
ABSTRACT
Evidence on influenza vaccine effectiveness from low and middle countries (LMICs) is limited due to limited institutional capacities; lack of adequate resources; and lack of interest by ministries of health for influenza vaccine introduction. There are concerns that the highest ethical standards will be compromised during trials in LMICs leading to mistrust of clinical trials. These factors pose regulatory and operational challenges to researchers in these countries. We conducted a community-based vaccine trial to assess the efficacy of live attenuated influenza vaccine and inactivated influenza vaccine in rural north India. Key regulatory challenges included obtaining regulatory approvals, reporting of adverse events, and compensating subjects for trial-related injuries; all of which were required to be completed in a timely fashion. Key operational challenges included obtaining audio-visual consent; maintaining a low attrition rate; and administering vaccines during a narrow time period before the influenza season, and under extreme heat. We overcame these challenges through advanced planning, and sustaining community engagement. We adapted the trial procedures to cope with field conditions by conducting mock vaccine camps; and planned for early morning vaccination to mitigate threats to the cold chain. These lessons may help investigators to confront similar challenges in other LMICs.
Evidence on effectiveness from low and middle income countries (LMICs) is limited.Citation1,Citation2 The paucity of research is due to limited institutional capacities in laboratory and field research and lack of adequate resources. There are also concerns among the public and policymakers about the ethics of conducting trials in LMICs, and potential violations of standards, especially among vulnerable groups, given that there is a large power differential between investigators and trial participants. This social climate can result in a mistrust of clinical trials,Citation3,Citation4,Citation5 which in some cases has resulted in lawsuits and legal recommendations to evoke stringent measures to protect the rights of research participants.Citation6
These factors pose unique challenges to researchers who are conducting community-based trials in LMICs and need to manage ethically compliant, locally acceptable, and scientifically meaningful studies in relatively resource-constrained settings. Though researchers have outlined challenges in conducting large vaccine trials for meningococcal and pneumococcal vaccines elsewhere,Citation7,Citation8 influenza vaccine trials pose many distinct challenges. These include the relatively low and variable influenza vaccine effectiveness compared with other pediatric vaccines, routine changes in the vaccine virus components, need for annual vaccination, and variability of influenza seasonality in tropical and monsoon settings. In addition, influenza vaccination is not currently provided through government funded immunization programmes in India, including the Expanded Programme on Immunization (EPI); and although the Government of India (GoI) recommends influenza vaccine for children 6 months-8 years, coverage is low.Citation9 Since adding influenza vaccination to the EPI schedule is currently unlikely in India or other LMICs, conducting research studies on influenza vaccines is also lower priority compared with other vaccines which are closer to roll-out. We present the challenges of conducting a community-based influenza vaccine trial in India, due to regulatory and operational issues, through a case study of a Phase IV community trial to assess the efficacy of live attenuated influenza vaccine (LAIV) and inactivated influenza vaccine (IIV) among children 2–10 years in rural north India.
Description of the vaccine trial
A community-based triple-blinded Phase IV randomized controlled trial was conducted to assess the absolute and relative efficacy of LAIV and IIV in comparison with control groups. The trial was conducted in six villages of the Ballabgarh block (Faridabad district) in the northern state of Haryana, India. The trial was intended to provide evidence for the utility of LAIV when produced locally by the Serum Institute of India (SII), which could be cost-effective if LAIV production occurred in India and production capacity could be more easily augmented during influenza epidemics or pandemics. The trial had two intervention arms: one dose of nasally administered LAIV or either one or two doses of IIV (as appropriate for age), administered through the intra-muscular route, with a second dose given after four weeks. The control arms included either one dose of intra-nasal placebo to mirror LAIV or one or two doses of inactivated polio vaccine (IPV) to mirror IIV. Both IIV and IPV were produced by Sanofi Pasteur SA, France and marketed by the Indian arm. Vaccines or intra-nasal placebo were administered for two consecutive years, 2015 and 2016, during June and July before the monsoon. Three thousand and forty-two eligible children between 2–10 years were randomized to receive LAIV, IIV, intra-nasal placebo, or IPV in the allocation ratio of 2:2:1:1. House-to-house weekly visits for illness surveillance began immediately after the first dose vaccination camps were completed and continued for the study duration. The second dose was given four weeks after the first through vaccination camps and house visits for children who were not covered during camps. For illness surveillance, episodes of febrile acute respiratory infection (FARI) were identified through weekly household surveillance of children, and nasal and throat swabs were obtained from children who reported FARI. Swabs were tested for influenza virus by polymerase chain reaction at the All India Institute of Medical Sciences (AIIMS), New Delhi laboratory. An assessment of vaccine safety was conducted through home visits on days 1, 3, 7, 14, 21, 28, and 42 after each vaccination for each of the four groups. The study was approved by the institutional ethics committee of AIIMS, New Delhi and Institutional Review Board of Centers for Disease Control and Prevention (CDC), Atlanta. The trial was registered in the clinical trial registry of India (Clinical Trial Registry No: CTRI/2015/06/005902).
Regulatory and ethical issues
Approvals
Drug Controller General of India (DCGI) is the licensing authority for vaccine trials in India (http://www.cdsco.nic.in/forms/default.aspx). The Drugs and Cosmetics Rules 1945, which governs the clinical trials conduct in India, was amended in response to recommendations from the Supreme Court of India in 2013 to introduce a multi-layered vetting of trials, including to strengthen the informed consent process, introduce no-fault compensation, register ethics committees, and discourage placebo-controlled trials.Citation10 While this process helps ensure the protection of vulnerable subjects in vaccine trials, the amended rule has made the clearance process for clinical trials much more complex.Citation11 One such issue in our trial was the use of intra-nasal placebo. Though the rules do not explicitly prohibit the use of placebo, they state that “use of placebo in placebo-controlled trials in case the standard care, though available, was not to be provided to the subject as per the clinical trial protocol will be deemed as trial related injury.”Citation12 In our study, there was no standard of preventive care available other than vaccination. The DCGI also mandates that investigators, ethics committees, and regulators must ensure that the use of placebo is “appropriate, efficient, and ethical.” Since no active control for intranasal influenza vaccine was available for our trial, there was no other choice but to use a intra-nasal placebo. However, this design decision required extensive communication and discussion with the DCGI, including personal visits and presentations, to receive the necessary approvals.
Regulatory approval is further complicated for influenza vaccine studies because each year, licensed LAIV and IIV are considered “new” vaccines when their formulation is updated. Thus, the DCGI must clear the first batch of any influenza vaccine before its use. Southern hemisphere composition of influenza vaccines is determined by the World Health Organization through a routine influenza vaccine strain selection process in September every year; thus, the influenza vaccines used in our trial was decided in September 2014. Since our study was to commence in June 2015, before the monsoon onset which is the peak influenza season in the study area, there were only 9 months during which we needed to obtain all approvals and procure vaccines after the strain selection process was completed, which potentially threatened successful initiation of the trial.Citation13 These approvals were also needed before mandatory insurance could be garnered for trial-related injuries, as required by the good practices guidelines of the Central Drugs Standards and Control Organization (CDSCO), further delaying study initiation. A Gantt chart depicting the approval timelines is presented in .
Figure 1. Gantt chart depicting the timeline of approvals for a vaccine trial in Ballabgarh, India.
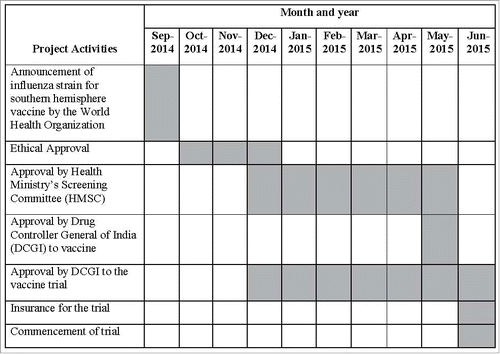
The time needed to obtain appropriate regulatory approvals and garner insurance can threaten vaccine trial timeliness, because trials must commence based on local influenza seasonality and also stay within research funding cycles. Hence, we suggest that investigators allocate at least 9 months to obtain all approvals required for similar vaccine trials in India; this timeline could be longer if the trial was for a completely novel product. This may differ for other countries depending on country-specific regulatory requirements where the trial will be conducted. Sustained engagement with various regulatory authorities was of paramount importance. Simultaneously working on the preparatory phase of the trial while waiting for approvals, such as finalizing standard operating procedures, ordering supplies, recruiting and training staff, and engaging with the community helped optimize the timeliness of our trial once approvals were received.
Adverse event reporting
Investigators were required to notify the institutional ethics committee and the DCGI for each serious adverse event (SAE), whether related or not to vaccination. Initial SAE report submission was required within 24 hours and a detailed report was required within 14 days as per The Drugs and Cosmetics Rules, 1945. SAEs included any death, life threatening condition, disability, congenital defect, or hospitalization anytime during the follow-up period after vaccination; in our trial, this was the study period of two years. In our trial, 14 hospitalizations or deaths occurred in the first year and were reported as SAEs, all of which were judged to be unrelated to vaccination by the DCGI; we reported all but one SAE within the stipulated time frame (median 12 days). The study participants were provided the mobile numbers of study physicians for appropriate clinical guidance for any adverse event any time during the study period. This process helped to detect SAEs early. In future studies, follow-up should be designed to ensure compliance with local reporting timeframes. Continuous community engagement and provision of dedicated communication channels helped us meet the required SAE reporting timelines which was an essential regulatory requirement.
Assessment and compensation for trial-related injuries
The final determination of whether an injury is related or unrelated to vaccination by the DCGI usually takes >6 months, and is based on input from the investigators and ethics committee; and may also include independent investigation by the DCGI. However, as per the Drugs and Cosmetics (Third Amendment) Rules 2014, in case of any injury to the subject during the clinical trial, free medical management must be provided as long as required or until it is established that the injury was unrelated to the vaccination, whichever is earlier.Citation14 Hence, in practice, the investigator or sponsor is liable to bear the cost of all illness management, including hospitalization for major co-morbidities whether related or not to the trial. Trial insurance covers only SAEs which are deemed “related” to the trial, thus investigators or sponsors bear a large cost due to a delayed assessment of the “relatedness” of the injury to the vaccination which puts excess financial burden on investigators or sponsors of trials. To mitigate this burden, we conducted outpatient clinics in the study area four days a week, providing free treatment to all study subjects for all common co-morbidities.
Informed consent
Written informed consent from parents or legal guardians and written assent from children > 7 years was obtained. As per the DCGI guidelines, audio-visual recording of consent and assent is mandatory in India for all vaccine trials,Citation14which necessitated additional manpower and equipment; and prolonged the consent process by at least 20 minutes for each consent. There were also challenges in obtaining consent in a community setting, as mothers were reluctant to provide consent without permission from the child's father or grandfather, partly due to poor health literacy and the household power structure where men often authorize such decisions. This further delayed consent as men were often away during daytime for work. There were also concerns regarding privacy during the audio-visual consent as most of consenters were mothers or other female guardians who were uncomfortable being video recorded. The required audio-visual consent and assent is time-consuming and resource-intensive, and these factors must be accounted for during clinical trial planning in India.
Operational issues
Participant enrollment
While the community attitude towards immunization was largely positive, there were some pockets of resistance with manifest apprehension about the potential vaccine adverse event. Overall, 4% of all eligible subjects refused consent and there was 19.5% study attrition before vaccination; this was higher in one village where 6.1% subjects refused consent and there was 45% attrition. The higher attrition in this village was because of more mistrust due to religious and cultural differences compared with the other villages. High attrition can reduce power in vaccine trials, and jeopardizes the ability to determine differences between groups. In our study, because there were multiple villages within the study area, there were enough participants from other villages to achieve the required sample size. In order to achieve this, we had to allay further apprehension within the community, by routinely engaging with parents of study participants, community leaders, and influencers such as community health volunteers and school teachers through regular community meetings over two years in each of our study villages.
Vaccine supplies and cold chain
Regular follow-up with manufacturers/suppliers was conducted to ensure timely vaccine supplies, including placebo. Study vaccines required storage at 2–8°C; thus, cold chain maintenance was challenging as Ballabgarh has an erratic power supply and temperatures during June can reach > 40–45°C. Vaccines were supplied from manufacturers by courier at a constant temperature between 4–5°C and stored in an ice-lined refrigerator and vaccine cabinet at the Ballabgarh field office. A continuous power supply was ensured with generators for backup during power cuts. Once outreach vaccination camps started, vaccines were transported to the community sites in cold boxes with ice gel packs. Multiple simulations were conducted before deciding on the number of gel packs needed for each cold box. Temperature inside the cold box during storage at Ballabgarh, transport to field camps, and at vaccination camps was monitored using digital temperature loggers with indicators for out of range temperature. Cold boxes were supplemented with extra gel packs when the logger showed the temperature was near 8°C or 2°C to ensure that the temperature remained acceptable (i.e. 2–8°C). Vaccination camps were also held from 7 AM to 10 AM, when ambient temperatures were lower. Cold chain maintenance is critical, and more so for live vaccines, and thus planning for continuous monitoring of the cold chain is essential for vaccine trials conducted during hot temperatures.
Vaccination
Vaccination camps were conducted at public settings such as village health centres and schools to achieve maximum participation. This allowed for maximum vaccine coverage within two weeks of starting the campaign. Before conducting an actual camp, four mock camps were held at the community sites to troubleshoot issues related to eligibility assessment, randomization, vaccination, and observation for immediate adverse events. Based on lessons learned from the pilot period, each household with recruited children was given an appointment card for vaccination one day before camp, with details about time and location. Two to three camps were held per day in each of six villages to cover all eligible children. Since camps were held in June when schools were in recess and many children were out of the village, we conducted a second round of vaccination campaigns after a two-week lag to cover all children. Due to extreme heat in June in the study area, camps started at 7 AM and approximately 80% of participants received vaccine by 12 noon.
Randomization to different study arms was performed on-site at the vaccine camps to avoid attrition after allocation. This included individual-level randomization and allocation concealment by trained study staff at each camp site. This was done by sequential allocation of study participants, as they reported to the vaccination camps, to vaccine or placebo arm through a randomization code concealed within a sealed envelope kept at the study site. However, random allocation to different arms among children from the same households and the community perception that vaccines must be injectable resulted in some apprehension among guardians regarding nasal vaccines. In such cases, the physician supervising the camp provided appropriate explanations which helped reduce further attrition.
Meticulous planning, innovative approaches, use of public space for vaccination, continuous engagement with families, and flexibility in the field allowed us to achieve our target in an efficient manner.
Summary of the lessons learned
Challenges in large community-based clinical trials in LMICs, including India, range from regulatory clearances and subject recruitment to logistical challenges in administering interventions and challenges in tracking and reporting adverse events. The trial regulatory requirements make them time-consuming and resource-intensive, during both preparatory and implementation phases. Advanced planning, understanding regulatory requirements, anticipation of community concerns, flexibility, and quick adaptations to field conditions can help investigators overcome some of these challenges. Investigators should develop standard operating procedures keeping in mind the field conditions in which the intervention will be administered, especially in operationally demanding conditions ().
Table 1. Summary of lessons learned during a vaccine trial in Ballabgarh, India.
Despite many challenges and several unanticipated delays, our study team successfully carried out the initial trial plan, including achieving sample size. Strategies that contributed to these successes included early and sustained community engagement, extensive pilot testing of procedures and mock vaccine camps, and adopting flexible approaches such as early morning vaccination to mitigate threats to the cold chain. These lessons learned may help investigators in LMICs prepare for such trials, which are essential to develop the evidence base for influenza and other novel vaccines in India and other LMICs, where the respiratory disease burden is highest.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of All India Institute of Medical Sciences or Centers for Disease Control and Prevention.
Authors' contribution
RK, RA, VVN, SS, KEL, SKK, LD, SJ and AK were all involved in the planning of the study and development of the study protocols, tools and manuals. RK, RA, VVN, SKK were involved in field implementation of the trial activities. LD led the team performing the virological studies. RK, SS, KEL, SJ and AK drafted the manuscript and incorporated the comments of all the authors. All the authors have read and approved the manuscript. AK is Guarantor of the Paper.
Acknowledgments
We acknowledge the support of all the investigators and co-investigators of this trial, our research staff, study participants and their parents and guardians, and other members of the community who helped us in conducting this trial. Clinical Trial Registry No: CTRI/2015/06/005902.
Additional information
Funding
References
- Breteler JK, Tam JS, Jit M, Ket JC, De Boer MR. Efficacy and effectiveness of seasonal and pandemic A (H1N1) 2009 influenza vaccines in low and middle income countries: a systematic review and meta-analysis. Vaccine. 2013;31:5168–77. doi:10.1016/j.vaccine.2013.08.056. PMID:24012574.
- Peasah SK, Azziz-Baumgartner E, Breese J, Meltzer MI, Widdowson MA. Influenza cost and cost-effectiveness studies globally–a review. Vaccine. 2013;31:5339–48. doi:10.1016/j.vaccine.2013.09.013. PMID:24055351.
- Nundy S, Gulhati CM. A new colonialism?–Conducting clinical trials in India. N Eng J Med. 2005;352:1633–6. doi:10.1056/NEJMp048361.
- Shetty P. Vaccine trial's ethics criticized. Nature. 474:427–8. doi:10.1038/474427a. PMID:21697918.
- Kummervold PE, Schulz WS, Smout E, Fernandez-Luque L, Larson HJ. Controversial Ebola vaccine trials in Ghana: a thematic analysis of critiques and rebuttals in digital news. BMC Public Health. 2017;17:642. doi:10.1186/s12889-017-4618-8. PMID:28784109.
- Supreme Court of India Record of Proceedings. Swasthya Adhikar Manch, Indore and Anr. v Union of India (UOI) and Others. WP(C) No. 33/2012 with WP(C) No. 79/2012. [accessed 2017 May 14]. http://cdsco.nic.in/writereaddata/SC%20Order%2030th%20Sept%202013.pdf
- Cutts FT, Enwere G, Zaman SM, Yallop FG. Operational challenges in large clinical trials: examples and lessons learned from the Gambia pneumococcal vaccine trial. PLoS Clin Trials. 2006;1:e16. doi:10.1371/journal.pctr.0010016. PMID:16871317.
- Marchetti E, Mazarin-Diop V, Chaumont J, Martellet L, Makadi MF, Viviani S, Kulkarni PS, Preziosi MP. Conducting vaccine clinical trials in sub-Saharan Africa: operational challenges and lessons learned from the Meningitis Vaccine Project. Vaccine 2012;30:6859–63. doi:10.1016/j.vaccine.2012.09.008. PMID:22989686.
- Directorate General of Health Services, Ministry of health and Family Welfare, Government of India. Seasonal Influenza: Guidelines for Vaccination with Influenza Vaccine. New Delhi April 2017 [accessed 2017 May 22]. https://mohfw.gov.in/sites/default/files/30580390001493710612.pdf.
- Ministry of Health and Family Welfare, Government of India. Drugs and Cosmetics (First Amendment) Rules. New Delhi: Government of India Press; 2013.
- Sugarman J, Bhan A, Bollinger R, Gupta A. India's new policy to protect research participants. BMJ. 2013;347:f4841. doi:10.1136/bmj.f4841. PMID:23903456.
- Ministry of Health and Family Welfare.GSR 292(E).Clinical Development Services Agency, India. 2014 Apr 24 [accessed on 2017 May 27]. http://www.cdsaindia.in/sites/default/files/GSR%20292%20(E)%20regarding%20compensation%20to%20clinical%20trial%20and%20changes%20in%20timelines%20for%20notification.pdf.
- Saha S, Chadha M, Shu Y, Group of Asian Researchers on Influenza (GARI). Divergent seasonal patterns of influenza types A and B across latitude gradient in Tropical Asia. Influenza Other Respir Viruses. 2016;10:176–84. doi:10.1111/irv.12372. PMID:26781162.
- Ministry of Health and Family Welfare.Order No GCT/20/SC/Clin/2013. Office of drug controller general of India. 2013 Nov 19. [accessed 2017 May 27] http://www.cdsco.nic.in/writereaddata/Office%20Order%20dated%2019.11.2013.pdf.