
ABSTRACT
Background: Under the WHO’s Global Action Plan for influenza vaccines, we conducted a phase 2–3 study of IVACFLU-S, a trivalent, seasonal inactivated influenza vaccine candidate.
Methods: In the phase 2 portion of the study, 252 participants received one dose of 15 mcg hemagglutinin (HA) vaccine per strain or placebo. Following determination of safety, 636 additional participants were randomized in phase 3 to receive vaccine or placebo. Immunogenicity was assessed in a subset of the participants in the phase 3 study.
Results: Higher proportion (70%) of participants in the IVACFLU-S arm reported solicited local adverse events (AEs) (p < .0001) as compared to placebo (25%). Mild injection site pain and tenderness were most common AEs seen in 55% and 60% of participants in the vaccine group. The solicited systemic AEs were comparable (p = .4149). The majority of solicited and unsolicited AEs were mild to moderate in severity. In the vaccine arm for the combined age group of 18–60 years of age, seroconversion against antigens A/H1N1, A/H3N2, and B was achieved in 70.3%, 76.1%, and 54.1% of participants respectively; seroprotection against antigens A/H1N1, A/H3N2, and B was achieved in 83.3%, 86.6%, and 60.3% of participants respectively; and the geometric mean fold rise for the hemagglutinin-inhibition (HI) antibody titers against antigen A/H1N1, A/H3N2, and B were 13.15, 11.85, and 5.87, respectively.
Conclusion: This study demonstrates the local reactogenicity, other safety, and immunogenicity of IVACFLU-S, first domestically produced influenza vaccine in Vietnam.
ClinicalTrials.gov number NCT03095599 (March 29, 2017)
1. Introduction
Influenza poses a major challenge to the global community; it imposes significant costs both in terms of morbidity and mortality due to annual epidemics and pandemics. Globally, seasonal influenza causes millions of cases of illness and up to 650,000 deaths each year.Citation1 The health impact of influenza is greatest in resource-limited countries due to underlying nutritional deficiencies and concomitant illness, limited access to health care, and the lack of widespread use of vaccines against common causes of bacterial pneumonia.
Vaccination is the most effective public health intervention to prevent influenza infection. However, public health programs in low-to-middle-income countries (LMIC) do not commonly feature influenza vaccine and its coverage remains low. This problem is compounded by the fact that vaccine production capacity is concentrated mostly in developed countries. The World Health Organization (WHO) initiated the Global Action Plan for Influenza Vaccines (GAP) in 2006 to promote increased utilization of seasonal influenza vaccine; encourage development of influenza vaccines with better yields and broader and longer protection; and augment global pandemic influenza vaccine production capacity.Citation2 Previous pandemic influenza outbreaks have been marked by significant shortfall in global vaccine supply due to insufficient vaccine production capacity. Global seasonal influenza vaccine production capacity indirectly affects the ability to produce pandemic vaccines; seasonal influenza vaccine production facilities can be used to produce pandemic influenza vaccine and the sales of seasonal vaccines would provide resources to stockpile pandemic vaccines.
Under WHO’s GAP program, vaccine manufacturers in selected resource constrained countries received financial support from the Biomedical Advanced Research and Development Authority (BARDA), part of the US Department of Health and Human Services’ Office of the Assistant Secretary for Preparedness and Response, and technical assistance from PATH for development of influenza vaccines. This involved improving technology, scaling-up, and carrying out preclinical and clinical studies on locally manufactured influenza vaccines. The Institute of Vaccines and Medical Biologics (IVAC) in Vietnam is one recipient of this assistance. The goal is to have a licensed, Vietnamese-made seasonal influenza vaccine and through that expertise sustain the capacity for pandemic influenza vaccine production.
The phase 1 trial of the IVAC seasonal trivalent inactivated split-virion influenza vaccine (IVACFLU-S), completed in March 2016, identified no safety concerns and demonstrated the vaccine to be immunogenic.Citation3 This study was conducted in 60 participants 18–45 years of age and evaluated 15 mcg of IVACFLU-S or sterile saline placebo in a 1:1 randomization. Given its promising findings, a phase 2/3 study followed to expand on the safety data and to confirm the immunological findings. In the phase 2/3 study, the participant age range was expanded to 60 years with the goal of seeking regulatory approval based on Vietnam Ministry of Health Guidance on Clinical Trial of Influenza Vaccine.Citation4 This paper reports the IVACFLU-S safety and immunogenicity results.
2. Results
2.1 Disposition of participants
Participant disposition is depicted in . In total, of the 1,399 participants who were screened, 888 participants received study product (740 participants to IVACFLU-S and 148 participants to placebo). Two participants did not complete the study. The reasons for study discontinuation were voluntary withdrawal and lost to follow-up. Immunogenicity testing was done only in phase 3 and prevaccination blood samples were collected from 252 participants. One participant did not provide a postvaccination sample; therefore, 251 participants were included in the immunogenicity analysis.
Figure 1. Participant disposition
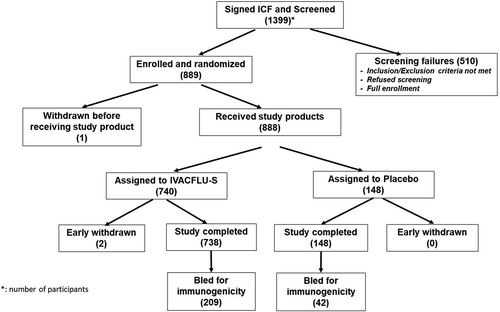
2.2 Demographic and other baseline characteristics
Baseline demographic characteristics of enrolled participants are shown in . The mean age of the participants was 42.8 years (range: 18–60 years). A slightly higher proportion of females (58.6%) were enrolled in the study. No significant differences in baseline ethnicity, race, or age were noted between the study groups.
Table 1. Summary of demographic and baseline characteristics
2.3 Immunogenicity assessment
The seroconversion analysis for HI antibody titers is summarized in . In the combined age group for IVACFLU-S arm, seroconversion against antigen A/H1N1, A/H3N2, and antigen B was achieved in 70.3%, 76.1%, and 54.1% of participants respectively. High seroconversion rates were seen in both 18- to 45-year and 46- to 60-year age groups. For antigen A/H1N1, the seroconversion rate was similar in both age groups. For antigen A/H3N2 and antigen B, the seroconversion rates were lower in the 18–45 age group than in the 46–60 age group. In the placebo group only one participant seroconverted for antigen B.
Table 2. Seroconversion rates for the HI titers by age group (day 22)
The analysis of seroprotection for HI antibodies is summarized in . In the overall age group, high postvaccination seroprotection rates against antigens A/H1N1 (83.3%) and A/H3N2 (86.6%) were observed. The seroprotection rates were relatively lower for antigen B (60.3%). For A/H1N1 and A/H3N2, seroprotection was achieved in a larger proportion of participants in the 18–45 age group than in the 46–60 age group. For antigen B, the seroprotection was achieved in a larger proportion of participants in the 46–60 age group than in the 18–45 age group. A considerable number of participants in the placebo arm had seroprotective titers for A/H1N1 and A/H3N2 strains in the prevaccination serum in both age groups.
Table 3. Seroprotection rates for the HI titers by age group (day 22)
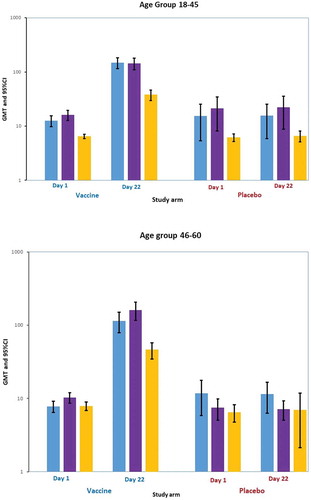
The GMT plot for the HI antibody titer at days 1 and 22 is presented in by age group. There were higher baseline titers to A/H1N1 and A/H3N2 as compared to B, especially in the 18–45 age group. For IVACFLU-S recipients the GMTs increased significantly against all three strains of antigens by day 22 postvaccination. The GMTs were marginally higher for A/H1N1 in the 18–45 age group while the response was higher for A/H3N2 and B in the 46–60 age group. GMFR analysis for the HI antibody titers is summarized in . The GMFR for the HI antibody titers was higher for antigen B in the 18–45 age group while the titers were higher for A/H1N1 and A/H3N2 in the 46–60 age group.
Table 4. Geometric mean fold rise for the HI titers by age group
Figure 2. Geometric mean titer (GMT) plot for HI titer at day 1 and day 22 by age group
2.4 Safety assessment
The safety analyses were based on the combined data from phase 2 and phase 3. An overall incidence of solicited adverse events (AEs) from day 1 to day 7 after vaccination is summarized in . The majority of the solicited AEs were reported on days 1 and 2 after vaccination. Higher proportion of participants in the IVACFLU-S arm reported solicited local AEs (p < .0001). The most frequently reported solicited local AE was tenderness (67%) followed by pain (62%). Majority (60% for tenderness and 55% for pain) of these events were mild. Overall most solicited local AEs were mild (~88%) to moderate (~11%) in severity. Solicited systemic AEs were less commonly reported and rates were comparable to placebo arm (p = .4149). The most frequently reported solicited systemic AE was fatigue/malaise, followed by generalized muscle aches. Majority of solicited systemic AE were mild (~78%) to moderate (~20%) in severity. Upper respiratory tract infection (1.5%) was the most frequently identified unsolicited AE. The majority of the unsolicited AEs were mild to moderate in severity and were considered not related to the study product by the investigator. Six serious adverse events (SAEs) were reported in the study. None of the SAEs reported were considered related to the study product by the investigator. No deaths were reported in this study.
Table 5. Incidence of solicited local and systemic adverse events from day 1 to day 7
3. Discussion
The results for this phase 2/3 study demonstrate that IVACFLU-S was safe and immunogenic in healthy adults between 18 and 60 years of age. The clinical trial requirements for influenza vaccines in Vietnam are guided by Vietnam Ministry of Health Guidance on Clinical Trial of Influenza Vaccine that requires only one of the three criteria (seroprotection, seroconversion, and GMT increase) to be met to qualify for success.Citation4 The serological criteria requirements in Vietnam are summarized in . The criteria have been adapted from EMEA guidelines for licensure of influenza vaccines with one noteworthy difference: the age groups required to evaluate immune response in the Vietnamese guidelines are 18–45 years and > 45 years, whereas the EMEA guidelines identify age groups between 18 and 60 years and ≥ 60 years.Citation5
Table 6. Vietnamese Ministry of Health guidance criteria for evaluation of influenza vaccine
IVACFLU-S met the Vietnam MOH seroconversion criteria for evaluation of influenza vaccine for all three antigens in both age groups. Seroconversion rates were > 70% for antigens A/H1N1 and A/H3N2, and > 50% for antigen B in both age groups on day 22 postvaccination. Seroprotection rates were > 75% for A/H1N1 and A/H3N2 in both age groups. The seroprotection rates for antigen B were 53.8% in age group 18–45 years and 66.7% in age group 46–60 years. IVACFLU-S met the seroprotection licensure criteria for all antigens in both age groups except for antigen B in age group 18–45 years. The seroprotection rates for antigen B were relatively lower in the 18–45 years age group as compared to 46- to 60-year age group. A possible reason for this could be that a higher number of participants in the 46- to 60-year age group had preexisting antibody titers of > 10 for antigen B.
The GMFR for the HI antibody titers was > 5.8 for all three strains of antigens in both age groups. The GMFR was higher for antigen B in 18–45 age group while the GMFR was higher for A/H1N1 and A/H3N2 in 46–60 age group. The Vietnam MOH GMFR criteria were met by IVACFLU-S for all three antigens in both age groups. Significant immune responses were observed in both immunologically naïve participants and in those with preexisting antibodies to influenza. The immunogenicity results are in line with other licensed vaccines.Citation6–Citation8 The immunogenicity of the B antigen was relatively low as compared to that of H1N1 and H3N2, though the immunogenicity of the B strain in the 18–45 age group met two out of three criteria, including the most important one, seroconversion rate. Low immunogenicity of B strain has also been seen with some of the previously licensed vaccines.Citation9–Citation11
Vaccination with a single intramuscular dose of IVACFLU-S seasonal trivalent split virion, inactivated influenza vaccine was generally safe and well-tolerated. The frequency of local solicited reactions was higher in the IVACFLU-S group as compared to placebo. Pain and tenderness were reported in higher proportion of participants as compared to other licensed vaccines. The systemic solicited events were similar to other licensed inactivated influenza vaccines.Citation12–Citation14 The majority of solicited and unsolicited AEs were mild to moderate in severity. The rate of unsolicited events was similar in vaccine and placebo groups. No SAEs were considered related to the study products and no immediate postvaccination AEs, such as shock and anaphylaxis, were reported.
A limitation of this study was the skewed distribution of participants between the vaccine and placebo arm. This limited the safety comparison between the two arms because of fewer numbers in the placebo group. However, there is ample demonstration of widespread safety with seasonal influenza vaccines worldwide. Immunogenicity licensure criteria have been the basis for approval of seasonal influenza vaccines worldwide but immune responses are a surrogate indicator of clinical protection. Performing an effectiveness study will truly illustrate the clinical value of this vaccine as high baseline titers for H1N1 and H3N2 detected in this study indicate widespread prevalence of influenza in Vietnam (). To expand the benefits of this vaccine to people who need them the most, future studies may involve individuals at higher risk, such as the elderly, individuals with preexisting conditions, and children.
Table 7. Prevaccination titers
In summary, though mild local reactions like pain and tenderness were significantly higher in the vaccine group, the vaccine was otherwise safe with respect to systemic reactions, unsolicited AEs and SAEs. This phase 2/3 randomized, double-blind study demonstrated local reactogenicity, other safety and immunogenicity of IVACFLU-S in adult volunteers from Vietnam. This study is a good foundation toward licensure of the first domestically produced seasonal influenza vaccine in Vietnam and based on this study, IVACFLU-S received licensure in January 2019. Licensure of this vaccine will significantly contribute to the mandate of WHO’s GAP by promoting development of a domestic influenza vaccine and consequently augmenting global pandemic influenza vaccine production capacity.
4. Methods
4.1 Study design and implementation
This trial was a phase 2/3, double-blind, randomized, placebo-controlled study to evaluate the safety and immunogenicity of a single intramuscular dose of IVACFLU-S seasonal trivalent inactivated split-virion influenza vaccine in male and female participants aged 18–60 years. Because of predictable immunogenicity, dose finding studies (phase 2) of traditional, nonadjuvanted inactivated influenza are often not required. In agreement with National Regulatory Authorities (NRAs), several manufacturers have initiated pivotal licensure phase 3 inactivated influenza vaccine trials after encouraging safety and immunogenicity data from phase 1 without conducting phase 2 studies. However, based on the recommendation of the Vietnamese Ministry of Health (MOH), a phase 2 study was incorporated in a seamless phase 2/3 design to assess the safety of the vaccine. In this design, transition from phase 2 to 3 was based on the blinded review of safety data by investigators, sponsors, and Ethics Review Committees (ERCs) at the clinical site (Pasteur Institute Ho Chi Minh City [PI HCMC])) and the MOH.
Placebo was used as a control in the study to blind participants and investigators for safety assessments rather than as a comparator for immunogenicity between the interventions. There is no immunogenicity data on any licensed seasonal influenza vaccine in Vietnamese population. Seasonal influenza vaccine is not commonly used in Vietnam especially in rural areas where this trial was conducted. These vaccines are available in private market and usually administered as fee-based services in big cities.
The study ran from March to October 2017 at PI HCMC. ERCs at WHO, PI HCMC, and the Vietnamese MOH reviewed and approved the study protocol. The study was conducted in accordance with the principles of the Declaration of Helsinki, Good Clinical Practice, and Vietnamese regulatory requirements. The study is registered in the National Institutes of Health registry (www.clinicaltrials.gov) with registration number NCT03095599.
A total of 888 healthy adults were ultimately included in the phase 2/3 study, and each participant provided written informed consent before enrollment. Pregnant women, breast feeding women, children, and people older than 60 years of age were not eligible for participation. Volunteers excluded from study participation included: those who received seasonal influenza vaccine in the previous 6 months, or any nonstudy vaccine within 4 weeks prior to enrollment; and those with current or recent (within 2 weeks of enrollment) acute severe illness with or without fever. A permuted-block randomization was used, and participants were stratified according to age (18–45 years and 46–60 years) and study site (only in phase 3). All study products were administered intramuscularly into the deltoid muscle.
In the phase 2 part of the study, 252 eligible participants were randomly assigned in a ratio of 5:1 (vaccine: placebo) to receive one dose of 15 mcg hemagglutinin (HA) vaccine per strain (a total of 45 mcg HA) or placebo at one study site. Study products were administered intramuscularly into the deltoid muscle. Safety data until day 8 postvaccination were reviewed by internal protocol safety review committee (comprising of investigators and medical officers from PATH, IVAC, and clinical research organization) and ERCs from MOH and clinical site.
Following determination that it would be safe to proceed, enrollment in the phase 3 part of the study commenced. In the phase 3 portion of the study, 636 participants were randomized to receive either IVACFLU-S or placebo at a ratio of 5:1 at two study sites. In both phases three scheduled clinic visits took place on days 1, 8, and 22. Safety monitoring home visits/telephone calls took place on days 2, 5, and 91. Safety was assessed in all phase 2 and phase 3 participants through day 91. Immunogenicity was assessed only in a subset of phase 3 participants, all enrolled at one site. Blood samples for immunogenicity testing were collected at day 1 before vaccination and 21 days after vaccination in 252 randomized participants (210 vaccine recipients and 42 placebo recipients) to ensure evaluable samples from at least 200 vaccine recipients (100 each from both age groups) and 40 placebo recipients (20 each from both age groups).
4.2 Investigational product
IVACFLU-S is a seasonal, inactivated, split virion, trivalent influenza vaccine (A/H3N2, A/H1N1, and B) grown in embryonated chicken eggs. The vaccine contains strains recommended by WHO for the 2016–2017 northern hemisphere influenza season: A/California/07/2009 (H1N1), A/Hong Kong/4801/2014 (H3N2), and B/Brisbane/60/2008 (B). The vaccine is prepared from virus grown in the allantoic cavity of embryonated eggs, split by Triton X-100, inactivated by formaldehyde and then purified. IVACFLU-S is formulated to contain 15 µg HA from each of the three vaccine strains in 0.5 mL of phosphate buffered saline (PBS) at a pH of 7.2 and filled in single-dose vials. No adjuvants were included in the vaccine formulation. The placebo was phosphate buffered saline (PBS), pH 7.2, which was available in 0.5 mL single-dose vials identical to the study vaccine vials. The study products were stored at a temperature between +2°C and +8°C, which yielded a shelf life of 12 months. Staff monitored and documented storage temperature daily. The study vaccine and placebo were not allowed to freeze. The study products were labeled and packaged at IVAC by a pharmacist who was not involved in the conduct of the study and were administered at the study clinics by blinded study personnel according to the sequential randomization code.
4.3 Immunological assessment
Study staff tested serum specimens for the presence of HI antibodies to the influenza strains in the vaccine VisMederi srl laboratory in Siena, Italy performed this testing using a validated assay.Citation15 Serum specimen collection occurred on days 1 and 22. Day 1 sample collection took place prior to administration of the study product. Serum specimens were stored at –20°C or below before being shipped to the laboratory. Seroprotection rates were defined as percentage of participants with HI titers ≥ 1:40 following vaccination. Seroconversion rates were defined as percentage of participants achieving an increase in HI titer from < 1:10 prevaccination to ≥ 1:40, or at least a fourfold postvaccination increase in titer from a prevaccination titer ≥ 1:10. Geometric mean fold rises of serum HI antibodies were defined as postvaccination GMT/prevaccination GMT. Subjects with HI antibody titers ≥ 1:10 were considered to be seropositive.
4.4 Safety assessment
Solicited AEs were assessed within 30 min (immediate reactogenicity) and continuously for 7 days after vaccination. Participants were given diary cards to record the occurrence and severity of local reactions including erythema/redness, swelling/induration, injection site pain, and systemic reactions including fever, fatigue/malaise, generalized muscle aches, joint aches, chills, nausea, vomiting, and headache. All solicited reactions were considered to be vaccine-related. Unsolicited AEs were assessed for 21 days after vaccination. Investigators also conducted causality assessments and graded severity of unsolicited AEs. Serious AEs were assessed throughout the study (i.e. till 90 days postvaccination). All AEs were coded by preferred term (PT) and primary system organ class (SOC) using the Medical Dictionary for Regulatory Activities (MedDRA).
4.5 Statistical considerations
The total sample size for the combined phase 2/3 study was 888 participants. Participants were randomized to receive either IVACFLU-S or placebo at a ratio of 5:1; this assigned 740 participants to the IVACFLU-S arm and 148 participants to the placebo arm. The sample size for the combined study was selected in response to the MOH’s requirement for additional safety data from phase 2 before proceeding to phase 3 to satisfy the MOH guidance for evaluation of influenza vaccines for adults.Citation4 The total sample size for the phase 2 was approximately 250 participants (with at least 200 randomized to receive vaccine and 40 randomized to receive placebo). The phase 3 sample size was approximately 650 participants (with at least 500 randomized to receive vaccine and 100 randomized to receive placebo). The two age groups (18–45 and 46–60 years of age) were to be equally distributed. Block randomization was stratified by site and age group. To account for potential dropouts, the sample size allowed for an overage of approximately 5%.
The MOH’s guidance on influenza vaccine clinical trials is based on the point estimate of immune responses in the vaccine group rather than on comparison to a control. No formal statistical testing to compare the immune response between vaccine and placebo was performed. Data were presented along with two-sided exact 95% confidence intervals (CIs) calculated using the Clopper-Pearson method. The safety analyses were based on the combined data from phase 2 and phase 3, pooled across study sites. The safety profile of IVACFLU-S was evaluated by the number and proportion of participants experiencing AEs by severity and relatedness to vaccination. For solicited events, Fisher’s exact test or the Cochran-Mantel-Haenszel test were used to compare the proportions of participants experiencing such events across the two treatment groups. No formal hypothesis testing with multiplicity adjustment was performed. No statistical testing was performed for unsolicited AEs, including SAEs. Antibody titers below the lowest limit of quantitation (reported as < 10) were set to half that limit (10/2 = 5). The data analysis was conducted using SAS® version 9.4.
Disclosure of potential conflicts of interest
The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and the Response; Biomedical Advanced Research and Development Authority. Le Van Be and Duong Huu Thai are employed with Institute of Vaccine and Medical Biologics (IVAC), which manufactured the vaccine.
Acknowledgments
The authors wish to acknowledge the support from: (1) Staff members of District Health Center of Ben Luc: Nguyen Nguyen Huynh Phuong, Phan Tan Thanh; District Health Center of Long Thanh: Nguyen Thi Van Van, Pham Thanh Hai; Provincial Preventive Medicine Center of Long An: Ngo Van Hoang; Provincial Preventive Medicine Center of Dong Nai: Bach Thai Binh. (2) Pasteur Institute Ho Chi Minh city: Nguyen Dieu Thuy, Truong Thi Thuy Dung, Nguyen Thi Huyen Trang, Nguyen Quoc Kien, Nguyen Thi Thanh Thao, Huynh Thi Kim Loan, Nguyen Thi Tuong Vy and Nguyen Thanh Long. (3) Site investigator: Phan Cong Hung. (4) PATH, Vietnam: Mona Byrkit, Nguyen Mai Huong, Hoang Thi Thi, and Le Thanh Hai. (5) PATH, USA: Joseph Chiu, Rahnuma Wahid, Muriel Socquet, Kristin Bedell, and Katie Regan. (6) IQVIA: team involved in project management, monitoring, medical writing, data management and statistical support. (7) World Health Organization (WHO): Erin Sparrow, Guido Torelli and Florence Barthelemy. (8) Biomedical Advanced Research and Development Authority (BARDA): Chuong Huynh, Karen Biscardi, James King, Michael Ohara, Frank Arnold, Sheng Li and Corrina Pavetto.
Additional information
Funding
Related Research Data
References
- Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, Cohen, C, Gran, JM, Schanzer, D, Cowling, BJ, et al. Global seasonal influenza-associated mortality collaborator network. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet. 2018 Mar 31;391(10127):1285–300. doi:10.1016/S0140-6736(17)33293-2.
- World Health Organization. Global pandemic influenza action plan to increase vaccine supply. [accessed 2018 Apr 5]. http://apps.who.int/iris/bitstream/10665/69388/1/WHO_IVB_06.13_eng.pdf2006
- Anh DD, Thiem VD, Anh NTH, Huong VM, Nga NT, Thang TC, Thai, DH, Chien, VC, Holt, R, Wahid, R, et al. Randomized safety and immunogenicity trial of a seasonal trivalent inactivated split virion influenza vaccine (IVACFLU-S) in healthy young Vietnamese adults. Vaccine. 2016 Oct 26;34(45):5457–62. doi:10.1016/j.vaccine.2016.08.052.
- Guidance on Clinical Trial of Influenza Vaccine. Ministry of health (Administration of science technology and training). Hanoi (Vietnam); 2013.
- The European Agency for the Evaluation of Medicinal Products. Note for guidance on harmonization of requirements for influenza vaccines, 1997. [accessed 2018Apr 6]. http://www.ema.europa.eu/docsen_GB/document_library/Scientific_guideline/2009/09/WC50000394.pdf
- Flublok package insert. accessed 2018Apr 10. https://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM524684.pdf
- Flucelvax quadrivalent prescribing information. [accessed 2018Apr 10]. https://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM502899.pdf
- Flulaval prescribing information. [accessed 2018 Apr 10]. https://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM112904.pdf
- [accessed 2018 Apr 12]. http://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM329134.pdf
- [accessed 2018 12 Apr]. http://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM305089.pdf
- [accessed 2018 12 Apr]. http://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM335392.pdf
- Cox MM, Izikson R, Post P, Dunkle L. Safety, efficacy, and immunogenicity of Flublok in the prevention of seasonal influenza in adults. Ther Adv Vaccines. 2015 Jul;3(4):97–108. doi:10.1177/2051013615595595.
- Roggelin L, Vinnemeier CD, Meyer S, Witte K, Marx L, Theeß W, Burchard, GD, Rolling, T and Cramer, JP. A 2013/2014 northern hemisphere season surface antigen inactivated trivalent influenza vaccine–assessing the immunogenicity and safety in an open label, uncontrolled study. Hum Vaccin Immunother. 2015;11(10):2370–75. doi:10.1080/21645515.2015.1064570.
- Haugh M, Gresset-Bourgeois V, Macabeo B, Woods A, Samson SI. A trivalent, inactivated influenza vaccine (Vaxigrip®): summary of almost 50 years of experience and more than 1.8 billion doses distributed in over 120 countries. Expert Rev Vaccines. 2017 Jun;16(6):545–64. doi:10.1080/14760584.2017.1324302.
- Manual for the laboratory diagnosis and virological surveillance of influenza. WHO Global Influenza Surveillance Network; 2011. [accessed 2018 Apr 6]. http://apps.who.int/iris/bitstream/10665/44518/1/9789241548090_eng.pdf