
ABSTRACT
Development of cancer vaccines targeting tumor self-antigens is complex and challenging due to the difficulty of overcoming immune tolerance to self-proteins. Vaccination against tumor self-protein D52 (D52) has been successful, although complete protection appears impaired by immune regulation. Our previous studies suggest that vaccine elicited CD8 + T cells producing interleukin 10 (IL-10) may have a negative impact on tumor protection. Understanding the role CD8+ IL-10 + T cells play in the immune response following vaccination with D52 could result in a more potent vaccine. To address this, we vaccinated IL-10 deficient mice with the murine orthologue of D52; vaccination of wild type (wt) C57BL/6J served as a control for comparison. In separate experiments, D52 vaccinated wt mice were administered IL-10R-specific mAb to neutralize IL-10 function. Interestingly, we observed similar protection against primary tumor challenge in the experimental groups compared to the controls. However, individual IL-10 deficient mice that rejected the primary tumor challenge were re-challenged 140 days post-primary challenge to access vaccine durability and immunologic memory against tumor recurrence. Mice deficient in IL-10 demonstrated a memory response in which 100% of the mice were protected from secondary tumor challenge, while wt mice had diminished recall response (25%) against tumor recurrence. These results with analysis of vaccine-elicited CD8 + T cells for tumor-specific killing and regulatory cell marker expression, add further support to our premise that CD8+ IL-10 + T cells elicited by D52 tumor-self protein vaccine contribute to the suppression of a memory CTL responses and durable tumor immunity.
KEYWORDS:
Introduction
Cancer immunotherapy is an effective and expanding field, however, more preventative approaches, such as vaccines, lag in development.Citation1,Citation2 Discovering tumor antigens suitable for targeting by vaccination is complex and challenging,Citation3-Citation6 therefore, various targets have been used in different vaccination strategies.Citation7 One tumor self-protein that provides promise of being an effective target in preclinical studies is tumor protein D52 (D52).Citation8-Citation13 Tumor protein D52 is a naturally expressed intracellular protein present at low but detectable levels in healthy cells and tissues where its normal function has yet to be defined. Increased expression of D52 has been demonstrated in association with prostate cancer as well as numerous other human malignancies. The murine orthologue of D52 (mD52) is ~86% identical to human D52 (hD52) at the amino acid level. Previous work from our laboratory demonstrated that over expression of mD52 in normal murine fibroblast cells induced anchorage independent growth in vitro and spontaneous lung metastasis in vivo. We also demonstrated that reduction of hD52 expression via RNAi resulted in increased apoptosis in human breast cancer cells and hD52 over-expression correlated with decreased survival in human breast cancer patients (reviewed in 11). Interestingly, shRNA reduction of mD52 expression abrogates spontaneous metastasis associated with murine 3T3.mD52 sarcoma cells (unpublished observation). Thus, D52 is actively involved in transformation, leading to increased cell proliferation and metastasis. Involvement in oncogenesis suggests that these antigens may be critical for tumor survival, making the overexpressed tumor self-protein D52 an excellent candidate for a cancer vaccine target.
Vaccination against this self-protein has been successful in murine models without inducing autoimmunity.Citation8-Citation11 D52 is a naturally expressed intracellular protein detectable in healthy cells, albeit at low levels, that has demonstrable oncogenic functions.Citation14-Citation16 In many cancers, D52 is overexpressed and required for tumor progression, indicating D52 as a good target for vaccinationCitation10,Citation11,Citation16-Citation22; however, 100% protection in preclinical models has yet to be obtained likely due to immune regulation.Citation10,Citation23 Our previous studies suggest CD8 + T cells producing interleukin 10 (IL-10) may contribute to the lack of complete protection from tumor challenge.Citation9,Citation12,Citation13,Citation23 CD8+ IL-10 + T cells may behave more like suppressor cells than effector T cells and inhibit immunity to an over expressed tumor self-protein, like D52.Citation23-Citation25
To test whether the CD8+ IL-10 + T cells elicited by D52 vaccination belong to a subset of previously described suppressor CD8 + T cells,Citation26-Citation30 we depleted CD122hi T cells, the specific marker found on previously described CD8 + T regulatory cells, in combination with mD52 vaccination. Depleting these CD122hi T cells did not augment vaccine-mediated tumor protection, and expression studies confirmed that these IL-10+ CD8 + T cells do not express Foxp3.Citation12 This indicated that the CD8+ IL-10 + T cells elicited in our vaccine models might be a different subset than those previously described. Understanding the role these potentially unique CD8+ IL-10 + T cells play in the immune response to D52 vaccination could contribute to the creation of a more potent and effective vaccine. Herein, we tested the hypothesis that CD8+ IL-10 + T cells elicited by D52-vaccination contribute to suppression of protective tumor immunity in a D52 vaccine murine cancer model.
Materials and methods
Mice and cell lines
Male, 6-week-old C57Black/6J wild type (wt) and IL-10 deficient (B6/129P2-IL10tm1cgn/J, IL-10 KO) mice were purchased from Jackson Labs (Bar Harbor, ME). All animals were cared for and treated according to institutional guidelines at Texas Tech University Health Sciences Center. The autochthonous C57Black/6J TRAMP-C2 tumor cell line and the BALB/c MKSA tumor cell line were maintained as previously described.Citation8,Citation31
Vaccination
Individual mice were vaccinated every 7–10 days with 50 μg of plasmid DNA containing full-length mD52 cDNA (mD52-DNA) in PBS, i.m. for a total of four injections (previously determined empirically). Empty vector DNA was used as a negative control vaccine. In some experiments, murine IL-10 Receptor-specific monoclonal antibody (mAb) (BioXCell, Lebanon, NH) was injected (100 μg i.p.) into wt mice 1 day prior to vaccination, with each of the four mD52 vaccinations, at tumor challenge, and after primary tumor challenge every 3 days for an additional four injections. Isotype-matched Ig Ab (BioXCell, Lebanon, NH) was used as a control.
Tumor challenge
Mice were challenged with autochthonous TRAMP-C2 tumor cells s.c. 14 days following the last vaccination. Tumor cells were harvested, counted, resuspended in PBS, and injected with a tumorigenic dose (5 × 105 cells/100 μL determined empirically) subcutaneously in the right flank of each mouse. The same procedure was followed for the secondary tumor challenge, in the opposite flank, approximately 140 days post-primary tumor challenge. Tumor growth was determined by perpendicular tumor measurements (a × b2)/2, where b is the smaller of the two measurements.Citation9,Citation12
Multiplex RT-PCR
Using Lympholyte-M® (Cedarlane Labs, Burlington, NC) gradient separated spleen-derived lymphocytes from vaccinated and tumor challenged mice, T cells were isolated using magnetic beads according to manufacturer’s instructions (LifeTech, Thermo Fisher, Waltham, MA) then in vitro stimulated with CD3/CD28 activator beads (Dynabeads, LifeTech, Thermo Fisher, Waltham, MA) and interleukin-2 (IL-2) (~10 ng/ml) (Peprotech) for 6 days. Magnetic beads specific for isolating untouched CD8 + T cells (InVitrogen, Waltham, MA) were used for purification of CD8 + T cells when appropriate. RNA was extracted using RNeasy Mini kit (Qiagen, Hilden, Germany) from combined total of 5 × 106 CD4+ and CD8 + T cells or 5 × 106 purified CD8 + T cells. For multiplex RT-PCR, cDNA was generated using 250 ng or 1 μg of DNA free RNA and oligo-dT primer. PCR reactions were performed according to manufacturer’s instructions (Qiagen, Hilden, Germany). Primer sets were purchased from InVitrogen (Waltham, MA) the sequences are described in . PCR conditions for 26 or 30 cycles were as follows: 94°C for 1 min, 60°C for 1 min 30 sec, and 72°C for 1 min 30 sec. All results were visualized on 3% agarose gels containing ethidium bromide. Amplification of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA served as an internal reference control.
Table 1. Primer sequences used for reverse transcription-polymerase chain reaction.
T cell culture and ELISAs for cytokine production
Untouched CD8 + T cells from IL-10 deficient immunized mice were isolated and stimulated in vitro by culturing Lympholyte-M® (Cedarlane Labs, Burlington, NC) gradient separated spleen-derived lymphocytes with CD3/CD28 activator beads (Dynabeads, LifeTech, Thermo Fisher, Waltham, MA) and IL-2 (~10 ng/mL) for 6 days. T cells were then pooled and cultured for 24 h with tumor targets and supernatants used for cytokine analyses. Conditions included medium alone, compared to T cells cultured with various tumor cell targets. Experimental targets were TRACMP-C2 tumor cells (H-2b+, mD52+). mKSA (H-2d+, mD52+) tumor cells, served as a control MHC mismatch, antigen-positive target. To confirm MHC-I restricted tumor recognition, blocking assays were performed by incubating tumor cells with anti-H-2b or anti-H-2d (negative control) mAb, prior to incubation with T cells. Assessment of cytokine secretion by tumor-specific T cell cultures were accomplished by applying culture supernatants to commercially available sandwich ELISA’s for IL-10 (R&D Systems, Minneapolis, MN) as per the manufacturer’s instructions. We performed all assays with the manufacturer’s provided internal controls, from which standard curves were generated in order to determine concentration of cytokines produced in experimental sets for ELISA detection of IL-10.Citation9,Citation10
Analysis of cytotoxic T lymphocyte (CTL)-mediated tumor cell lysis
T cells from spleens of IL-10 deficient mice that survived tumor challenge were isolated and subjected to standard cytotoxic T lymphocyte (CTL)-mediated tumor cells lysis analysis. CTLs were generated by culturing untouched CD8 + T cells with CD3/CD28 activator beads (Dynabeads, LifeTech, Thermo Fisher, Waltham, MA) and IL-2 (~10 ng/mL) for 6 days. CTLs were then cultured with tumor targets for 4 h prior to supernatants being collected. Specificity was evaluated by mixing various numbers of CTLs with a constant number of target cells (5 × 103 cells per well) in 96 well-round bottom plates. Specific lysis was determined by LDH-release method (Promega, Madison, WI) and measured using a Victor3 plate reader (Perkin Elmer, Boston, MA).
Statistical analysis
Tumor challenge data were analyzed with a paired t-test to determine whether significant differences existed between tumor-free mice at day 54 and day 90 post tumor challenge in IL-10 KO and wt mice using GraphPad, InStat. Statistical significance (p < .05) for repeated RT-PCR experiments was calculated by unpaired t-tests using GraphPad, InStat.
Results
IL-10 receptor blocking does not enhance D52-vaccine-elicited tumor immunity
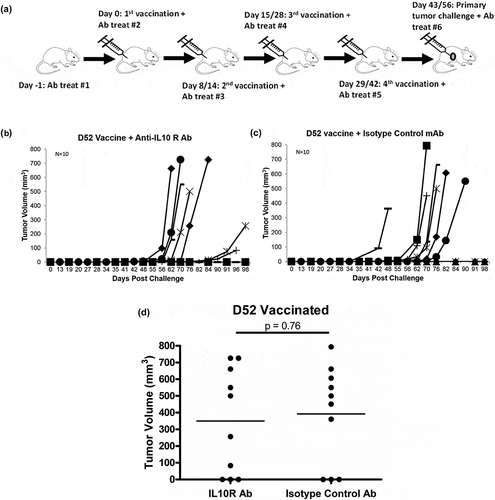
Previously we demonstrated protective tumor immunity ranging from 14% to 50% (mean of 30%) protection against tumor challenge following mD52 plasmid DNA i.m. vaccination. We observed CD8+ IL-10 + T cells that may be a unique undefined subset of suppressor CD8 + T cells preventing complete tumor protection following mD52 vaccination.Citation11,Citation12 In the present study, we used this same model to test the effect of inhibiting or blocking IL-10 function on enhancing tumor protection following vaccination against mD52. We vaccinated wt mice with mD52 plasmid DNA (i.m.) along with administration of anti-IL-10 receptor (IL-10R) mAb (i.p.) (, repeat experiments, data were combined for graphing purposes). Of the 10 mice vaccinated with mD52 and administered anti-IL-10R mAb 30% (3/10) were protected from primary tumor challenge with TRAMP-C2 tumor cells (). Tumor protection in the group administered isotype control Ab was also 30% (3/10) (). Administering anti-IL-10R mAb in an attempt to block IL-10 function in conjunction with mD52 vaccination did not enhance tumor protection. There was no significant difference between experimental and control immunized and treated groups of mice (). To evaluate the induction of immunologic memory a secondary tumor challenge was administered in the opposite flank to mice form experimental and control groups that survived the primary challenge (3/10). In both, the anti-IL-10R mAb and the isotype control Ab groups, approximately 30% (1/3) of the remaining mice were protected (not shown), demonstrating that IL-10R blocking was not effective. Vaccination with empty vector as a control provided no protection (not shown).
Figure 1. Tumor growth in mice vaccinated with murine D52 (mD52) as a plasmid DNA vaccine and challenged with autochthonous TRAMP-C2 tumor cells. (a) C57Bl/6J mice received four vaccinations with mD52 plasmid DNA i.m. with administration of either anti-IL-10 receptor mAb or isotype antibody i.p. All mice were challenged with 5 × 105 TRAMP-C2 s.c. in the right flank 2 weeks after the final vaccination. Shown are the schedules for two independent experiments Day exp 1/exp 2 (indicated by Day 8/14; Day 15/28; Day 29/42; tumor challenge Day 43/56). (b) Tumor growth in mice vaccinated four times, injected i.p. with anti-IL-10R mAb and challenged with TRAMP-C2 tumor cells. Each graph line represents an individual mouse. (c) Tumor growth in mice vaccinated four times, injected i.p. with isotype control Ab and challenged with TRAMP-C2 tumor cells. Each graph line represents an individual mouse. (d) Comparison of day 98 tumor growth in vaccinated mice treated with anti-IL-10R mAb (IL10R Ab) or isotype control Ab (Isotype Ab). Tumor volume (mm3) was determined by perpendicular measurements (axb2)/2, where b is the smaller of the two measurements. Data are representative of two repeated experiments (N = 10). Significance was determined using a paired t-test (p < .05).
IL-10 deficiency improves D52-vaccine-elicited tumor immunity
It is widely known that targeting IL-10 in vivo is challengingCitation32-Citation34; as such, results in the previous experiment were not surprising. To overcome this hurdle, we utilized IL-10 deficient mice (IL-10 KO) to evaluate the effect IL-10 has on tumor protection following mD52 plasmid DNA vaccination (). IL-10 KO mice and wild type (wt) mice were vaccinated with mD52 i.m. and challenged with TRAMP-C2 tumor cells as described in the Methods section. Vaccination of both IL-10 KO mice and wt mice elicited 40% (4/10) protective tumor immunity following primary tumor challenge (). Interestingly, mice deficient in IL-10 had a significant delayed onset of tumor burden while wt mice did not. Comparing tumor burden in vaccinated IL-10 deficient mice at an early time point (day 56) versus an ending time point (day 90) there was a significant difference (p = .0368) in tumor-free mice. In contrast, wt mice showed no significant difference (p = .1679) in tumor-free mice at the same time points (). Vaccination with vector control provided no protection from tumor challenge (not shown). This delayed tumor onset in IL-10 KO mice over time indicates an increase in anti-tumor immunity in the absence of IL-10.
Figure 2. Tumor growth in IL-10 deficient (KO) mice and wild type (WT) mice vaccinated with murine D52 (mD52) as a plasmid DNA vaccine and challenged with TRAMP-C2 tumor cells. (a) C57Bl/6J mice received four vaccinations with mD52 plasmid DNA i.m. All mice were challenged with TRAMP-C2 s.c. in the right flank 2 weeks after the final vaccination. Shown are the schedules for two independent experiments Day exp 1/exp 2 (indicated by Day 8/14; Day 16/28; Day 30/43; primary tumor challenge Day 44/56; secondary tumor challenge Day 184/189). (b) Tumor growth in IL-10 deficient (KO) mice vaccinated four times and challenged with TRAMP-C2 tumor cells. Each graph line represents an individual mouse. (c) Tumor growth in wild type (WT) mice vaccinated four times and challenged with TRAMP-C2 tumor cells. Each graph line represents an individual mouse. (d) Comparison of day 98 tumor growth in vaccinated wild type (WT) and vaccinated IL-10 deficient (IL10 KO) mice. Data are presented as percent tumor free (% Tumor Free) in a specific group. Tumor volume (mm3) was determined by perpendicular measurements (axb2)/2, where b is the smaller of the two measurements. Data are representative of two repeated experiments (N = 10). Significance was determined using a paired t-test (p < .05).
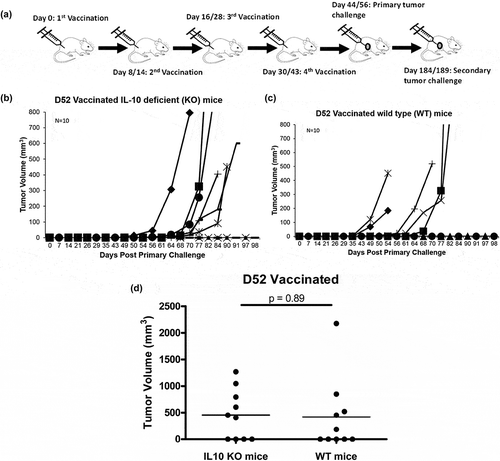
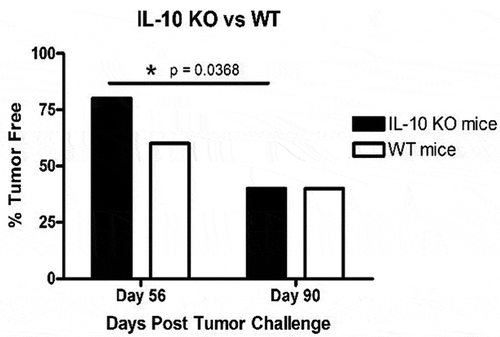
Figure 3. Comparison of autochthonous TRAMP-C2 tumor growth in vaccinated wild type (WT) and IL-10 deficient (IL-10 KO) mice at day 56 and day 90 post-primary tumor challenge. Data are presented as percent tumor free (% Tumor Free) in a specific group. Significance was determined using a paired t-test (p < .05).
IL-10 deficiency augments D52-vaccine elicited memory immune response
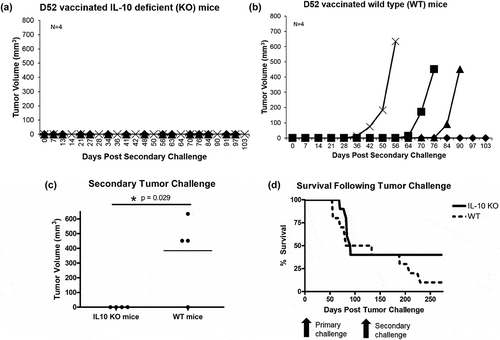
To test mD52 vaccine durability and immunologic memory, mD52 vaccinated IL-10 KO mice that rejected a primary tumor challenge with TRAMP-C2 tumor cells were re-challenged with the same dose of TRAMP-C2 cells in the opposite flank approximately ~140 days after rejection of the primary challenge (184–189 days after the final mD52-DNA vaccination) (). Strikingly, mD52 vaccinated mice deficient in IL-10 that rejected a primary tumor challenge were 100% (4/4) protected from secondary tumor challenge (), while mD52 vaccinated mice with functional IL-10 (wt) that rejected a primary tumor challenge were only 25% (1/4) protected (). Complete rejection of secondary tumor challenge (as a model of cancer recurrence) by mD52 vaccine-induced tumor immunity in IL-10 KO mice was significant (p = .029) compared to wt mice (), and the overall survival of mD52 vaccinated IL-10 KO mice (40%) was markedly greater than that of wt mice (10%) (). These results corroborate our previous studies exploring D52-based vaccination in murine cancer models.Citation8-Citation12 Importantly these results demonstrate generation of a potent and durable memory response in the absence of IL-10, suggesting an immune suppressive role for IL-10 induced by vaccination with the oncogenic tumor-self antigen D52.
Figure 4. Autochthonous TRAMP-C2 tumor growth in IL-10 deficient (KO) and wild type (WT) mice vaccinated with murine D52 (mD52) as a plasmid DNA vaccine and inoculated with a secondary tumor cell challenge ~140 days after rejection of the primary challenge (; 184–189 days after the final vaccination). (a) Secondary TRAMP-C2 tumor growth in IL-10 deficient (KO) mice vaccinated four times and capable of rejecting a primary tumor challenge with TRAMP-C2 tumor cells. Each graph line represents an individual mouse. (b) Secondary TRAMP-C2 tumor growth in wild type (WT) mice vaccinated four times and capable of rejecting a primary tumor challenge with TRAMP-C2 tumor cells. Each graph line represents an individual mouse. (c) Comparison of secondary tumor growth on day 103 post-secondary tumor challenge with TRAMP-C2 cells in vaccinated IL-10 deficient (IL10 KO) and wild type (WT) mice. Tumor volume (mm3) was determined by perpendicular measurements (axb2)/2, where b is the smaller of the two measurements. Significance was determined using a paired t-test (p < .05). (d) Comparison of overall survival between vaccinated IL-10 deficient (IL10 KO) and wild type (WT) mice following primary and secondary TRAMP-C2 tumor challenges (). Data are presented as percent survival (% Survival) in a specific group over time (Day 0 represents primary tumor challenge and Day 133 represents secondary tumor challenge in the opposite flank relative to primary tumor challenge of Day 0).
D52 vaccine-elicited CD8 + T cells are a source of IL-10
Our previous studies indicated that CD8+ IL-10 producing T cells are elicited in response to vaccination with tumor self-protein D52.Citation9,Citation10,Citation12 This was confirmed by multiplex RT-PCR analysis of D52 vaccine-induced T cell cultures containing either CD4+ and CD8 + T cells or CD8 + T cells alone.Citation23 We analyzed T cells from mD52 vaccinated mice that rejected tumor challenge compared with T cells from mD52 vaccinated mice that had delayed tumor onset. T cells were isolated, in vitro stimulated for 6 days with IL-2 or IL-15 (in separate experiments 10 ng/ml of IL-15 was substituted to maintain memory T cells and increases anti-tumor effect of previously defined CD8 + T regs).Citation35 cDNA generated from RNA isolated from the cultured T cells was analyzed by multiplex PCR using specific primers, including IL-10, IFN-γ, PD-1, and Blimp-1. In RNA samples isolated from combined CD4+ and CD8 + T cells, IL-10 was not detectable using multiplex RT-PCR. However, when RNA isolated from purified CD8 + T cells was analyzed, IL-10 expression was detected. This further confirms that mD52 vaccine-elicited CD8 + T cells are a source of the IL-10 detected. Interestingly, when vaccination failed to inhibit tumor growth, faint Blimp-1 mRNA expression was detected in CD8 + T cells alone, but not detected in combined CD4+ and CD8 + T cell co-cultures. Blimp-1 is known to play a role in IL-10 production in T regulatory cells,Citation36,Citation37 further supporting our rationale that mD52 vaccine-induced CD8 + T cell are a source of IL-10 and may represent a unique regulatory population.
To expand our findings we analyzed CD8 + T cells isolated from wt or IL-10 deficient mice that were mD52 vaccinated and tumor challenged as described in the methods section. CD8 + T cells from mice of the same vaccine groups were either pooled for analysis, or in separate experiments CD8 + T cells were analyzed from individual mice. All isolated CD8 + T cells were stimulated in vitro for 6 days in the presence of IL-2 (substitution of IL-15 in previous studies yielded results that were not different from IL-2). RNA was isolated from 5 × 106 CD8 + T cells and multiplex RT-PCR was carried out using the specific primer sets optimized for multiplex RT-PCR (). IFN-γ expression was detected in pooled CD8 + T cells isolated from vaccinated wt mice exhibiting delayed onset tumor growth following tumor challenge (, WT pool), as well as in vaccinated wt and IL-10 deficient mice that completely rejected tumor challenge (, WT, IL10 KO, IL10 KO pool). However, as expected IL-10 mRNA expression was not detected in 5 × 106 CD8 + T cells isolated from IL-10 KO mice (, IL-10 KO; IL-10 KO pool) or by ELISA (not shown). Faint IL-10 was detected in 5 × 106 CD8 + T cells isolated form vaccinated wt mice ( wt, wt pool) corroborating our previously published findings.Citation9,Citation10,Citation12 CTLA-4 expression was slightly, but significantly, increased in vaccinated IL-10 deficient mice compared to vaccinated wt mice, though both groups expressed detectable levels of CTLA-4 message (). Expression of IL-10 in vaccinated wt mice did not differ between mice that were protected from tumor challenge or demonstrated delayed tumor onset suggesting that other immune regulation mechanisms may also be responsible for the lack of complete tumor protection, as previously described by our group.Citation10,Citation12,Citation23
Figure 5. Relative expression of multiple T cell-associated genes in T cells from D52 vaccinated and tumor challenged mice. (a) Representative 30 cycle RT-PCR showing mRNA expression of target genes expressed in CD8 + T cells from wildtype (WT) and IL-10 deficient (IL10 KO) mice. CD8 + T cells were isolated from splenic mononuclear cells and kept individually (WT or IL10KO) or pooled (WT pool or IL10KO pool) with like mice and analyzed for mRNA expression. From the left, the first two lanes are 1Kb and 100 bp ladders, respectively. The H20 lanes represent no template controls. The markers bands of interest are indicated in white text within the gel image and include IFN-γ, IL-10, CD4, CD8, CD3, GAPDH, PD-1, Blimp-1. (b) Graphic presentation showing relative differences in RT-PCR mRNA expression of target genes expressed in CD8 + T cells from wildtype (WT) and IL-10 deficient (IL10 KO) mice. Expression is shown as target gene as a ratio of GAPDH expression (Primer/GAPDH). Values were calculated using the integrated density value (IDV) ratio compared to GAPDH (as a housekeeping reference gene) following 30 cycles of RT-PCR. The genes of interest are indicated on the X-axis and include IFN-γ, IL-10, CD4, CD8, CD3, GAPDH, PD-1, Blimp-1 and CTLA-4, and the associated IDV values are represented on the Y-axis. Significance was determined using an unpaired t-test (p < .05).
D52 vaccination of IL-10 deficient mice elicits tumor-specific CTLs
CD8 + T cells isolated from vaccinated, IL-10 deficient mice that rejected tumor challenge with TRAMP-C2 autochthonous tumor cells were tested for in vitro killing of TRAMP-C2 as a relevant tumor target, compared to mKSA BALB/c tumor cells as an antigen-positive, MHC-I miss-match control. CD8 + T cells specifically lysed TRAMP-C2 tumor targets but not mKSA tumor cells (). This finding was not surprising and corroborated our previously published findings in vaccinated wt C57BL/6J mice.Citation10,Citation12 Of note, 70% specific killing of relevant tumor cells by CD8 + T cells from vaccinated IL-10 deficient mice at effector to tumor cells ratios of 10:1 was approximately 2–3 fold higher than previously observed by our group for vaccinated wt mice.Citation9 This suggests that absence of IL-10 may facilitate more effective killing of relevant tumor targets by CD8 + T cells isolated from vaccinated IL-10 deficient mice than we previously observed for CD8 + T cells isolated from vaccinated wt mice.
Figure 6. Cytotoxic T cell response from IL-10 Deficient D52 vaccinated and TRAMP-C2 tumor challenged mice. CD8 + T cells were isolated from splenic mononuclear cells of vaccinated mice that rejected tumor challenge and pooled. Line graph depicting specific lysis of autochthonous TRAMP-C2 tumor cells at various effector-to-target cell ratios. The C57Bl/6J mD52-expressing tumor cell TRAMP-C2 served as a MHC-matched target. The BALB/c-derived mD52-expressing tumor cell line mKSA served as a MHC-mismatched control target.
Discussion
Our previous studies exploring multiple vaccination approaches with the tumor self-protein D52 demonstrated success in inducing an immune response capable of rejecting tumor formation.Citation11 Importantly, autoimmunity does not occur in mice vaccinated with the tumor self-protein D52, even when classic CD4+ CD25+ Treg cells are depleted with anti-CD25 mAb administered with D52 vaccination.Citation9,Citation10 Although various vaccination approaches were successful, reaching 80% protection from tumor challenge, complete protection (100%) from tumor challenge has not yet been achieved.Citation11 We postulate that this may be due to the self-nature of the vaccine antigen D52 eliciting a unique subset of CD8+ IL-10 + T cells with regulatory potential. When T cells from D52 vaccinated mice that were completely protected from tumor challenge were compared with those from vaccinated mice that failed to reject tumor challenge, CD8+ IL-10 + T cells were more evident in mice that failed to reject the tumor.Citation12,Citation23 In addition to IFN-γ, the only other cytokine produced by CD8 + T cells was IL-10,Citation12,Citation23 which was consistent with observations from our previous D52-based vaccine studies in two mouse strains.Citation9,Citation10,Citation23
In the study described herein, we tested the hypothesis that a population of CD8+ IL-10 + T cells elicited by D52-vaccination may impact protective tumor immunity. We addressed this initially through blocking IL-10 function by targeting the IL-10 receptor with a monoclonal antibody administered with D52 vaccination (); and secondly, by vaccinating IL-10 deficient (IL10 KO) mice, followed by tumor challenge (). Blocking the IL-10R with specific mAb did not augment overall, protective tumor immunity; 30% protection was observed in both experimental and control groups (). This could be due to commonly accepted difficulties in targeting the effects of IL-10 by blocking its receptor with mAb due to multiple cellular sources of IL-10 and the systemic nature of the receptor expression. Many different cells produce IL-10 leading to a large amount of cytokine that is difficult to inhibit with receptor-specific mAb administered in a passive systemic approach.Citation34 The amount of IL-10 produced could outcompete the monoclonal antibody administered for the IL-10 receptor; thus allowing IL-10 to continue as a suppressive cytokine produced by the immune system to dampen its response to the tumor self-protein D52. This is also the widely accepted case for administration IL-10 cytokine-specific mAb. Our second approach, in which IL-10 deficient mice were vaccinated with D52, gave similar results to the previous experiment in overall tumor immunity following primary tumor challenge (). This is likely due to other mechanisms of regulation to include CD4 + T regs, as previously reported by our group and others.Citation10,Citation16,Citation24,Citation38 However, a significant delay in tumor growth was observed in vaccinated IL-10 deficient mice following primary tumor challenge (). Interestingly, when mice were tested for vaccine durability by a secondary tumor challenge in the opposite flank, mice deficient in IL-10 had a 100% recall of protective immunity, while only 25% of wt mice (those with functional IL-10) were protected (). From this, we can conclude that IL-10 has an impact on suppression of a memory response after vaccination to a tumor self-protein, such as D52. In further analyses, mRNA from either a total of 5 × 106 combined CD4+ and CD8 + T cells or 5 × 106 CD8 + T cells alone was interrogated for IL-10 by RT-PCR. IL-10 message expression was only detected in purified CD8 + T cells; further supporting that CD8 + T cells are a source of IL-10 as we previously reported ().Citation10,Citation12 Interestingly, when vaccination failed to inhibit tumor growth, Blimp-1 expression (known for inducing IL-10 expression in Tregs)Citation36,Citation37,Citation39 was detected in purified CD8 + T cells, but not detected in total T cells comprised of both CD4+ and CD8 + T cells (). These CD8 + T cells could be inhibiting tumor immunity after vaccination to the tumor self-protein D52, indicating a regulatory function. With the present study we have expanded support for the uniqueness of these CD8+ IL-10 + T cells in more detail by employing IL-10 knockout mice with D52 vaccination with analysis of IFN-γ, IL-10, and Blimp-1 as well as CTLA-4 and PD-1 expression in CD8 + T cells elicited by D52 vaccination.
T regulatory cells that produce IL-10 have been reported as CD4+ CD25 + T cells or CD8+ CD122 hi (IL-2R β chain) T cells.Citation29,Citation40 Previously, we depleted CD25+ Treg cells and CD122hi Treg cells alone and together in combination with D52-targeted vaccination. CD25+ Treg depletion augmented tumor immunity,Citation10,Citation12 however depletion of CD122 hi T cells did little to aid in tumor immunity, and in fact exacerbate tumor growth. This suggests that CD4+ CD25+ Tregs play a role in our system while CD8+ CD122 hi Treg cells do not, and that anti-CD122 mAb is likely targeting CD122 on effector cells in our model. Together our data support the notion that the IL-10 producing CD8 + T cells in our system may be a unique subset of CD8 + T cells. We do not believe that CD122hi expression is a marker for the CD8 + T cells that produce IL-10 describe herein, since in vivo depletion of CD122hi T cells did not augment tumor immunity as one would expect from other reports,Citation29 but instead exacerbated tumor growth.Citation12 In addition, the CD8+ IL-10 + T cells elicited in our vaccine model are restricted by classical MHC-ICitation8-Citation12 and therefore distinct from Qa-1-restricted CD8 Tregs.Citation41 Therefore, our data support the conclusion that these CD8+ IL-10 + T cells are potentially unique for the reasons stated, but also because our system is neither a model of autoimmunity nor a model of viral infection. Ours is a vaccine model against a ubiquitously expressed self-protein that is aberrantly over-expressed in numerous cancers. In this vaccine model CD8+ IL-10 + T cells are elicited in the absence of tumor or any disease state (tumor challenge coming after induction of immunity to D52 via vaccination), supporting the hypothesis that they may indeed represent a unique subset of CD8 + T cells that use IL-10 and apoptotic proteins to suppress T cell responses against self-proteins. Of note, we have observed these CD8+ IL-10 + T cells following D52 vaccination in more than one mouse strain, suggesting a conserved role for their function. It is also interesting that these CD8+ IL-10 + T cells are elicited by mD52 (fully self) vaccines and hD52 (partially xenogeneic) vaccinesCitation11 suggesting that their epitope specificity may lie within the region of the D52 protein that is conserved between species.
The notion that IL-10 and its receptor play a role in inhibiting anti-tumor immunity is not new. Vicari et al. have shown that administration of CpG immunostimulatory oligonucleotide in conjunction with anti-IL-10 receptor antibody elicited robust antitumor therapeutic responses marked by anti-tumor immune memory in a mouse treatment model of pre-established tumors.Citation42 Similarly, our model employs the use of a molecular adjuvant comprised of CpG immunostimulatory oligonucleotide contained within the plasmid DNA-based vaccine that encodes the full-length cDNA for D52. We also demonstrate an effect on immune memory when anti-IL-10 receptor antibody is administered with the DNA-based D52 vaccine, where ~30% of mice that survived primary tumor challenge survived a secondary challenge. A key difference in the study by Vicari et al. and our study is they use a 7 day established tumor treatment model and assess infiltrating DC function in the tumor microenvironment. Our model does not include an established tumor microenvironment prior to vaccination/treatment as such the DCs involved in our model are resident in the lymph nodes that drain the vaccine site and appear to be normal mature in phenotype.
Others have reported a role for IL-10 receptor blocking in human cell lines in vitro.Citation43,Citation44 These studies though supportive of our claims in a mouse vaccine model are markedly different as they use primary human tumor cell cultures and commercially available human tumor cells lines or a single human monocyte cell line as in vitro models to assess IL-10 receptor function. The goal of these studies is to establish dose-dependent tumor cell killing mediated by blocking of IL-10 receptor on tumor cells, and proteomic analyses of macrophage survival signals associated with IL-10 or IL-10 receptor blocking the human monocyte-derived cell line U937. In contrast, we focused on in vivo vaccine-induced tumor rejection mediated by specific CD8 + T cells and a simultaneous role for a unique population of CD8+ IL-10 + T cells with regulatory potential.
To further explore this uniqueness we plan to interrogate CD8 + T cells isolated from IL-10 deficient mice, IFN-γ deficient mice, and wt mice that have been vaccinated with D52 without tumor challenge. Since these mice will not be challenged with tumor cells, cross-priming to other antigens in the tumor cells is not possible, thus focusing the activation and specificity on D52 elicited CD8 + T cells (activated T cells isolated from these mice will be specific for D52). This will allow for the analysis of an early focused and specific CD8 + T cell response to the tumor self-protein D52 and compare those CD8 + T cells elicited that only produce IFN-γ (IL-10 KO) with those that only produce IL-10 (IFN-γ KO).
It will also be important to assess the presence infiltrating immune cells (CD8 and CD4 T cells, dendritic cells and perhaps macrophages) in the tumors of mice that failed to respond to the vaccine and did not reject tumor challenge. Mice that survive no longer have detectable evidence of tumor and therefore have no tumor microenvironment to resect or analyze. Regrettably, for this study, we were unable harvest tumors from mice that failed to reject tumor challenge to make tissue sections and stain for infiltrating immune cells. We will in future studies harvest tumors and analyze the tumor microenvironment from vaccinated mice that do not reject tumor challenge, and extend our studies to therapy where all mice receive tumor before vaccination and assess treatment of established tumors to include analysis of the tumor microenvironment.
An additional, interesting and important question specifically relates to in vivo adaptive responses elicited following tumor killing and secondary anti-tumor responses as opposed to vaccine-induced initial/primary tumor killing. Our past and current focus relative to tumor immunity in this model is a deeper understanding of the CD8 + T cell response elicited by our vaccine that specifically targets the overexpressed oncogenic tumor-self protein D52. D52 is a modest-sized soluble intracellular protein that is neither expressed on the tumor cell surface nor secreted outside the tumor cell. This precludes an initial/primary anti-tumor response mediated by vaccine-elicited antibodies. Vaccine-induced primary tumor killing by antibodies has been shown function through ADCC or target antigen crosslinking, resulting in inhibitory signaling in the tumor cell much like anti-HER-2/neu Ab therapy. We have previously confirmed that adaptive immune responses to our vaccine result in primary tumor challenge rejection that is mediated completely by T cells. We also confirmed previously that our vaccine approach herein, namely plasmid DNA encoding cDNA for D52, does not elicit D52-specific IgG antibody responses. However, if the vaccine is comprised of recombinant D52 protein in adjuvant anti-D52 IgG antibodies are produced, but do not participate in primary tumor rejection due to the absence of D52 on the tumor cell surface. None the less, it is very likely that antibody-mediated tumor cell killing augments complete rejection of secondary challenge contributing to T cell-mediated vaccine-elicited memory and durability reported by our group. We hypothesize that the antibody response in this case would be induced by cross-presentation against additional tumor antigens that are surface expressed. Cross priming to additional tumor antigens is not addressed in our present study, but will be explored in future studies.
In conclusion, we vaccinated IL-10 deficient mice with the murine orthologue of D52; vaccination of wild type (wt) C57BL/6J mice served as a control for comparison. In some experiments, D52 vaccinated wt mice were administered IL-10R-specific mAb to neutralize IL-10 function 1 day prior to vaccination, on the day of each vaccination, as well as at the time of tumor challenge. D52 vaccination in conjunction with IL-10R mAb administration did little to augment tumor immunity. IL-10 deficient mice exhibited similar tumor immunity when compared to wt mice in response to a primary tumor challenge. However, T cell memory responses to the vaccine and tumor were magnified in vaccinated IL-10 deficient mice. Specifically, individual IL-10 deficient mice that rejected primary tumor challenge were re-challenged (secondary tumor challenge) 140 days post-primary challenge, to access vaccine durability and immunologic memory against tumor recurrence. Mice deficient in IL-10 demonstrated a memory response in which 100% of the mice were protected from secondary tumor challenge, while wt mice had diminished recall response against tumor recurrence (25%). These results with the subsequent analysis of vaccine-elicited CD8 + T cells for tumor-specific killing and regulatory cell marker expression, add further support to our premise that CD8+ IL-10 + T cells elicited by D52 tumor-self protein vaccine contribute to the suppression of a memory CTL responses and durable tumor immunity.
Abbreviations
| TPD52 | = | tumor protein D52 |
| hD52 | = | human D52 |
| mD52 | = | murine D52 |
| Treg | = | T regulatory cell |
| IL-10 | = | interleukin 10 |
| IFN-γ | = | interferon gamma |
| mAb | = | monoclonal Ab |
| wt | = | wild type |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
Related Research Data
References
- Ogi C, Aruga A. Clinical evaluation of therapeutic cancer vaccines. Hum Vacc Immunother. 2013;9:1049–57.
- Weiner M, Surana R, Murray J. Vaccine prevention of cancer: can endogenous antigens be targeted? Cancer Prev Res. 2010;3(4):410–15. doi:10.1158/1940-6207.CAPR-10-0040.
- Cavallo F, Calogero RA, Forni G. Are oncoantigens suitable targets for anit-tumour therapy? Nat Rev Cancer. 2007;7(9):707–13. doi:10.1038/nrc2208.
- Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen surviving. Clin Cancer Res. 2007;13(20):5991–94. doi:10.1158/1078-0432.CCR-07-0686.
- Beatty GL, Vonderheide RH. Telomerase as a universal tumor antigen for cancer vaccines. Expert Rev Vaccines. 2008;7(7):881–87. doi:10.1586/14760584.7.7.881.
- Schreiber TH, Raez L, Rosenblatt JD, Podack ER. Tumor immunogenicity and responsiveness to cancer vaccine therapy: the state of the art. Semin Immunol. 2010;22(3):105–12. doi:10.1016/j.smim.2010.02.001.
- Lewis JD, Reilly BD, Bright RK. Tumor-associated antigens: from discovery to immunity. Int Rev Immunol. 2003;22:81–112. doi:10.1080/08830180305221.
- Payton LA, Lewis JD, Byrne JA, Bright RK. Vaccination with metastasis-related tumor associated antigen TPD52 and CpG/ODN induces protective tumor immunity. Cancer Immunol Immunother. 2008;57:799–811. doi:10.1007/s00262-007-0416-y.
- Lewis JD, Sullivan LA, Byrne JA, de Riese W, Bright RK. Memory and cellular immunity induced by a DNA vaccine encoding self antigen TPD52 administered with soluble GM-CSF. Cancer Immunol Immunother. 2009;58:1337–49. doi:10.1007/s00262-009-0659-x.
- Bright JD, Schultz HN, Byrne JA, Bright RK. Injection site and regulatory T cells influence durable vaccine-induced tumor immunity to an over-expressed self-tumor associated antigen. OncoImmunol. 2013;7:(e25049)1–11.
- Bright RK, Bright JD, Byrne JA. Overexpressed oncogenic tumor-self antigens. Hum Vacc Immunother. 2014;10(11):3297–305. doi:10.4161/hv.29475.
- Bright JD, Aldrich JF, Byrne JA, Bright RK. Vaccination with the prostate cancer over-expressed tumor self-protein TPD52 elicits protective tumor immunity and a potentially unique subset of CD8+ T cells. Austin J Clin Immunol. 2014;1:13.
- Mirshahidi S, Kramer VG, Whitney JB, Essono S, Lee S, Dranoff G, Anderson KS, Ruprecht RM. Overlapping synthetic peptides encoding TPD52 as breast cancer vaccine in mice: prolonged survival. Vaccine. 2009;27(12):1825–33. doi:10.1016/j.vaccine.2009.01.089.
- Wang R, Xu J, Mabjeesh N, Zhu G, Zhou J, Amin M, He D, Marshall FF, Zhau HE, Chung LW. PrLZ is expressed in normal prostate development and in human prostate cancer progression. Clin Cancer Res. 2007;13(20):6040–48. doi:10.1158/1078-0432.CCR-07-0640.
- Shehata M, Bièche I, Boutros R, Weidenhofer J, Fanayan S, Spalding L, Zeps N, Byth K, Bright RK, Lidereau R, et al. Nonredundant functions for tumor protein D52-like proteins support specific targeting of TPD52. Clin Cancer Res. 2008;14(16):5050–60. doi:10.1158/1078-0432.CCR-07-4994.
- Lewis JD, Payton LA, Whitford JG, Byrne JA, Smith DI, Yang L, Bright RK. Induction of tumorigenesis and metastasis by the murine orthologue of tumor protein D52. Mol Cancer Res. 2007;5(2):133–44. doi:10.1158/1541-7786.MCR-06-0245.
- Balleine RL, Fejzo MS, Sathasivam P, Basset P, Clarke CL, Byrne JA. The hD52 (TPD52) gene is a candidate target gene for events resulting in increased 8q21 copy number in human breast carcinoma. Genes Chromosom Cancer. 2000;29:48–57. doi:10.1002/1098-2264(2000)9999:9999<::AID-GCC1005>3.0.CO;2-O.
- Rubin MA, Varambally S, Beroukhim R, Tomlins SA, Rhodes DR, Paris PL, Hofer MD, Storz-Schweizer M, Kuefer R, Fletcher JA, et al. Over expression, amplification, and androgen regulation of TPD52 in prostate cancer. Cancer Res. 2004;64:3814–22. doi:10.1158/0008-5472.CAN-03-3881.
- Byrne JA, Balleine RL, Fejzo MS, Mercieca J, Chiew YE, Livnat Y, St Heaps L, Perters GB, Byth K, Karlan BY, et al. Tumor protein D52 (TPD52) is over expressed and a gene amplification target in ovarian cancer. Int J Cancer. 2005;117:1049–54. doi:10.1002/ijc.21250.
- Largo C, Alvarez S, Saez B, Blesa D, Martin-Subero JI, Gonzalez-Garcia I, Brieva JA, Dopazo J, Siebert R, Calasanz MJ, et al. Identification of overexpressed genes in frequently gained/amplified chromosome regions in multiple myeloma. Haematologica. 2006;91:184–91.
- Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354(23):2419–30. doi:10.1056/NEJMoa055351.
- Loukopoulos P, Shibata T, Katoh H, Kokubu A, Sakamoto M, Yamazaki K, Kosuge T, Kanai Y, Hosoda F, Imoto I, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98:392–400. doi:10.1111/j.1349-7006.2007.00395.x.
- Bright RK, Mamula MJ. Regulation of adaptive immune responses to self-antigens in cancer and autoimmunity. Current Trends Immunol. 2014;15:47.
- Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler T, Thompson TC, Old LJ, Wang R. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13(23):6947–58. doi:10.1158/1078-0432.CCR-07-0842.
- Endharti AT, Rifa’l M, Shi Z, Fukuoka Y, Nakahara Y, Kawamoto Y, Takeda K, Isobe K, Suzuki H. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-γ production and proliferation of CD8+ T cells. J Immunol. 2005;175:7093–97. doi:10.4049/jimmunol.175.11.7093.
- Saitoh O, Abiru N, Nakahara M, Nagayama Y. CD8+CD122+ T cells, a newly identified regulatory T subset, negatively regulate Graves’ hyperthyroidism in a murine model. Encocrinol. 2007;148(12):6040–46. doi:10.1210/en.2007-0300.
- Lee YH, Ishida Y, Rifa’I M, Shi Z, Isobe K, Suzuki H. Essential role of CD8+CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis. J Immunol. 2008;180(2):825–32. doi:10.4049/jimmunol.180.2.825.
- Suzuki H, Shi Z, Okuno Y, Isobe K. Are CD8+CD122+ cells regulatory T cells or memory T cells? Hum Immunol. 2008;69:751–54. doi:10.1016/j.humimm.2008.08.285.
- Wang L-X, Li Y, Yang G, Pang P-Y, Haley D, Walker EB, Urba WJ, Hu H-M. CD122+ CD8+ Treg suppress vaccine-induced antitumor immune responses in lymphodepleted mice. Eur J Immunol. 2010;40:1375–85. doi:10.1002/eji.200839210.
- Sun J, Dodd H, Moser EK, Sharma R, Braciale TJ. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by anti-viral CTLs. Nat Immunol. 2011;12(4):327–34. doi:10.1038/ni.1996.
- Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–30.
- Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:685–765. doi:10.1146/annurev.immunol.19.1.683.
- O’Garra A, Vieira P. Th1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7:425–28. doi:10.1038/nri2097.
- Trinchieri G. Interleukin-10 production by effector T cells: Th1 cells show self control. J Emp Med. 2007;204:239–43.
- Motegi A, Kinoshita M, Inatsu A, Habu Y, Saitoh D, Seki S. IL-15-induced CD8+CD122+ T cells increase antibacterial and anti-tumor immune responses: implications for immune function in ages mice. J Leukoc Biol. 2008;84(4):1047–56. doi:10.1189/jlb.0807530.
- Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, Hodgkin PD, Nutt ST. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol. 2006;7(5):466–74. doi:10.1038/ni1321.
- Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Annu Rev Immunol. 2008;26:133–69. doi:10.1146/annurev.immunol.26.021607.090241.
- O’Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. 2004;10(8):801–05. doi:10.1038/nm0804-801.
- Martins GA, Cimmuno L, Shapiro-Shelef M, Szabolcs M, Herron A, Magnusdottir E, Calame K. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol. 2006;7:457–65. doi:10.1038/ni1320.
- Rifa’i M, Shi Z, Zhang SY, Lee YH, Shiku H, Isobe K, Suzuki H. CD8+ CD122+ regulatory T cells recognize activated T cells via conventional MHC class I-alpha betaTCR interaction and become IL-10-producing active regulatory cells. Int Immunol. 2008;20:937–47. doi:10.1093/intimm/dxn052.
- Lu L, Cantor H. Generation and regulation of CD8+ regulatory T cells. Cell Mol Immunol. 2008;5:401–06. doi:10.1038/cmi.2008.50.
- Vicari AP, Chiodoni C, Vaure C, Aït-Yahia S, Dercamp C, Matsos F, Reynard O, Taverne C, Merle P, Colombo MP, et al. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med. 2002;196:542–49. doi:10.1084/jem.20020732.
- Ni G, Chen S, Yuan J, Cavezza SF, Wei MQ, Li H, Pan X, Liu X, Wang T. Comparative proteomic study reveals the enhanced immune response with the blockade of interleukin 10 with anti-IL-10 and anti-IL-10 receptor antibodies in human U937 cells. PLoS One. 2019;14(3):e0213813. doi:10.1371/journal.pone.0213813.
- Beguelin W, Sawh S, Chambwe N, Chan FC, Jiang Y, Choo J-W, Scott DW, Chalmers A, Geng H, Tsikitas L, et al. IL10 receptor is a novel therapeutic target in DLBCLs. Leukemia. 2015;29:1684–94. doi:10.1038/leu.2015.57.