilled, single-dose, disposable syringes. It was a split virion vaccine, inactivated with Beta-propiolactone (BPL) and the virion split with Triton X-100. The vaccine contained 15 μg of hemagglutinin antigen of each virus strain recommended for the 2014–15 season in the northern hemisphere: X-181 reassortant of A/California/7/2009 (H1N1); X-223A reassortant of A/Texas/50/2012 (H3N2), and NYMC BX-51B reassortant of B/Massachusetts/2/2012. The placebo, which was also manufactured by the Torlak Institute, consisted of phosphate-buffered saline (PBS), pH 7.4, also supplied in prefilled, single-dose, disposable syringes. The vaccine and placebo were stored in a refrigerator at a temperature between 2 °C and 8 °C. The shelf-life was estimated to be 12 months under these conditions. Storage temperature was monitored daily and documented. Back-up power or storage was available in case of primary power failure. The vaccine and placebo syringes were identically packaged and labelled so that the study could be conducted in a double-blind manner. Randomization was performed by an individual not involved in the conduct of the study. In the case of any unblinding, researchers were required to report this in writing to the overseeing Ethics Committee. The allocation code of the product injected into each subject was recorded on the case report form.
Safety assessment
Solicited local and systemic reactions and unsolicited adverse events (AEs) were collected and recorded. Solicited reactions are local or systemic AEs, typically associated with intramuscular influenza vaccination, that were specifically asked about during the 7-day post-vaccination period. Unsolicited events were reported by the subject or observed by study staff during clinic visits. Such events were recorded up to 21 days post-vaccination, except for serious adverse events (SAEs), which were recorded throughout the entire study period of 90 days. If a new solicited event was reported by the subject more than 7 days post-vaccination, it was recorded as an unsolicited AE. Blood was collected for basic clinical chemistry and hematology at screening and on day 7. Laboratory analyses included bilirubin, alanine aminotransferase (ALAT), creatinine, total protein, white blood cells, hematocrit, hemoglobin, and platelets. All clinically significant laboratory findings were reported as unsolicited adverse events. All clinical safety evaluations were made by a qualified clinician. Any clinical sign, symptom or laboratory finding that at any time met the established criteria for an SAE was categorized as such.
Any solicited local or systemic reaction that occurred during the 7-day period post-injection was regarded as related to product administration.
The study was conducted in accordance with the rules for good clinical practice (GCP), including International Conference on Harmonisation guidelines, directive 2001/20/EC of the European Parliament, and the most recent version of the Declaration of Helsinki.
Assessment of immunogenicity
Serum antibodies detected by hemagglutination inhibition
Hemagglutination inhibition (HAI) is the most frequently used serological test for determining immunological responses to influenza vaccination.Citation18 Serum specimens obtained before, and 21 days after, vaccination were tested at the Torlak Diagnostic Laboratory for the presence and titer of HAI antibodies to each of the influenza strains in the vaccine. They were re-tested by VisMederi, laboratory (Siena, Italy) using a validated assay. HAI antibody titers to the three influenza antigens (H1, H3 and B) were determined in duplicate on separate days, with paired specimens tested simultaneously. The HAI used 0.5% turkey red blood cells with the three separate antigens. Serum samples were treated with receptor-destroying enzyme and pre-adsorbed on human red blood cells before testing. HAI assays were performed at a starting dilution of 1:10, with subsequent serial twofold dilutions. Each antigen was diluted to eight HA units (with 50 µl). Titers were determined by identifying the last well with a total lack of agglutination. If the results for duplicate samples were more than 2-fold different, the test was repeated. Samples for which the titer was greater than the last dilution were diluted further and retested. The final results are given as the reciprocal titer of the lowest duplicate value.
Serum antibodies detected by microneutralization
The serum samples were tested for the presence of neutralizing antibodies to each of the influenza strains in the vaccine using a microneutralization (MN) test. For this, serial dilutions of serum, along with fixed amount of live influenza vaccine virus, were added to a culture of Madin-Darby canine kidney (MDCK) cells. Virus-infected cells exhibit a cytopathic effect that can be detected by staining. Titers of neutralizing antibodies are expressed as the greatest dilution of serum inducing 50% neutralization of the cytopathic effect in tissue culture (TCID50). At the end of the incubation period, each well of the 96-wells microtiter plate is checked under an optical microscope to assess the presence of local lesions (Cytopathic effects ‘CPE’) in the cell lawn. Cytopathic effects (CPE) were defined as complete destruction of the cell lawn in the well or the presence of hole(s) in the cell lawn, surrounded by destroyed cells. An incomplete or not overgrown cell lawn with hole(s) surrounded by intact cells is not regarded as CPE. Cell control was characterized by an intact cell lawn, meaning that the cells have not been infected. When compared with the cell control, the wells which show CPE are regarded as “positive” (infected), while the wells which don't show CPE are defined as “negative” (protected).
The method used to perform the microneutralization test is based on the production of serum dilution series and on the fact that each dilution step of the single serum is repeated in 8 wells into the single microtiter plate (8 repetitions). This results in a very reliable determination of the neutralization titre (Nt).
The Nt in this particular test is defined as the serum dilution by means of which 50% of the wells are protected against a virus-induced cytopathogenic effect (CPE). The Spearman-Kärber formula is used to calculate the NT, providing correct results where:
– a lowest dilution with 100% protection;
– a highest dilution with 0% protection, and
– at least one dilution with over 0% but less than 100% protection are involved.
In the event that no well exhibits protection at all (meaning that all wells exhibit CPE) the neutralization titre is determined as the exact half of the titre that would result if one single well was protected.
Statistical methods
The primary objective of the study was to evaluate the safety of one dose of the vaccine. The sample size was selected based on logistical considerations. Immunogenicity requirements were not considered in the choice of sample size since this was a secondary objective. Sample size for this study was 60 healthy male and female adults, 18 to 45 years old, 30 subjects per group.
Safety analysis
The vaccine safety profile was assessed in terms of the number and percentage of subjects with: (1) adverse events occurring within 30 minutes after vaccination; (2) solicited local and systemic reactions within seven days post-vaccination; (3) unsolicited adverse events within 21 days post-vaccination (including changes in clinical laboratory parameters); and (4) SAEs occurring within 90 days post-vaccination. Two-sided exact 95% confidence intervals (CIs) were calculated. For solicited AEs, Fisher's exact test was used to compare the proportions of reactions in the two treatment groups.
Immunogenicity analysis
Immunogenicity assessments included: number and percentage of subjects with a serum HAI antibody titer ≥ 1:40 (seroprotection titer) 21 days post-vaccination for each of the 3 antigens; number and percentage of subjects seroconverting for each of the 3 antigens (seroconversion was defined as a serum HAI titer on day 21 meeting one of the following criteria: (1) pre-vaccination titer <1:10 and post-vaccination titer ≥ 1:40, or (2) pre-vaccination titer ≥ 1:10 and at least a four-fold increase in post-vaccination titer); geometric mean titers (GMTs) of serum HAI antibodies pre- and post-vaccination for each of the three antigens; rise in GMTs of serum HAI antibodies for each of the three antigens; number and percentage of subjects with a fourfold rise in MN titer for each of the antigens; GMTs of MN antibodies for each of the three antigens.
The percentages of subjects with an immune response were calculated, along with the corresponding two-sided exact 95% (Clopper-Pearson) CIs. GMT was summarized by treatment group, along with the corresponding two-sided 95% CIs, by exponentiation the corresponding log-transformed means and their 95% CIs. No multiplicity adjustment to the error rate, alpha, was made because the exploratory nature of the study meant that no formal statistical hypothesis was being tested.
Disclaimer
The authors alone are responsible for the views expressed in this article, which do not necessarily represent the views, decisions or policies of the institutions with which the authors are affiliated.
Disclosure of potential conflicts of interest
Authors declare that there is no conflict of interest. ClinicalTrials.gov identifier: NCT02585700, October 13, 2015
Acknowledgments
We thank John Donnelly for his dedication to this program, David Taylor, Joseph Chiu and Iksung Cho for their work on protocol development, and Niraj Rathi for coordinating the development of this manuscript. We thank Zorica Nikolic and Vesna Obradovic of the Clinical Center of Serbia for their support as nurses during the trial. We also thank Comac Medical for providing the clinical monitoring, data management and analysis services.
For their support towards the project to establish influenza vaccine production in Serbia we thank Florence Barthelemy, Martin Friede, Miljana Grbic and Marie-Paule Kieny at the World Health Organization as well as Rick Bright, Sheng Li, Chuong Huynh and Julie Schafer at the Biomedical Advanced Research and Development Authority (BARDA), US Department of Health and Human Services.
KHVI_A_1415683_Supplemental.docx
Download MS Word (14.6 KB)Additional information
Funding
References
- Lafond KE, Nair H, Rasooly MH, Valente F, Booy R, Rahman M, Kitsutani P, Yu H, Guzman G, Coulibaly D, et al. Global Role and Burden of Influenza in Pediatric Respiratory Hospitalizations, 1982–2012: A Systematic Analysis. PLoS Med. 2016;13(3):e1001977. https://doi.org/10.1371/journal.pmed.1001977. PMID:27011229.
- Zhou H, Thompson WW, Viboud CG, Ringholz CM, Cheng PY, Steiner C, Abedi GR, Anderson LJ, Brammer L, Shay DK. Hospitalizations associated with influenza and respiratory syncytial virus in the United States, 1993–2008. Clin Infect Dis. 2012;54(10):1427-36. https://doi.org/10.1093/cid/cis211. PMID:22495079.
- Dawood FS, Fiore A, Kamimoto L, Nowell M, Reingold A, Gershman K, Meek J, Hadler J, Arnold KE, Ryan P, et al. Influenza-associated pneumonia in children hospitalized with laboratory-confirmed influenza, 2003–2008. Pediatr Infect Dis J. 2010;29(7):585-90. https://doi.org/10.1097/INF.0b013e3181d411c5. PMID:20589966.
- Nair H, Brooks WA, Katz M, Roca A, Berkley JA, Madhi SA, Simmerman JM, Gordon A, Sato M, Howie S, et al. Global burden of respiratory infections due to seasonal influenza in young children: a systematic review and meta-analysis. Lancet. 2011;378(9807):1917-30. https://doi.org/10.1016/S0140-6736(11)61051-9. PMID:22078723.
- Factsheet about seasonal influenza, European Centre for Disease Prevention and Control. https://ecdc.europa.eu/en/seasonal-influenza/facts/factsheet (accessed 4 February 2018).
- Paules C, Subbarao K. Influenza. Lancet. 2017;390(10095):697-708. https://doi.org/10.1016/S0140-6736(17)30129-0. PMID:28302313.
- Vaccines against influenza WHO position paper – November 2012. Weekly Epidemiological Record. World Health Organization. 2013;87(47):461-76. http://www.who.int/wer/2012/wer8747.pdf. PMID:23210147.
- Fineberg HV. Pandemic preparedness and response–lessons from the H1N1 influenza of 2009. N Engl J Med. 2014;370(14):1335-42. https://doi.org/10.1056/NEJMra1208802.
- Friede M, Palkonyay L, Alfonso C, Pervikov Y, Torelli G, Wood D, Kieny MP. WHO initiative to increase global and equitable access to influenza vaccine in the event of a pandemic: supporting developing country production capacity through technology transfer. Vaccine. 2011;29(Suppl 1):A2-7. https://doi.org/10.1016/j.vaccine.2011.02.079. PMID:21684422.
- Grohmann G, Francis DP, Sokhey J, Robertson J. Challenges and successes for the grantees and the Technical Advisory Group of WHO's influenza vaccine technology transfer initiative. Vaccine. 2016;34(45):5420-4. https://doi.org/10.1016/j.vaccine.2016.07.047. PMID:27506497.
- Wahid R, Holt R, Hjorth R, Berlanda Scorza F. Chemistry, manufacturing and control (CMC) and clinical trial technical support for influenza vaccine manufacturers. Vaccine. 2016;34(45):5430-5. https://doi.org/10.1016/j.vaccine.2016.07.046. PMID:27484011.
- WHO vaccine-preventable diseases: Monitoring system. 2017 global summary, selected for: Immunization schedule for 3 vaccines (Influenza, Influenza_Adult, Influenza_Pediatric). http://apps.who.int/immunization_monitoring/globalsummary/schedules (accessed 11 September 2017).
- Unpublished studies. Immunogenicity (mice); Toxicity: single dose, repeated dose (rats); Local Tolerance (rabbits), 2014–2015, Institute of Virology, Vaccines and Sera “Torlak”, Belgrade, Serbia (data on file).
- WHO influenza vaccines. http://www.who.int/influenza/vaccines/about/en/ (accessed 11 September 2017).
- WHO FluNet. Influenza Laboratory Surveillance Information by the Global Influenza Surveillance and Response System (GISRS) http://gamapserver.who.int/gareports/Default.aspx?ReportNo=1 (accessed 15 May 2017).
- Lee GM, Greene SK, Weintraub ES, Baggs J, Kulldorff M, Fireman BH, Baxter R, Jacobsen SJ, Irving S, Daley MF, et al. H1N1 and seasonal influenza vaccine safety in the vaccine safety datalink project. Am J Prev Med. 2011;41(2):121-8. https://doi.org/10.1016/j.amepre.2011.04.004. PMID:21767718.
- Delore V, Salamand C, Marsh G, Arnoux S, Pepin S, Saliou P. Long-term clinical trial safety experience with the inactivated split influenza vaccine, Vaxigrip. Vaccine. 2006;24(10):1586-92. https://doi.org/10.1016/j.vaccine.2005.10.008. PMID:16271424.
- European Medicines Agency: 21 July 2016 EMA/CHMP/VWP/457259/2014 Committee for Medicinal Products for Human Use. Guideline on Influenza Vaccines Non-clinical and Clinical Module. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211324.pdf (accessed 11 September 2017).
- Anh DD, Thiem VD, Anh NTH, Huong VM, Nga NT, Thang TC, Thai DH, Chien VC, Holt R, Wahid R, et al. Randomized safety and immunogenicity trial of a seasonal trivalent inactivated split virion influenza vaccine (IVACFLU-S) in healthy young Vietnamese adults. Vaccine. 2016;34(45):5457-62. https://doi.org/10.1016/j.vaccine.2016.08.052. PMID:27567493.