
ABSTRACT
A therapeutic vaccine for human Chagas disease (American trypanosomiasis caused by Trypanosoma cruzi) is under development based on the success of vaccinating mice with DNA constructs expressing the antigens Tc24 and Tc-TSA-1. However, because DNA and nucleic acid vaccines produce less than optimal responses in humans, our strategy relies on administering a recombinant protein-based vaccine, together with adjuvants that promote Th1-type immunity. Here we describe a process for the purification and refolding of recombinant TSA-1 expressed in Escherichia coli. The overall yield (20–25%) and endotoxin level of the purified recombinant TSA-1 (rTSA-1) is suitable for pilot scale production of the antigen for use in phase 1 clinical trials. Mice infected with T. cruzi were treated with rTSA-1, either alone or with Toll-like receptor 4 (TLR-4) agonist adjuvants including monophosphoryl lipid A (MPLA), glucopyranosyl lipid A (GLA, IDRI), and E6020 (EISEI, Inc). TSA-1 with the TLR-4 agonists was effective at reducing parasitemia relative to rTSA-1 alone, although it was difficult to discern a therapeutic effect compared to treatment with TLR-4 agonists alone. However, rTSA-1 with a 10 ug dose of MPLA optimized reductions in cardiac tissue inflammation, which were significantly reduced compared to MPLA alone. It also elicited the lowest parasite burden and the highest levels of TSA-1-specific IFN-gamma levels and IFN-gamma/IL-4 ratios. These results warrant the further evaluation of rTSA-1 in combination with rTc24 in order to maximize the therapeutic effect of vaccine-linked chemotherapy in both mice and non-human primates before advancing to clinical development.
Introduction
Chagas disease, also known as American trypanosomiasis, is a zoonotic disease caused by Trypanosoma cruzi parasites, which are transmitted by blood-feeding triatomine bugs. The disease is responsible for a major burden of illness, particularly in the Americas, affecting an estimated 7.2 million people who currently live with T. cruzi infection,Citation1 and causing up to 10,600 annual deaths,Citation2–Citation4 as well an estimated $7.2 billion in annual economic losses.Citation3 However, additional information from the BENEFIT randomized clinical trial indicates that the mortality rate from Chagas disease may in fact be far higher than current estimates indicate.Citation5 Drug treatments are effective during the acute phase of the infection as well as in children, but efficacy appears questionable for patients in the chronic phase, with a very high variability in treatment outcomes.Citation6–Citation10 Thus, complementary or alternative tools are urgently needed for a better care of infected patients, and vaccines may represent an attractive strategy for the treatment, prevention, or control of T. cruzi infections.
Economic modeling pointed out that both a preventive or therapeutic vaccine would provide not only savings in health care costs, but also a positive return on investment.Citation11,Citation12 In addition, many proof-of-principle studies have now clearly demonstrated that different vaccine formulations can control a T. cruzi infection in mice (reviewed inCitation13). These are based on DNA vaccines or recombinant virus expressing parasite antigens as well as on recombinant proteins with adjuvants.
In particular, trypomastigote surface antigen (TSA-1) and Tc24 parasite antigens have emerged as very promising candidates for further vaccine development.Citation14 Indeed, DNA vaccines expressing these antigens, alone or in combination, have been found to be able to control a T. cruzi infection in a variety of mouse modelsCitation15–Citation18 as well as in dogs.Citation19,Citation20 However, currently there are no licensed DNA vaccines for humans due in part to the inability of DNA to elicit robust immune responses in humans as they do in mice.Citation21 Therefore we have embarked on the development of a recombinant protein-based vaccine and begun process development for the large-scale production these two antigens as recombinant proteins and testing of their potential for the control of T. cruzi infection. Production of recombinant Tc24 and its variants has been described and its efficacy to induce an immune response and control infection in different formulations has been demonstrated.Citation22–Citation24 There is additional evidence for Tc24 and TSA-1 combinations to enhance vaccine efficacy.Citation12
Here we focused on developing a production process for TSA-1 and testing its efficacy as a therapeutic vaccine against T. cruzi in mice, as a new critical step towards the development of a multi-antigen vaccine. TSA-1 is 85 kDa parasite protein belonging to the trans-sialidase family of surface proteins, playing an important role in the scavenging of sialic acid by the parasite.Citation25,Citation26 Several members of this protein family have been found to be effective vaccine candidates against T. cruzi in a wide range of formulations.Citation15,Citation27–Citation32 In particular, the amino-terminal moiety of the protein is the most immunogenic and able to confer protection against infection, whereas the carboxy-terminal part, appears to mask protective epitopes from the amino-terminal part of the proteins.Citation33 We thus developed a scalable expression and purification process, for the large-scale production of rTSA-1, and tested its efficacy as a therapeutic vaccine formulated with several adjuvants, for the control of a T. cruzi infection in mice.
Results
Expression, purification and refolding of rTSA-1
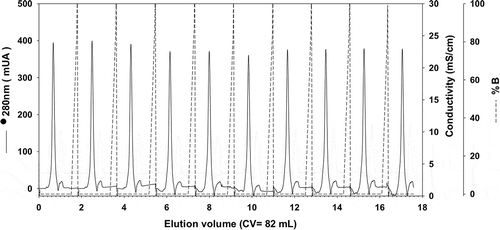
We first developed an expression, purification and refolding process for rTSA-1. The expression was induced for 18 h at 30°C in a 10 L batch culture with a yield up to 270 mg of rTSA-1/L (). The recombinant protein was found in the insoluble fraction as inclusion bodies (IB) and one purification step using Ni-affinity chromatography was sufficient to recover up to 90% of rTSA-1 with up to 95% purity (). The best refolding yields (up to 75%) were obtained by SEC with a protocol where a linear gradient of 0 to 8 M urea was introduced in the 0.3CV before the sample injection followed by a 1.2 CV of elution step with refolding buffer without urea. shows the chromatographic profile of a single SEC refolding batch of rTSA-1 in an 82 ml column. The left shoulder of elution peak (a) contains few rTSA-1 aggregates. The main elution peak (0.67 CV) eluted up to 75% of the refolded rTSA-1 (b) without urea and only small amount of the recombinant protein co-eluted with a lower concentration of urea (c) (). Similar results were obtained when the refolding protocol was scaled up to 475 ml column ( and ). The chromatographic profile of ten consecutive SEC refolding batches using the 82 ml column () indicates that this SEC refolding protocol is reproducible. The refolded rTSA-1 (0.2–0.3 mg/ml) (fraction b) was concentrated, and the refolding buffer exchanged to PBS by dialysis or Gel filtration chromatography. The estimated overall yield was about 20–25% of the expressed rTSA-1 with an endotoxin level less than 10 EU/mg of protein. The purified TSA-1 from inclusion bodies and refolded by Size Exclusion Chromatography had a final purity of 94–96%.
Figure 1. Expression of the recombinant TSA-1 in E. coli BL21 (DE3).
A fermenter containing 10L 2xYT medium (supplemented with kanamycin 50 µg/mL) was inoculated with E. coli BL21 (DE3)/pET41TSA1. When the optical density (OD600) reached 0.6, the expression of the recombinant protein was induced with 1 mM IPTG (USB) at 30°C maintaining the OD at 30% saturation. A) SDS-PAGE analysis of the induction of rTSA-1 in a batch fermentation. Total cell extract before and after the addition of 1 mM IPTG (2-18h) and without IPTG (N). Molecular marker (M). B) The rTSA-1 expressed as inclusion bodies was purified under denaturing conditions by Ni2+ Affinity chromatography. SDS-PAGE analysis of the total cell extract (lane 1), solubilized inclusion bodies (lane 2) and purified rTSA-1 (lane 3).
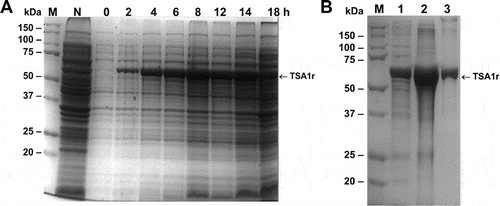
Figure 2. Chromatographic refolding of rTSA-1.
Chromatographic profile of the refolding of rTSA-1 by SEC using 82 mL (A) or 475 mL (C) volume (Column Volume) of Sepharose 6FF (GE Healthcare, UK) packed into a 1.5 x 50 cm or 2.6 X 100 cm columns respectively. Fractions were collected and pooled in (a), (b) and (c) samples, and analyzed by SDS-PAGE under non-reducing condition; 82 mL column (B) and 475 mL column (D). Molecular marker (M), samples (a), (b) (c). See Materials and methods for details.
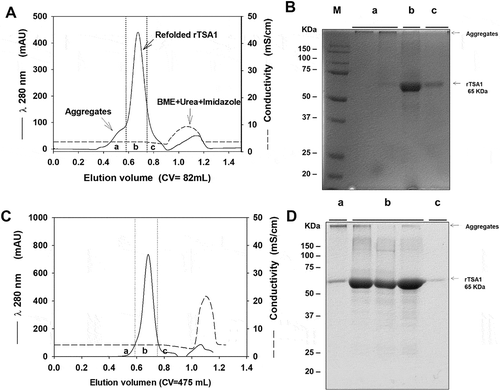
Figure 3. Consecutive chromatographic refolding batches of rTSA-1.
Chromatographic profile of ten consecutive SEC refolding batches of denatured rTSA-1 using a 82 mL volume (Column Volume) of Sepharose 6FF (GE Healthcare, UK) packed into a 1.5 x 50 cm. Absorbance at 280 nm (solid lane) and 0 to 8 M urea gradient (dashed lane). See Materials and methods for details.
Therapeutic efficacy of r TSA-1 against T. cruzi infection
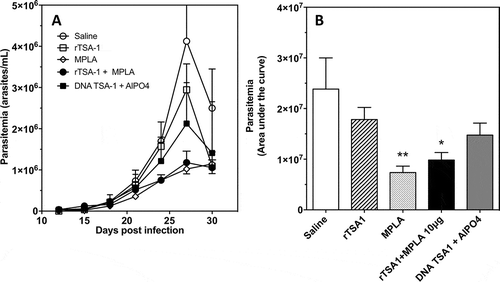
Balb/c mice were infected with T. cruzi, and treated at days 7 and 14 post-infection with rTSA-1 alone, or formulated with monophosphoryl Lipid A (MPLA) as adjuvant. Control mice received saline solution or the MPLA adjuvant alone. An additional experimental group was treated with a DNA vaccine encoding TSA-1. Parasitemia was measured during 30 days post-infection, and confirmed that control mice receiving saline solutions presented a high parasitemia, which peaked at day 27 post-infection (). Significant differences were observed among the different groups (ANOVA F = 4.01, P = 0.01). Treatment with unadjuvanted rTSA-1 had a negligible effect on parasitemia, but formulation of the recombinant protein with MPLA as adjuvant resulted in a significantly lower parasitemia (Dunn’s post-hoc test, P < 0.05), with a 4-fold reduction in the peak parasitemia. Interestingly, therapeutic vaccination with TSA-1 DNA vaccine was less effective than the recombinant protein plus MPLA in controlling parasitemia, while administration of MPLA alone had a similar effect as TSA-1 plus MPLA (Dunn’s post-hoc test, P < 0.01) (). Only 57% of mice from the saline control group survived the acute infection, while survival of all vaccinated groups was 85–100%, but this difference was not significant (P = 0.12). We also measured circulating IgG antibodies against rTSA-1. Vaccinated mice tended to have higher levels of IgG, but this did not reach statistical significance, and IgG2a/IgG1 ratios remained similar in all groups (Supplementary Figure 1).
Figure 4. Effect of therapeutic vaccination on parasitemia of T. cruzi infected mice.
Balb/c mice were infected with T. cruzi, and treated at days 7 and 14 post-infection with the indicated vaccines (N = 7 mice per group). Control mice received saline solution. Parasitemia was measured over time (A), and overall parasitemia was estimated as the area under the curve (B) in arbitrary units. There was a significant difference among groups (ANOVA F = 4.01, P = 0.01), and * and ** indicate a significant differences with the saline group as determined by post-hoc Dunn’s test (P < 0.05 and P < 0.01, respectively). The experiment was performed twice.
Mice were sacrificed at day 50 post-infection, to collect cardiac tissue biopsies, and we further assessed the effect of therapeutic vaccination on T. cruzi infection by measuring cardiac tissue inflammation and parasite burden. There were significant differences in inflammatory cell density among groups (Kruskal-Wallis, P = 0.036), and therapeutic vaccination with TSA-1 plus MPLA as adjuvant resulted in a significant 2-fold reduction in inflammatory cell density in the heart compared with control mice (Dunn’s test, P < 0.05) (, and Supplementary Figure 2). Other vaccinated goups, including MPLA alone, presented small but not significant decreases in inflammation. Similarly, measurement of cardiac parasite burden indicated that control mice presented a high parasite burden, while mice that had been treated with rTSA-1 with MPLA or MPLA alone had a 30–40 fold reduction in parasite burden compared to the saline treated mice (Kruskal-Wallis, P = 0.031, Dunn’s test P < 0.05) ().
Figure 5. Effect of therapeutic vaccination on cardiac tissue inflammation and parasite burden in T. cruzi infected mice.
Mice were sacrificed at day 50 post-infection (N = 7 mice per group) and cardiac biopsies were processed for histopathologic analysis (A) and measurement of parasite burden (B). Micrographs of tissue sections were stained with hematoxylin and eosin, and inflammatory cell density was determined by image analysis using Multispec. There were significant differences in inflammatory cell density among groups (Kruskal-Wallis, P = 0.036). Cardiac parasite burden was measured by qPCR, and showed significant differences among groups (Kruskal-Wallis, P = 0.031). * indicates a significant difference with the saline group (Dunn’s test, P < 0.05). The experiment was performed twice.
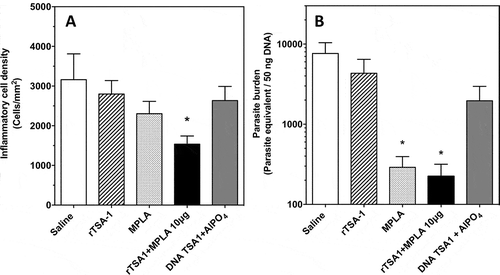
Taken together, these data clearly showed that therapeutic vaccination with rTSA-1 with MPLA as adjuvant was able to control T. cruzi infection in mice, as indicated by lower parasitemia, cardiac inflammation and parasite burden. MPLA alone had also a significant therapeutic effect on parasitemia and parasite burden, although it did not reduce cardiac tissue inflammation compared to the rTSA-1-MPLA combination.
Optimization of therapeutic vaccine formulation
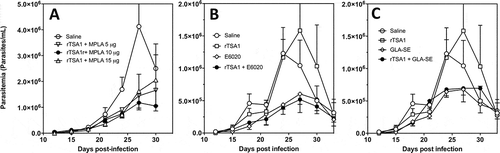
Given the key role of MPLA adjuvant in potentiating rTSA-1 antigen, we tested different vaccine formulations, to evaluate MPLA at lower (5 µg) and higher (15 µg) doses compared to the initial dose tested (10 µg). Balb/c mice were infected with T. cruzi and treated with two doses of the various vaccine formulations at days 7 and 14 post-infection, while parasitemia was measured every 3 days as before. The therapeutic efficacy of vaccine treatment tended to be reduced when rTSA-1 was formulated with a lower or a higher MPLA dose, suggesting that 10 µg was the optimum dose with this adjuvant ( and Supplementary Figure 3). We also evaluated the alternative TLR-4 agonists GLA-SE and E6020, at doses of 5 µg. Antigen formulation with GLA-SE and E6020 resulted in a comparable reduction in parasitemia as MPLA in spite of the lower adjuvant dose ( and , respectively, and Supplementary Figure 3). As with MPLA, treatment with both adjuvants alone was similarly effective in reducing parasitemia.
Figure 6. Effect of rTSA-1 formulation on therapeutic vaccine control of parasitemia.
rTSA-1 antigen was formulated with MPLA at different doses (A), or with E6020 (B) or GLA-SE (C) as adjuvants, and infected mice were treated with two doses of vaccine at days 7 and 14 post-infection (N = 4 to 7 mice per group). Parasitemia was measured to assess vaccine efficacy in controlling infection. (A) Formulation with MPLA at 10 µg/dose was the best formulation compared to MPLA at 5 µg and 15 µg (ANOVA, F = 2.67, P = 0.04). (B) Formulation of rTSA-1 with E6020 and E6020 alone also allowed to control parasitemia (ANOVA, F = 10.26, P < 0.001). (C) Similarly, formulation of rTSA1 with GLA-SE and GLA-SE alone significantly reduced parasitemia (ANOVA, F = 4.89, P = 0.009). * indicates a significant differences with the saline group as determined by post-hoc Dunn’s test (P < 0.05).
To further understand the role of MPLA dose in vaccine efficacy, we analyzed the cellular immune response at the end of the acute phase. Splenocytes were isolated and stimulated in vitro with rTSA-1 to detect antigen-specific responses by flow cytometric analysis. We observed significant differences in the TSA-1 specific activation of CD4+ and CD8+ T cells producing IFNγ and IL-4 (). Therapeutic immunization with TSA-1 alone did not affect cytokine production by both T cell populations, in agreement with the poor efficacy of the antigen alone to control T. cruzi infection. Also as expected, the administration of MPLA alone had no detectable effect on TSA-1 specific immune response, in spite of its significant effect on T. cruzi infection. The addition of a 5 µg dose of MPLA to rTSA-1 was insufficient to significantly alter TSA-1-specific immune response. On the other hand, doses of 10 and 15 µg of MPLA allowed to significantly increase antigen-specific cytokine production by CD4+ ( and ) and CD8+ T cells ( and ). Importantly, the dose of 10 µg lead to a more polarized response as it preferentially stimulated IFNγ production, and did not activate significant IL-4 production, while the dose of 15 µg resulted in a less polarized response with a strong stimulation of both IFNγ and IL-4 production. Indeed, only the 10 µg dose allowed to reach a significant increase in the ratio of IFNγ/IL-4 CD4+ and CD8+ T cells (from 0.24 ± 0.02 to 0.82 ± 0.18 for CD4+ and from 0.35 ± 0.04 to 0.64 ± 0.12 for CD8+ T cells, respectively). The immune profile observed thus closely matched the protection level of each vaccine formulation, and indicates that an intermediate dose of 10 µg of MPLA is sufficient to elicit a significant activation of IFNγ producing T cells, with no activation of IL-4 producing cells, leading to improved infection control.
Figure 7. Effect of vaccine formulation on T cell phenotype.
T. cruzi-infected mice were treated with the indicated vaccine formulations, including saline solution, rTSA-1, MPLA, combinations of rTSA-1+ MPLA at doses of 5, 10 and 15 µg, and TSA-1 DNA (N = 4 to 7 mice per group). Splenocytes were harvested at 50 days post-infection, and stimulated in vitro with rTSA-1. Antigen-specific CD4+ (A and C) and CD8+ (B and D) T cells were analyzed for their production of IFNγ (A and B) and IL-4 (C and D) cytokines. Vaccine treatment resulted in significant differences in IFNγ-producing CD4+ (Kruskal-Wallis = 25.24, P < 0.001) and CD8 + T cells (Kruskal-Wallis = 25.96, P < 0.001), as well as in IL-4-producing CD4+ (Kruskal-Wallis = 13.51, P < 0.035) and CD8 + T cells (Kruskal-Wallis = 25.86, P < 0.001). * and ** indicate a significant difference with the saline control group (Dunn’s post-hoc test, P < 0.05 and P < 0.01, respectively).
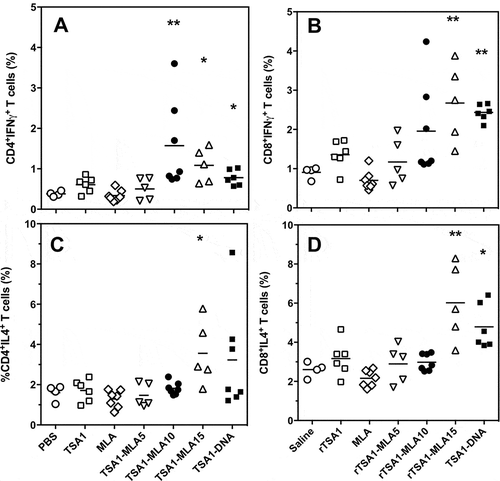
Finally, therapeutic vaccination with the DNA vaccine encoding TSA-1 induced an antigen-specific response that more closely resembled that induced by the recombinant protein vaccine with 15 µg of MPLA as adjuvant, with a significant stimulation of both IFNγ and IL-4 production by T cells. Again, this was in agreement with the limited control of T. cruzi infection afforded by this formulation of the DNA vaccine.
Discussion
A rTSA1 production process was developed in this study, from its expression in E. coli as inclusion bodies. rTSA-1 was obtained highly pure (95%) after the initial Ni+2 affinity purification of solubilized inclusion bodies. However, a critical step was to develop an efficient refolding protocol. Although, more than 2000 experimental refolding protocols can be consulted in the REFOLDdb database,Citation34 only few of them are suitable for large production scale. Here, it has been shown that rTSA-1 can be refolded with yields up to 75% by a protocol using SEC chromatography with either a 82 ml CV or 475 ml CV column. The refolding protocol is reproducible as is shown by the consecutive refolding batches and can be scaled up using the chromatographic system available at production scale.Citation35–Citation37 The overall yield (20–25%) and the endotoxin level of the purified rTSA-1 suggested that the expression, purification and refolding process developed in this study is suitable for the production of rTSA-1. Importantly, all the steps involved in the production process of TSA-1 reported here are susceptible to scaling-up for the GMP production of TSA-1.
The evaluation of the therapeutic efficacy of rTSA-1 formulated with MPLA indicated that it was able to significantly control a T. cruzi infection in mice, as evidenced by a lower parasitemia, cardiac tissue inflammation and parasite burden. These findings were also associated with high levels of antigen-specific IFNγ levels, as well as IFNγ/IL-4 ratios. Remarkably, this formulation of rTSA-1 was more effective that TSA-1 DNA in controlling T. cruzi infection, highlighting our ability to induce a protective immune response with a recombinant protein formulation. However, it has to be noted that the efficacy of the DNA vaccine was lower than that observed previously, suggesting variability in the protective efficacy of this vaccine when formulated with AlPO4.Citation15–Citation17 The rTSA-1 antigen is thus a potential candidate for further optimization of a therapeutic vaccine.
The efficacy of rTSA-1 in controlling T. cruzi infection was critically dependent on formulation with a TLR-4 adjuvant, as the protein alone had negligible effect on the infection. This is in agreement with many vaccine studies showing that recombinant proteins alone fail to induce the appropriate Th1 immune response needed for the control of T. cruzi. Moreover, we observed that several TLR-4 agonists may be used with rTSA-1, and E6020 and GLA-SE even seem to be more potent than MPLA as they provided a similar therapeutic effect with a lower dose of adjuvant. Thus, optimized synthetic TLR-4 adjuvants appear very promising for inclusion in vaccine formulations. Interestingly, all three adjuvants alone induced significant control of T. cruzi infection. This is likely due to the strong ongoing immune response against abundant T. cruzi antigens already initiated by infection, which may be reoriented in a non-specific manner by the adjuvant during the acute phase. A comparable reorientation of the immune response has been observed with therapeutic DNA vaccines.Citation16,Citation18 This immunotherapeutic effect of adjuvants appears similar to that of other immunotherapies such as CpG administration against Leishmania parasites, which can also promote a strong control of the infection.Citation38,Citation39 It is also very likely that the therapeutic effect of adjuvant alone may wane as infection progresses to the chronic phase and circulating parasite antigen is much less abundant for immune stimulation.
Importantly, the dose of adjuvant was also critical for an appropriate (re)-orientation of the immune response by the therapeutic vaccine: a low dose of MPLA adjuvant was ineffective to alter the immune response, but a high dose induced an immune response too broad as it included both IFNγ- and IL-4-producing T cells. An intermediate dose was thus best, as it preferentially activated IFNγ-producing T cells, but not IL-4 production, thereby promoting a more polarized reorientation of the immune response, which in turn allowed a better control of T. cruzi. Further optimization of rTSA-1 vaccine formulation may thus be possible to improve the therapeutic efficacy of this vaccine candidate.
The study presented some limitations. One limitation was that vaccination was only evaluated during the acute phase of the infection, when high parasitemia and parasite burden are likely causing a strong activation of the immune system, which may overshadow some of the effects of the vaccines tested. For example, we did not detect major changes in antibody levels against TSA-1 in vaccinated animals, possibly due to the already high level of these antibodies induced by the infection itself. The strong effect of the adjuvants alone, which prevented to see clear differences between the adjuvant and rTSA-1 + adjuvant formulations, is also likely related to the ongoing acute infection, which provided a strong immune stimulation by multiple parasite antigens. However, we did not measure the immune response to whole parasite lysate or additional antigens to analyze this process. Such effects may be minimized during the chronic phase of the infection, when circulating parasite levels are much lower.
In conclusion, we developed here a scalable expression, purification and refolding process for the large scale production of rTSA-1 vaccine candidate and showed that this antigen is effective for the control of an ongoing T. cruzi infection in mice when formulated with various TLR-4 adjuvants. This antigen thus represents a potential candidate, which combined with other antigens such as Tc24 can broaden the immune response against the parasite, while re-orienting it to a more polarized Th1 phenotype, for an optimum control of T. cruzi infection. Thus, rTSA-1, possibly in combination with rTc24, is part of promising vaccine candidates for the development of a human therapeutic vaccine against T. cruzi.
Materials and methods
Expression, purification and refolding of the recombinant TSA-1 (rTSA-1)
Expression of rTSA-1
The DNA sequence coding for the protective N-terminal fragment of TSA-1 (30–617 residues without signal peptide)Citation30 was amplified using the plasmid pcDNA3-TSA1 as a templateCitation40 and subcloned into a pET41a (Novagen) expression vector with GST-tag deleted, and with His-tag expressed in C-terminus, by using NdeI/XhoI restriction sites. After being confirmed for the correct insert and reading frame by double-stranded DNA sequencing using vector flanking primer T7 promoter and T7 terminator, the recombinant plasmid DNA was transformed into Escherichia coli BL21 (DE3), and the best expression clone was selected for making a seed stock with 20% of glycerol. The production of rTSA-1 was performed in a 310 Bio-Flo fermenter (Eppendorf) containing 10L 2xYT medium (supplemented with kanamycin 50 µg/mL) in a batch culture fermentation. The fermenter was inoculated with E. coli BL21 (DE3)/pET41TSA1 seed stock. Cells were grown at 37°C, 500 rpm, until the optical density (OD600) reached 0.6, and expression of the recombinant protein was induced with 1 mM IPTG (USB) for 18 h at 30°C maintaining the DO at 30% saturation. Then, cells were separated by centrifugation, resuspended in 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM PMFS, 1 mM DTT. The cells were incubated 15 min with 1 mg/mL lysozyme at 30°C and with DNase 1 µg/mL at 37°C for other 15 min and lysed by three freezing/thawing steps. The soluble fraction (SF) and insoluble fraction (IF) were separated by centrifugation (29,000 g) for 30 min at 4°C; the pellet containing the inclusion body of rTSA-1 was stored at −20°C.
Purification of rTSA-1
To purify the rTSA-1 expressed as inclusion bodies, the pellet was washed twice in each following washing buffers by centrifugation (29,000 xg for 20 min at 4°C): buffer A (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 M urea, 2% Triton X-100), buffer B (20 mM Tris-HCl pH 8.0, 1 mM DTT, 2 M guanidine hydrochloride (GndHCl)) and buffer C (20 mM Tris-HCl pH 8.0, 1 mM DTT, 1% Triton X-114). After one last washing step with 20 mM Tris-HCl pH 8.0, the inclusion bodies (IB) were resuspended in the solubilization buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 5 mM imidazole, 1 mM β-mercaptoethanol and 8 M urea) and incubated overnight at RT with mild agitation in an orbital shaker. The supernatant (solubilized IB) was separated by centrifugation at 15,000 g for 30 min, filtered through a 0.45 µm membrane and stored at 4°C. The solubilized protein was purified by Immobilized Metal Affinity Chromatography (IMAC) using two 5 mL Ni-Sepharose 6 fast flow columns (GE Healthcare) in tandem using the NGC Q Chromatographic System (Bio-Rad). The columns (10 ml total column volume- CV) were equilibrated with 10 CV of solubilization buffer and 20 mL of solubilized IB were loaded onto the column at 0.5 mL/min. After a washing step with 3 CV of solubilization buffer, the protein was eluted with 3 CV of solubilization buffer containing 500 mM imidazole.
Chromatographic refolding of rTSA-1
rTSA-1 refolding was performed by size exclusion chromatography (SEC) using a 1.5 x 50 cm or 2.6 × 100 cm column packed with 82 mL or 475 ml of Sepharose 6FF (GE Healthcare, UK) respectively. All the chromatographic refolding batches were done in a NGC Q chromatograph (Bio-Rad, USA) at a linear flow rate of 53.3 cm/h. The columns were equilibrated with 1.2 CV of refolding buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 5% glycerol, 0.4 mM GSSG, 0.2 mM GSH, 1 mM PMSF) followed by a urea gradient in 0.3 CV from 0 to 8 M. Then, 2 or 25 ml (2.5–3 mg/ml) of purified and denature rTSA-1 were loaded into the 82 or 475 ml column respectively. Protein was eluted with 1.2 CV of refolding buffer. To test the reproducibility of the refolding protocol up to ten consecutive refolding batches were performed in each column.Citation35 Then, refolded rTSA-1 was loaded to a Ni-Sepharose 6 fast flow column and the refolding buffer of the eluted protein was exchange to PBS either by dialysis or a gel filtration step in a 1.6 x 70 column packed with 120 ml of Sepachryl-200 (GE Healthcare).
Sample analysis
All samples were analyzed under non-reducing and reducing conditions by SDS-PAGE in 10% acrylamide gels and stained with Coomassie Brilliant Blue. Protein concentration was determined in denatured samples by a densitometric and spectrophotometric method in refolded samples (ξ = 116100 M−1cm−1, MW 65.693 kDa). The Endotoxin level in both purified inclusion bodies and refolded protein was determined with the chromogenic method Limulus Amebocyte Lysate (LAL) (Pierce-Thermo Scientific, USA) following the indication of the provider. The concentration was calculated with a standard curve performed under the same conditions as the samples (R2 = 0.994) and reported as endotoxin units per mg of protein (EU/mg). All samples were analyzed in triplicate. The purity of rTSA-1 was estimated by a densitometric analysis using a Image Lab software (Bio-Rad) of several lanes of 10% SDS-PAGE loaded with different amounts of TSA-1.
Mice and T. cruzi infection
Female BALB/c mice, 6–8 weeks old, housed in the institutional Animal Experimentation Unit of the “Dr. Hideyo Noguchi” Regional Research Center, were used in all experiments. They were infected via intraperitoneal with 500 blood parasites of the H1 strain and infection was monitored by measuring parasitemia every 3 days, from day 12 to day 30–40, using a Newbauer hematocytometer as in previous studies.Citation15,Citation17 The protocol was approved by the Institutional Bioethics Committee of the Regional Research Center “Dr. Hideyo Noguchi” of the Autonomous University of Yucatan (Reference #CBI-CIR-12–12).
Therapeutic vaccination and vaccine efficacy
Infected mice were divided into groups of 5–8 mice, and received two doses of the vaccine at days 7 and 14 post infection, as before,Citation16,Citation17 for the evaluation of vaccine therapeutic efficacy. Each vaccine dose consisted of 20 µg of rTSA-1, with 5, 10 or 15 µg of MPLA (Sigma-Aldrich), a potent TLR4 adjuvant,Citation41 administered via intramuscular. Additional groups of mice received rTSA antigen alone, or MPLA alone, or saline solution, as controls. One additional group of mice received two low doses of 20 µg TSA-1 DNA vaccine with AlPO4 as adjuvantCitation42 for comparison with the recombinant protein. Alternative adjuvants were also tested, including glucopyranosyl lipid A formulated in a stable emulsion (GLA-SE, IDRI, Seattle, WA) and E6020 (EISEI, Inc.),Citation43 both being synthetic TLR-4 agonists. The progression of infection was monitored by evaluating parasitemia, as detailed above. After 50 days of follow-up, surviving mice were euthanized and cardiac tissue removed to assess parasite burden by real-time qPCR in a Illumina® EcoTM thermocycler.Citation19,Citation22 Cardiac biopsies were embedded in paraffin, to perform tissue sections, which were stained with hematoxylin and eosin. Tissue micrographs were taken at 40x magnification, and analyzed using Multispec to determine the density of inflammatory cells as described before.Citation16
Antibody measurement
TSA-1-specific IgG, IgG1 and IgG2a were measured by ELISA. Briefly, 96-wells microplates were coated overnight at 4ºC with 0.125 µg of rTSA-1 in carbonate buffer. After washing, wells were blocked with 1% BSA for 1 h at 37ºC, washed five times, and 1:200 serum dilutions were added and incubated for 30 min at 37ºC. After five washes, a 1:1000 dilution of secondary antibody labeled with alkaline phosphatase was added (anti-IgG, or anti-IgG1 or anti-IgG2a) and incubated for 30 min at 37ºC. After washing five times, 1 mg/ml of p-nitrophenylphosphate substrate was added and the reaction was stopped by adding HCl 1N. Plates were read at 405 nm on an ELISA plate reader, and antibody levels were expressed as OD levels.
T cell phenotyping
We evaluated the immune correlates for parasite control by measuring CD4+ and CD8+ T cells antigen-specific cytokine production by flow cytometry. Briefly, spleens were removed from euthanized mice, macerated on a sterile mesh, and cells were resuspended in RPMI 1640 medium. Following centrifugation at 160 g for 10 min at 4ºC, the cells were resuspended in a lysis solution (0.17 M Tris pH 7.2, 0.16 M NH4Cl) and incubated for 5 min at 37ºC to remove erythrocytes. The remaining splenocytes were washed three times with RPMI and resuspended in RPMI supplemented with 20 mM sodium pyruvate, 5 x 10−2 mM mercaptoethanol, 10 mM glutamine, and 10% fetal bovine serum. Viability of cells was assessed by Trypan blue exclusion and cell number was determined in a Neubauer chamber. Splenocytes (2X105 cells/well in triplicate) were cultured for 24 h at 37ºC and 5% CO2, with or without stimulation with 50 µg/mL rTSA-1 or with 50 µg/ml of concanavalin A, respectively.
Following stimulation, cells were collected, washed with PBS and stained for 30 min at 4ºC with anti-CD3, anti-CD4 y anti-CD8 monoclonal antibodies labeled with Alexa fluor 647,PE-Cy7 and PerCP-Cy5.5, respectively (BD). After washing cells were permeabilized with 0.1% saponin during 30 min, washed again and intracellular staining was performed with anti-IFNγ and anti-IL-4 monoclonal antibodies labeled with Alexa fluor 488 and PE, respectively (BD). After two washes with FACS buffer, cells were fixed with 10% formaldehyde at 4ºC for 10 min, and resuspended in FACS buffer for analysis in a BD FACSVerse flow cytometer. A total of 100,000 events per sample were adquired and analyzed using FACSuite™ v1.0.5 software.
Briefly, lymphocytes were gated by size using a FSC-A vs SSC-A dot plot (Supplementary Figure 4), single cells were selected using a dot plot of SSC-A vs SSC-H. T cells were further identified by their expression of CD3 on a CD3-Alexa647 vs SSC-A dot plot. Next, CD8+ or CD4+ T lymphocytes were selected using a dot plot of SSC-A vs their cluster of differentiation. Finally, intracellular expression of cytokines (% of IFNγ+ or IL4+ cells) was evaluated using a dot plot of IFNγ-Alexa fluor 488 (or IL4-PE) vs SSC-A.
Statistical analysis
Data are presented as mean ± SEM, except otherwise indicated. Comparison among vaccine groups was performed by ANOVA or Kruskal-Wallis tests for multiple groups, depending on the distribution of the data, followed by Dunn’s post-hoc test. Differences in survival were assessed by Mantel-Cox log-rank test. All tests were run in Prism 6.0 software.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download MS Power Point (28.3 MB)Acknowledgments
This work was funded by the Consejo Nacional de Ciencia y Tecnologia of Mexico (CONACYT, grant #CB-2010-01-156513 to ED and INFR-2016-269657 to JOL) and grant #187714 from the Carlos Slim Foundation. L.E.V was supported by scholarship #319117 from CONACYT. We are thankful to the Infectious Disease Research Institute (IDRI, Seattle, WA, USA) for providing us with GLA-SE.
Supplementary material
Supplemental data for this article can be access on the publisher’s website.
Additional information
Funding
References
- Global Burden of Disease. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1211–1259. doi:10.1016/S0140-6736(17)32154-2.
- Global Burden of Disease. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi:10.1016/S0140-6736(14)61682-2.
- Hotez PJ, Alvarado M, Basanez MG, Bolliger I, Bourne R, Boussinesq M, Brooker SJ, Brown AS, Buckle G, Budke CM, et al. The global burden of disease study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl Trop Dis. 2014;8:e2865. doi:10.1371/journal.pntd.0002865.
- WHO. Chagas disease in Latin America: an epidemiological update based on 2010 estimates. Weekly Epidemiol Rec. 2015;90:33–43.
- Pecoul B, Batista C, Stobbaerts E, Ribeiro I, Vilasanjuan R, Gascon J, inazo MJ, Moriana S, Gold S, Pereiro A, et al. The BENEFIT Trial: where do we go from here?. PLoS Negl Trop Dis. 2016;10:e0004343. doi:10.1371/journal.pntd.0004343.
- Antunes AP, Ribeiro AL, Sabino EC, Silveira MF, Oliveira CD, Botelho AC. Benznidazole therapy for Chagas disease in asymptomatic Trypanosoma cruzi -seropositive former blood donors: evaluation of the efficacy of different treatment regimens. Rev Soc Bras Med Trop. 2016;49:713–720. doi:10.1590/0037-8682-0165-2016.
- Antinori S, Grande R, Bianco R, Traversi L, Cogliati C, Torzillo D, Repetto E, Corbellino M, Milazzo L, Gali M, et al. High frequency of adverse reactions and discontinuation with benznidazole treatment for chronic Chagas disease in Milano, Italy. Clin Infect Dis. 2015;60:1873–1875. doi:10.1093/cid/civ230.
- Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A, Jr., Rosas F, Villena E, Quiroz R, Bonilla R, Britto C, et al. Randomized trial of benznidazole for chronic chagas’ cardiomyopathy. New England J Med. 2015;373:1295–1306. doi:10.1056/NEJMoa1507574.
- Morillo CA, Waskin H, Sosa-Estani S, Del Carmen Bangher M, Cuneo C, Milesi R, Mallagray M, Apt W, Beloscar J, Gascon J, et al. Benznidazole and posaconazole in eliminating parasites in asymptomatic T. Cruzi carriers: the STOP-CHAGAS Trial. J Am Coll Cardiol. 2017;69:939–947. doi:10.1016/j.jacc.2016.12.023.
- Murcia L, Carrilero B, Ferrer F, Roig M, Franco F, Segovia M. Success of benznidazole chemotherapy in chronic Trypanosoma cruzi-infected patients with a sustained negative PCR result. Eur J Clin Micro & Infect Dis. 2016;35:1819–1827. doi:10.1007/s10096-016-2733-6.
- Lee BY, Bacon KM, Wateska AR, Bottazzi ME, Dumonteil E, Hotez PJ. Modeling the economic value of a chagas’ disease therapeutic vaccine. Hum Vaccines Immunother. 2012;8:1293–1301. doi:10.4161/hv.20966.
- Lee BY, Bacon KM, Connor DL, Willig AM, Bailey RR. The potential economic value of a Trypanosoma cruzi (Chagas disease) vaccine in Latin America. PLoS Negl Trop Dis. 2010;4:e916. doi:10.1371/journal.pntd.0000916.
- Quijano-Hernandez I, Dumonteil E. Advances and challenges towards a vaccine against Chagas disease. Human Vaccines. 2011;7:1184–1191. doi:10.4161/hv.7.11.17016.
- Dumonteil E, Bottazzi ME, Zhan B, Heffernan MJ, Jones K, Valenzuela JG, Kamhawi S, Ortega J, Rosales SP, Lee BY, et al. Accelerating the development of a therapeutic vaccine for human Chagas disease: rationale and prospects. Expert Rev Vaccines. 2012;11:1043–1055. doi:10.1586/erv.12.85.
- Dumonteil E, Escobedo-Ortegon J, Reyes-Rodriguez N, Ramirez-Sierra MJ, Arjona-Torres A. Immunotherapy of Trypanosoma cruzi infection with DNA vaccines in mice. Infect & Immun. 2004;72:46–53. doi:10.1128/IAI.72.1.46-53.2004.
- Limon-Flores AY, Cervera-Cetina R, Tzec-Arjona JL, Ek-Macias L, Sanchez-Burgos G, Ramirez-Sierra MJ, Cruz-Chan JV, VanWynsberghe NR, Dumonteil E. Effect of a combination DNA vaccine for the prevention and therapy of Trypanosoma cruzi infection in mice: role of CD4+ and CD8+ T cells. Vaccine. 2010;28:7414–7419. doi:10.1016/j.vaccine.2010.08.104.
- Sanchez-Burgos G, Mezquita-Vega G, Escobedo-Ortegon J, Ramirez-Sierra MJ, Arjona-Torres A, Rodrigues MM, Ouaissi A, Dumonteil E. Comparative efficacy of DNA vaccines encoding various Trypanosoma cruzi antigens. FEMS Microbiol Med Immunol. 2007;50:333–341. doi:10.1111/j.1574-695X.2007.00251.x.
- Zapata-Estrella H, Hummel-Newell C, Sanchez-Burgos G, Escobedo-Ortegon J, Ramirez-Sierra MJ, Arjona-Torres A, Dumonteil E. Control of Trypanosoma cruzi infection and changes in T cell populations induced by a therapeutic DNA vaccine in mice. Immunol Lett. 2006;103:186–191. doi:10.1016/j.imlet.2005.11.015.
- Quijano-Hernandez IA, Castro-Barcena A, Vazquez-Chagoyan JC, Bolio-Gonzalez ME, Ortega-Lopez J, Dumonteil E. Preventive and therapeutic DNA vaccination partially protect dogs against an infectious challenge with Trypanosoma cruzi. Vaccine. 2013;31:2246–2252. doi:10.1016/j.vaccine.2013.03.005.
- Quijano-Hernandez IA, Bolio-González ME, Rodríguez-Buenfil JC, Ramirez-Sierra MJ, Dumonteil E. Therapeutic DNA vaccine against Trypansomoma cruzi in dogs: a pilot clinical trial. Ann N Y Acad Sci. 2008;1149:343–346. doi:10.1196/annals.1428.098.
- Porter KR, Raviprakash K. DNA vaccine delivery and improved immunogenicity. Curr Issues Mol Biol. 2017;22:129–138. doi:10.21775/cimb.022.129.
- Martinez-Campos V, Martinez-Vega P, Ramirez-Sierra MJ, Rosado-Vallado M, Seid CA, Hudspeth EM, Wei J, Liu Z, Kwityn C, Hammond M, et al. Expression, purification, immunogenicity, and protective efficacy of a recombinant Tc24 antigen as a vaccine against Trypanosoma cruzi infection in mice. Vaccine. 2015;33:4505–4512. doi:10.1016/j.vaccine.2015.07.017.
- Barry MA, Wang Q, Jones KM, Heffernan MJ, Buhaya MH, Beaumier CM, eegan BP, Zhan B, Dumonteil E, Bottazzi ME, et al. A therapeutic nanoparticle vaccine against Trypanosoma cruzi in a BALB/c mouse model of Chagas disease. Human Vaccines & Immunother. 2016;12:976–987. doi:10.1080/21645515.2015.1119346.
- Konduri V, Halpert MM, Liang D, Levitt JM, Cruz-Chan JV, Zhan B, Bottazzi ME, Hotez PJ, Jones KM, Decker WK. Genetic adjuvantation of a cell-based therapeutic vaccine for amelioration of chagasic cardiomyopathy. Infect & Immun. 2017;85e00127-17.
- Pereira-Chioccola VL, Schenkman S. Biological role of Trypanosoma cruzi trans-sialidase. Biochem Soc Trans. 1999;27:516–518.
- Schenkman S, Eichinger D, Pereira ME, Nussenzweig V. Structural and functional properties of Trypanosoma trans-sialidase. Annu Rev Microbiol. 1994;48:499–523. doi:10.1146/annurev.mi.48.100194.002435.
- Wizel B, Nunes M, Tarleton RL. Identification of Trypanosoma cruzi trans-sialidase family members as targets of protective CD8+ TC1 responses. J Immunol. 1997;159:6120–6130.
- Costa F, Franchin G, Pereira-Chioccola VL, Ribeirao M, Schenkman S, Rodrigues MM. Immunization with a plasmid DNA containing the gene of trans-sialidase reduces Trypanosoma cruzi infection in mice. Vaccine. 1998;16:768–774.
- Costa F, Pereira-Chioccola VL, Ribeirao M, Schenkman S, Rodrigues MM. Trans-sialidase delivered as a naked DNA vaccine elicits an immunological response similar to a Trypanosoma cruzi infection. Braz J Med Biol Res. 1999;32:235–239.
- Fujimura AE, Kinoshita SS, Pereira-Chioccola VL, Rodrigues MM. DNA sequences encoding CD4+ and CD8+ T-cell epitopes are important for efficient protective immunity induced by DNA vaccination with a Trypanosoma cruzi gene. Infect & Immun. 2001;69:5477–5486. doi:10.1128/IAI.69.9.5477-5486.2001.
- Vasconcelos JR, Hiyane MI, Marinho CR, Claser C, Machado AM, Gazzinelli RT, Gazzinelli RT, Bruna-Romero O, Alvarez JM, Boscardin SB, et al. Protective immunity against Trypanosoma cruzi infection in a highly susceptible mouse strain after vaccination with genes encoding the amastigote surface protein-2 and trans-sialidase. Hum Gene Ther. 2004;15:878–886. doi:10.1089/hum.2004.15.878.
- Hoft DF, Eickhoff CS, Giddings OK, Vasconcelos JR, Rodrigues MM. Trans-sialidase recombinant protein mixed with CpG motif-containing oligodeoxynucleotide induces protective mucosal and systemic trypanosoma cruzi immunity involving CD8+ CTL and B cell-mediated cross-priming. J Immunol. 2007;179:6889–6900.
- Wrightsman RA, Dawson BD, Fouts DL, Manning JE. Identification of immunodominant epitopes in Trypanosoma cruzi trypomastigote surface antigen-1 protein that mask protective epitopes. J Immunol. 1994;153:3148–3154.
- Mizutani H, Sugawara H, Buckle AM, Sangawa T, Miyazono KI, Ohtsuka J, Nagata K, Shojima T, Nosaki S, Xu Y, et al. REFOLDdb: a new and sustainable gateway to experimental protocols for protein refolding. BMC Struct Biol. 2017;17:4. doi:10.1186/s12900-017-0074-z.
- Antonio-Perez A, Ramon-Luing LA, Ortega-Lopez J. Chromatographic refolding of rhodanese and lysozyme assisted by the GroEL apical domain, DsbA and DsbC immobilized on cellulose. J Chromatogr A. 2012;1248:122–129. doi:10.1016/j.chroma.2012.05.086.
- Jungbauer A, Kaar W, Schlegl R. Folding and refolding of proteins in chromatographic beds. Curr Opin Biotechnol. 2004;15:487–494. doi:10.1016/j.copbio.2004.08.009.
- Middelberg AP. Preparative protein refolding. Trends Biotechnol. 2002;20:437–443.
- Zimmermann S, Egeter O, Hausmann S, Lipford GB, Röcken M, Wagner H, Heeg K. CpG oligodeoxynucleotides trigger protective and curative Th1 responses in lethal murine leishmaniasis. J Immunol. 1998;160:3627–3630.
- Flynn B, Wang V, Sacks DL, Seder RA, Verthelyi D. Prevention and treatment of cutaneous leishmaniasis in primates by using synthetic type D/A oligodeoxynucleotides expressing CpG motifs. Infect & Immun. 2005;73:4948–4954. doi:10.1128/IAI.73.8.4948-4954.2005.
- Dumonteil E, Escobedo-Ortegon J, Reyes-Rodriguez N, Arjona-Torres A, Ramirez-Sierra MJ. Immunotherapy of Trypanosoma cruzi infection with DNA vaccines in mice. Infect & Immun. 2004;72:46–53. doi:10.1128/IAI.72.1.46-53.2004.
- Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi:10.1126/science.1138963.
- Rosado-Vallado M, Mut-Martin M, Garcia-Miss MR, Dumonteil E. Aluminium phosphate potentiates DNA vaccines against Leishmania mexicana. Vaccine. 2005;23:5372–5379. doi:10.1016/j.vaccine.2005.05.037.
- Ishizaka ST, Hawkins LD. E6020: a synthetic Toll-like receptor 4 agonist as a vaccine adjuvant. Expert Rev Vaccines. 2007;6:773–784. doi:10.1586/14760584.6.5.773.