
ABSTRACT
HPV remains the most common sexually transmitted disease worldwide, despite improvements in awareness, screening, prophylactic vaccination uptake, and surgical treatment. VGX-3100 is an immunotherapy that uses electroporation to introduce DNA encoding for modified HPV-16 and HPV-18, E6-and E7 proteins into myocytes to stimulate an effector T cell response. We now report immunogenicity and safety of VGX-3100 for a refrigeration-stable formulation, which improves patient-care setting usability.
This multi-arm, double-blinded, randomized trial enrolled 235 healthy men and women to receive either a refrigerated (RF) or frozen formulation (FF) of VGX-3100. Three doses were administered intramuscularly with electroporation at 0, 4, and 12 weeks. Non-inferiority of RF to FF was assessed by comparing the proportion of subjects who achieved a ≥2-fold increase from baseline to Week 14 in Spot Forming Units/106 PMBCs using an interferon-γ enzyme-linked immunospot assay.
There were no related SAEs. Injection site reactions were the most common adverse event (54%, RF; 66%, FF) the majority of which resolved within a few minutes following administration. The primary endpoint was met with 89.9% of RF recipients and 97.2% of FF recipients reaching a ≥2-fold rise in SFU/106 PBMC, 2 weeks following the last dose; RF was statistically non-inferior to FF (p = .022).
A systemic, immunologic approach has the potential to fill a critical gap in the ability to treat men and women with high grade HPV diseases. These safety and immunogenicity data are supportive of the continued development of a refrigerated formulation of VGX-3100.
KEYWORDS:
Introduction
VGX-3100 is an HPV immunotherapy in clinical development that is comprised of two synthetic DNA plasmids that encode and express antigen targets for anti-HPV humoral and cellular immune responses. Unlike gene replacement therapies, the DNA in VGX-3100 is not designed to integrate into the host genome. Non-integrating plasmids have been estimated to have 3000-fold lower mutation potential than the spontaneous mutation rate of mammalian genomes.Citation1,Citation2 Made with DNA SynCon™ technology, VGX-3100 contains both pGX3001 that encodes for a synthetic, non-oncogenic HPV-16 consensus E6 and E7 fusion gene and pGX3002 that encodes for a synthetic, non-oncogenic HPV-18 consensus E6 and E7 fusion gene. Both genes are driven by a cytomegalovirus (CMV) immediate-early promoter. VGX-3100 is delivered intramuscularly using the CELLECTRA™5P Device, which generates a localized electroporation field to enable DNA plasmid translocation to the nucleus. Transfection with the synthetic DNA leads to overexpression of the modified E6 and E7 HPV antigenic proteins. This induces a cellular and humoral immune response that is specific for HPV 16/18 infected dysplastic cells.Citation3
The CELLECTRA™5P device uses electroporation (EP) technology to increase the uptake and expression of plasmid DNA. Induction of an electrical field in the target tissue makes the cell membrane temporarily permeable to the large synthetic DNA molecules enabling entry and protein expression.Citation4,Citation5 DNA delivered with EP has also been shown to elicit a superior immunogenicity when compared to DNA delivered without EP.Citation5 The voltage generated by the CELLECTRA™5P is limited to 200 volts for safety. The device itself is equipped with safety features and error checking functions to prevent sudden voltage increases, to ensure that the treatment is being injected intramuscularly, and to maintain a constant current throughout administration.
HPV-101 was a phase 1b trial designed to compare a refrigerated formulation of VGX-3100 (RF) containing a saline sodium citrate (SSC) buffer to a frozen formulation of VGX-3100 (FF) containing water for injection and low molecular weight poly-L-glutamate (WFI+LGS) in healthy adult volunteers. The trial aimed to demonstrate that three doses of 6 mg RF has a non-inferior immune response compared with three doses 6 mg of FF when both are delivered intramuscularly followed by EP to healthy adults. The rationale for testing a refrigerator stable product was to increase accessibility for subjects with insufficient infrastructure for the management of frozen product storage and handling and to simplify the overall administration process.
Methods
Trial design, drug material, and participants
The HPV-101 trial was a multi-arm, double-blinded, randomized phase 1b trial conducted at an outpatient clinical setting in the US. The RF was comprised of 6mg of VGX-3100 DNA plasmids in a saline sodium citrate (SSC) buffer. The FF was comprised of 6mg of VGX-3100 DNA plasmids in water for injection and low molecular weight poly-L-glutamate (WFI+LGS). The DNA plasmid constituents of the RF and FF were derived from the same cell banks and identity was confirmed through restriction analysis. Study drug material was maintained under proper cold chain conditions at the study site for less than 5 weeks prior to use which is well within the product stability parameters that extended up to 48 months at 2–8°C for RF and 60 months at −20°C for FF.
Medically healthy female and male subjects aged 18–55 years with a BMI ≥ 18.5 kg/m2 with no clinically significant findings were eligible. Subjects who were pregnant, breast feeding, or considering becoming pregnant, had been immunized with a prophylactic HPV vaccine, had a history of a positive serologic test for human immunodeficiency virus, hepatitis B, hepatitis C, or any clinically significant disease were excluded from the trial. The protocol, subject informed consent form, and written information were approved by the Institutional Review Board. The trial was conducted in accordance with the ethical principles that have their origins in the Declaration of Helsinki and with Good Clinical Practice (GCP) guidelines as denoted in the International Council for Harmonization (ICH) E6 requirements. Each subject or their legally authorized representative gave written informed consent.
Randomization and masking
Patients were randomly assigned to receive either RF or FF by a computer-generated allocation schedule. Two hundred thirty-five subjects were enrolled. In the 18.5 to 25 kg/m2 BMI group, subjects were randomized to 1 of 2 treatment groups: FF with a 19mm array and RF with a 19mm array in a 1:1 ratio stratified by age (<25 and ≥25 years of age). In the > 25 kg/m2 BMI group, subjects were and randomized to 1 of 4 treatment groups: FF with a 19mm array, FF with a 25mm array, RF with a 19mm array, or RF with a 25mm array in a 1:1:1:1 ratio stratified by age (<25 and ≥25 years of age). The longer needle array (25 mm) was included in this trial to assess safety of use in cases where a longer needle may be required for proper intramuscular injection (i.e. overweight or obese individuals). The study was blinded to the formulation administered but open-label to the array size used. There was a dedicated unblinded staff member at each site that was responsible for blinding and dispensing the dose as per the subject’s randomization. Vials were labeled and unblinded personnel filled the correct dose into a blinded syringe with only the study identifier and transferred the syringe to the blinded personnel. An unblinded clinical research associate assigned by the sponsor was responsible for reviewing drug accountability within the pharmacy. Unblinded monitoring reports were reviewed by the sponsor’s designated clinical operations team member to ensure adequate oversite. No unblinded information was available to the sponsor personnel directly involved with the conduct of the study.
Procedures
Participants in the BMI 18.5 to 25 kg/m2 cohort received either 6 mg FF or 6 mg RF delivered intramuscularly using a 19mm array, both followed by EP with CELLECTRA™5P. Participants in the BMI ≥ 25 kg/m2 cohort received 6 mg FF or RF delivered intramuscularly using either a 19mm or 25mm array, all followed by EP with CELLECTRA™5P. Subjects received 3 doses of VGX-3100 over twelve weeks: Day 0, Week 4, and Week 12.
Subjects were monitored for laboratory abnormalities at screening, Week 16, and Week 40. Injection site reactions and systemic events for a week following each dose were followed using a participant diary. Any adverse events (AEs) and serious adverse events (SAEs) were reported to and reviewed by the sponsor. A visual analog scale (VAS) was used immediately after treatment as well as five minutes, and ten minutes following to assess injection site pain. Participants who received treatment were followed for safety assessments until Week 40. Laboratory evaluations included a complete blood count, serum chemistry, creatine phosphokinase (CPK), urinalysis, electrocardiogram, and pregnancy test for women of child-bearing potential. Injection sites was assessed prior to study treatment and 2 weeks post study treatment.
Laboratory AEs were graded according to the “Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventative Vaccine Clinical Trials,” issued in September 2007. Clinical AEs and SAEs were graded according to the “Common Toxicity Criteria for Adverse Events (CTCAE)” version 4.03. SAEs were reported to the sponsor and Ethics Committee within the reporting period.
The anti-HPV-16/18 E6 and E7 antibody responses induced by the formulations of VGX-3100 were assessed using a standardized binding enzyme-linked immunosorbent assay (ELISA): 96-well enzyme immunoassay plates were coated with HPV16 or HPV18 E7 proteins (1 μg/ml) (ProteinX Lab). Sera were diluted with 1% bovine serum albumin in PBS and tested in triplicate with an automated and calibrated plate washer and read on a kinetic microplate reader (Molecular Devices). The reported titer is the highest dilution where the average optical density (OD) was greater than 0.15 absorbance units and greater than the average OD plus 2.5 times SD at baseline at the same dilution. T-cell responses were assessed using an HPV IFN-γ ELISpot assay: Peripheral blood mononuclear cells (PBMC) were recovered from cryopreservation overnight in cell culture medium and spun, washed, and resuspended the following day. PBMCs were stimulated overnight with two sets of pooled peptides, each containing 15–amino acid residues overlapping by 8 amino acids representing the entire consensus E6/E7 fusion protein sequence of HPV16 or HPV18, at a concentration of 2 mg/ml per peptide. The average number of SFU counted in R10 wells was subtracted from the average in individual HPV peptide wells and then adjusted to 1 × 106 PBMCs for each HPV peptide pool. Flow cytometry was used to assess the ability of T-cells to synthesize lytic proteins following long-term exposure to an antigen (i.e. Lytic Granule Loading assay): Peripheral blood mononuclear cells (PBMC) were recovered from cryopreservation overnight in cell culture medium and spun, washed, and resuspended the following day. After counting, 106 PBMCs were plated into a 96-well plate in R10 medium. For antigen-specific responses, cells were stimulated 5 days with a combination of 15-mer peptides overlapping by 8 amino acid residues corresponding to HPV16 E6 and E7 or HPV18 E6 and E7 that had been pooled at a concentration of 2 mg/mL, while an irrelevant peptide was used as a negative control (OVA) and concanavalin A was used as a positive control (Sigma-Aldrich). All peptides were resuspended using DMSO. No costimulatory antibodies or cytokines were added to cell cultures at any point. At the end of the 5-day incubation period, plates were spun to pellet cells and all samples were washed with PBS and subjected to staining for CD3-APCH7, CD4-PerCPCy5.5, CD14-Pacific Blue, CD-16 Pacific Blue, CD137-APC, Granulysin-FITC (BD Biosciences), CD8-BV605, Granzyme A-AF700 (BioLegend), CD-19 Pacific Blue, granzyme B-PETR (Invitrogen), and perforin-PE (Abcam). Staining for extracellular markers (CD4, CD8, CD137) occurred first, followed by permeabilization to stain for the remaining markers. CD3 was stained intracellularly to account for downregulation of the marker following cellular activation. Prepared cells were acquired using an LSRII flow cytometer equipped with BDFACSDiva software (BD Biosciences). Acquired data were analyzed using the FlowJo software version X.0.7 or later (Tree Star).
Outcomes
The primary objective was to demonstrate that there is a non-inferior immune response following three 6 mg doses of RF relative to FF administered via a intramuscular (IM) injection followed by EP with CELLECTRA™5P. This objective was measured by comparing the proportion of subjects who achieved ≥ a 2-fold increase in Spot Forming Units (SFUs) per 106 peripheral blood monocular cells (PMBCs) using an interferon-γ enzyme-linked immunospot (IFN-γ ELISpot) assay from baseline to Week 14 in each of the groups. The secondary objectives were to evaluate the safety and tolerability of the formulations of VGX-3100 when delivered IM followed by EP in healthy adults with a BMI of 18.5 to 25 kg/m2 with electrodes 19mm in length as well as in healthy adults with a BMI of ≥25 kg/m2 with electrodes 19mm vs. 25mm in length. The incidence of injection site reactions and pain, AEs and SAEs, and changes in laboratory safety parameters were evaluated to determine safety.
Statistical analyses
With 116 subjects, there was approximately 80% power to declare non-inferiority of RF to FF based on an assumed true response of 79% for the primary endpoint in both groups, a non-inferiority margin of 15 percentage points, a 1-sided 5% type I error and 80% evaluability. Target enrollment was 242 participants. Immunogenicity was analyzed using the modified intention to treat (mITT) population which consisted of participants who had received 1 or more doses of the treatment with EP and had available valid immunologic endpoint data. Statistical testing of non-inferiority was performed based on the primary immunogenicity endpoint using the method of Miettinen and Nurminen. ELISA titers (log-transformed) were analyzed between treatment groups using t-tests and corresponding confidence intervals. Increases from baseline to Week 14 in VGX-3100 specific CD8+/CD137+ PBMCs that were perforin+ were analyzed with the Wilcoxon rank-sum test (with exact Monte-Carlo estimation).The population used to evaluate safety consisted of subjects who received 1 or more doses of the treatment with EP. Statistical comparisons of the proportions of subjects who experienced adverse events were performed using the method of Miettinen and Nurminen.Citation6 All statistical analyses were done using SAS statistical software Version 9.3.
Results
Participants
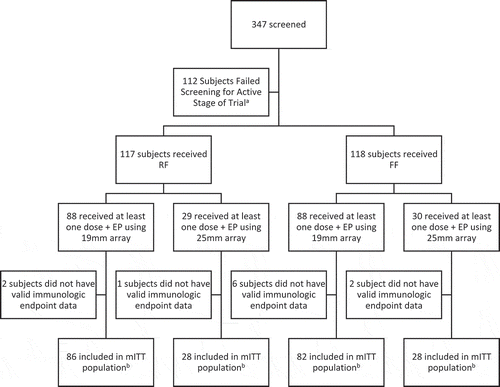
Three hundred forty-seven subjects were screened. Ten subjects were entered into a safety lead-in phase. The safety and tolerability of the 25mm electrode array was deemed acceptable in this safety lead-in phase which used a formulation buffer. Two hundred thirty-five subjects, including nine from the safety lead-in phase, were then randomized for treatment. One hundred seventeen subjects received RF and 118 subjects received FF, both followed by EP, stratified by age and BMI categories. shows the complete trial profile by group. The trial ended at the 40-week study period as outlined in the protocol.
Figure 1. Trial profile.
Demographics
Eighty-nine percent of subjects completed all 3 doses of treatment (92% in the RF group and 86% in the FF group), 3% of subjects received only 2 doses, and 9% of subjects received only 1 dose. Twenty-seven participants discontinued study treatment: 15 were lost to follow-up, 1 discontinued after a SAE of a ruptured ectopic pregnancy, 8 were based on an unspecified subject decision, and 1 was due to death caused by trauma after a physical altercation (two reasons were not recorded). Nine subjects discontinued treatment early but remained on the study for reasons including pregnancy (2 subjects), subject withdrawal (5 subjects), 1 subject who became ineligible due to receipt of travel vaccines and 1 subject who became ineligible due to participating in another study. None of the subjects discontinued treatment due to an AE. Eighty-five percent of subjects completed all 3 doses of treatment and follow-up visits. Participants were followed for 40 weeks. summarizes the demographic and clinical characteristics. The majority of subjects were female (63%) and separately ≥25 years old (87%). Ninety-five percent of participants were included in the mITT population including 97.4% (114/117 subjects) of the RF group and 93.2% (110/118 subjects) of the FF group. summarizes the demographic and clinical characteristics for the > 25 kg/m2 BMI group according to use of 19mm and 25mm arrays.
Table 1. Baseline demographics and clinical characteristics of safety population.
Table 2. Baseline demographics and clinical characteristics of subjects with BMI > 25 kg/m2.
Safety
There were no related deaths or SAEs. Eighty percent of subjects experienced one or more treatment emergent adverse event (TEAE) during the study with each such subject having 4 TEAEs on average in both formulation groups ().
Table 3. Overall summary of adverse events by formulation group in the safety population.
Seventy-eight percent of subjects in the RF group and 81% of subjects in the FF group experienced a TEAE, of which 62% and 73% were treatment-related, respectively. Most TEAEs occurred within 7 days of injection.
Eighty-three percent of the TEAEs were Grade 1, 15% were grade 2, and 2% were Grade 3 or higher. There were 7 subjects who experienced a transient Grade 3 or 4 increase in blood creatine phosphokinase with the highest observed value being 8416 U/L, and one Grade 4 (1738 U/L) in the RF group was considered related to study treatment. All other events Grade 3 or higher were considered unrelated to study treatment. In the RF group, there were 3 subjects that experienced a treatment-emergent SAE and in the FF group, there was 1 subject that experienced a treatment-emergent SAE. Treatment-emergent SAEs included 1 injury, 1 craniocerebral injury, 1 ruptured ectopic pregnancy, and 1 spontaneous abortion. The Investigator did not classify any of the SAEs as treatment-related. One death as a result of injury following a physical altercation was reported in the RF group, but it was not considered related to study treatment. There were no clinically significant changes in vital sign parameters, physical examination findings, and ECG results. summarizes the adverse events for the > 25 kg/m2 BMI group according to use of 19mm and 25mm arrays.
Table 4. overall summary of adverse events by formulation group for subjects with BMI > 25 kg/m2 in the safety population.
Injection site reactions were the most common TEAEs (). Fifty-four percent of subjects in the RF group and 66% of subjects in the FF group reported injection site pain. Other common TEAEs reported for the RF and FF groups respectively were fatigue (27% and 32%), headache (16% and 16%), injection site pruritus (11% and 15%), myalgia (10% and 12%), arthralgia (7% and 13%), upper respiratory tract infection (18% and 16%), and nausea (9% and 14%). Between the two formulation groups, there were no statistically significant differences in the proportion of subjects who experienced any preferred term adverse events. There were statistically significant differences in the system organ classes of general disorders and administration site conditions (57% for the RF group and 73% for the FF group) and skin and subcutaneous tissue disorders (1% for the RF group and 7% for the FF group).
Table 5. Treatment-Emergent Adverse Events (TEAEs) ≥2% by preferred term,comparison of VGX-3100 formulations in the safety population.
There were no meaningful differences in VAS scores between the two formulation groups.
Seven pregnancies were reported with outcomes of: 3 healthy deliveries, 1 spontaneous abortion, 1 ruptured ectopic pregnancy, 1 elective abortion, and 1 unknown outcome. Three subjects discontinued study treatment due to pregnancy including 1 due to an SAE of ruptured ectopic pregnancy that was assessed as unrelated to study treatment. Another subject experienced an SAE of a spontaneous abortion that was assessed as unrelated to study treatment.
Immunogenicity
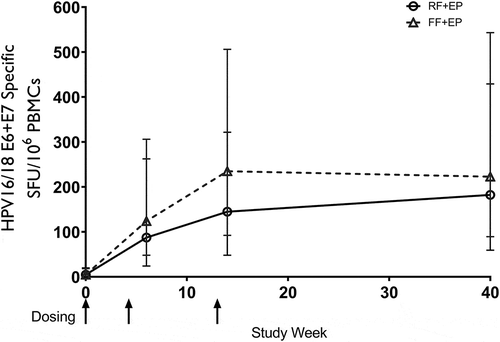
The study met its primary endpoint with 89.9% of RF recipients and 97.2% of FF recipients reaching a ≥2-fold rise in SFU per 106 PBMC at week 14 over baseline as demonstrated by the ELISpot assay ().
Table 6. Primary immune response at week 14: 2-Fold or Higher Rise in Combined HPV-16/18 E6/E7 SFU per 106 PBMCs from Baseline to Week 14 in IFN-gamma ELISpot for mITT population.
RF was shown to be statistically non-inferior to FF (p = .022). There were no significant differences between formulation groups when controlling for a) weight b) array size among those in the >25 kg/m2 BMI group, or c) gender. shows that in both the FF and the RF groups, T-cell responses were still persistent through Week 40.
Figure 2. HPV-16/18 E6/E7 IFN-gamma ELISpot results (SFU/106 PBMC) for mITT population by visit.
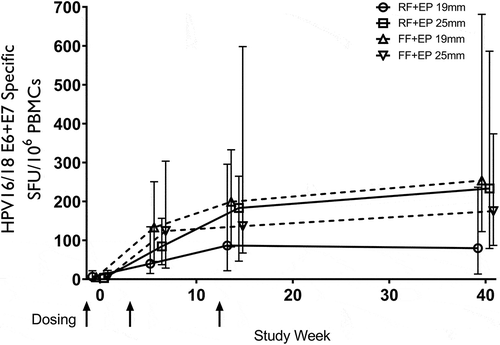
shows the T-cell responses for the >25 kg/m2 BMI group according to use of 19 mm and 25 mm arrays.
Figure 3. HPV-16/18 E6/E7 IFN-gamma ELISpot results (SFU/106 PBMC) for subjects with BMI >25 in the mITT population by visit.
The ELISA titers showed both groups had an induced humoral response against the HPV-16/18 E7 antigens with no statistically significant differences between groups at the primary time point visit, Week 14 ().
Table 7. E7 Specific ELISA titer results for mITT population by visit.
PBMCs were also assayed for activated antigen specific CD8+ T cells co-expressing markers of lytic potential at baseline and 2 weeks following the third immunization in a lytic granule loading assay. The frequency of perforin positive CD8+ CD137+ T cells increased similarly above baseline in both groups with no statistically significant differences between formulation groups, shown in .
Table 8. Increase from baseline to week 14 in Frequency of VGX-3100 Specific perforin+CD8+ CD137+ (co-positive) PBMCs by formulation for mITT population.
Discussion
In this randomized formulation trial, RF was shown to be non-inferior to FF, and the safety profiles were also comparable, which supports the continued development of the refrigerated, liquid formulation in clinical trials. Both RF and FF had an adequate safety profile based upon AE and SAE monitoring, subject-reported local pain reporting, physical examination findings, vital signs, laboratory testing, and ECG testing. Acute administration site pain was the most commonly reported event in both groups. The majority of trial participants completed all three doses. The safety and immunogenicity data also support the use of a 25mm array in subjects with a BMI > 25 kg/m2.
The treatment of cervical HSIL has been without innovative improvements for decades, with the mainstay of treatment being surgery, which has several limitations and risks. Treatment of women with cervical HSIL usually consists of either surgical removal of the affected tissue by cold knife conization (CKC), loop electrical excision procedure (LEEP), ablative therapy via laser, or cryotherapy. Invasive cervical cancer may be treated by radical hysterectomy, and/or combination radiation and chemotherapy. All treatments for cervical HSIL are surgical procedures and are associated with a variety of short and long term risks which include pain, heavy bleeding, decreased fertility, and pre-term birth.Citation7,Citation8 Most importantly, none of the currently available surgical treatments for HSIL eradicates the underlying cause of the high grade cervical dysplasia, which is persistent infection with one or more of the high-risk HPV types. Patients are, therefore, left at risk for recurrent disease, which occurs in 10–16% for cervical HSILCitation9,Citation10 and up to 50% for vulvar and anal HSIL.Citation11-Citation13 It is also known that HPV infection can exist in multiple locations (typically oropharyngeal and anogenital). VGX-3100 therefore is being developed as a systemic immunotherapy which could also address the underlying HPV infection etiology even if multifocal.
HSIL is preventable with use of the licensed prophylactic vaccines; however, the number of US-eligible teenagers who are up-to-date with the prophylactic vaccination series remains low, 51.1% in 2018 in US girls ages 13–17, which leaves a vulnerable, under-protected population.Citation14,Citation15 Younger women who are already infected with HPV are relegated to monitoring due to the higher expected rates of spontaneous regression, which are particularly observed for non-HPV 16/18 types,Citation16 and the negative impacts of surgery upon reproductive health, which leaves an infected population with the psychosocial stress of having an untreated STD potentially for years and also enables continued transmission of HPV within the population.
A limitation to this study is that it was conducted in a healthy population and therefore does not evaluate the efficacy of viral and histological regression of HPV infections or lesions, respectively. The immune responses observed were both humoral and cellular and systemic in nature. The efficacy of VGX-3100 has been shown previously in women with cervical HSIL,Citation17 and efficacy studies of anal and vulvar HSIL are ongoing. An additional limitation is that the study population may have included individuals who had preexisting or existing HPV infection and/or disease, which could alter immune responsiveness. VGX-3100 has the potential to become a safer therapeutic option than the standard of care (i.e. surgical excision) for HPV-related lesions. Development of VGX-3100 will continue using the refrigerated formulation with the CELLECTRA™ electroporation device with the aim of simplifying and automating the procedure in further studies.
Disclosure of potential conflicts of interest
RKH, MD, KK, and PB are or have been employees of Inovio Pharmaceuticals.
Acknowledgments
The authors wish to acknowledge Dr. Scott White, MD of Inovio Pharmaceuticals, and the clinical trial staff of Miami Research Associates for their support of trial site conduct, and the work of the Inovio Research and Development group under the supervision of Dr. Kate Broderick, PhD for formulation development.
Additional information
Funding
Related Research Data
References
- Martin T, Parker SE, Hedstrom R, Le T, Hoffman SL, Norman J, Hobart P, Lew D. Plasmid DNA malaria vaccine: the potential for genomic integration after intramuscular injection. Hum Gene Ther. 1999;10:759–68. doi:10.1089/10430349950018517.
- Nichols WW, Ledwith BJ, Manam SV, Troilo PJ. Potential DNA vaccine integration into host cell genome. Ann N Y Acad Sci. 1995;772:30–39. doi:10.1111/nyas.1995.772.issue-1.
- Morrow MP, Kraynyak KA, Sylvester AJ, Shen X, Amante D, Sakata L, Parker L, Yan J, Boyer J, Roh C, et al. Augmentation of cellular and humoral immune responses to HPV16 and HPV18 E6 and E7 antigens by VGX-3100. Mol Ther Oncolytics. 2016;3:16025. doi:10.1038/mto.2016.25.
- Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16:867–70. doi:10.1038/nbt0998-867.
- Gehl J, Mir LM. Determination of optimal parameters for in vivo gene transfer by electroporation, using a rapid in vivo test for cell permeabilization. Biochem Biophys Res Commun. 1999;261:377–80. doi:10.1006/bbrc.1999.1014.
- Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4:213–26. doi:10.1002/(ISSN)1097-0258.
- Santesso N, Mustafa RA, Wiercioch W, Kehar R, Gandhi S, Chen Y, Cheung A, Hopkins J, Khatib R, Ma B, et al. Systematic reviews and meta-analyses of benefits and harms of cryotherapy, LEEP, and cold knife conization to treat cervical intraepithelial neoplasia. Int J Gynaecol Obstet. 2016;132:266–71. doi:10.1016/j.ijgo.2015.07.026.
- Kyrgiou M, Athanasiou A, Paraskevaidi M, Mitra A, Kalliala I,Martin-Hirsch P, Arbyn M, Bennett P, Paraskevaidis E. Adverse obstetric outcomes after local treatment for cervical preinvasive and early invasive disease according to cone depth: systematic review and meta-analysis. BMJ. 2016;354:i3633.
- Xi LF, Kiviat NB, Wheeler CM, Kreimer A, Ho J, Koutsky LA. Risk of cervical intraepithelial neoplasia grade 2 or 3 after loop electrosurgical excision procedure associated with human papillomavirus type 16 variants. J Infect Dis. 2007;195:1340–44. doi:10.1086/522476.
- Nobbenhuis MA, Meijer CJ, van den Brule AJ, Rozendaal L, Voorhorst FJ, Risse EK, Verheijen RHM, Helmerhorst TJM. Addition of high-risk HPV testing improves the current guidelines on follow-up after treatment for cervical intraepithelial neoplasia. Br J Cancer. 2001;84:796–801. doi:10.1054/bjoc.2000.1689.
- Committee Opinion No.675. Management of vulvar intraepithelial Neoplasia. Obstet Gynecol. 2016;128:e178–82. doi:10.1097/AOG.0000000000001713.
- Frega A, Sopracordevole F, Scirpa P, Biamonti A, Lorenzon L, Scarani S, De Sanctis L, Pacchiarotti A, Moscarini M, French D, et al. The re-infection rate of high-risk HPV and the recurrence rate of vulvar intraepithelial neoplasia (VIN) usual type after surgical treatment. Med Sci Monit. 2011;17:Cr532–5. doi:10.12659/MSM.881941.
- Long KC, Menon R, Bastawrous A, Billingham R. Screening, surveillance, and treatment of anal intraepithelial Neoplasia. Clin Colon Rectal Surg. 2016;29:57–64. doi:10.1055/s-00000049.
- Walker TY, Elam-Evans LD, Yankey D, Markowitz LE, Williams CL, Mbaeyi SA, Fredua B, Stokley S. National, Regional, State, and selected local area vaccination coverage among adolescents aged 13–17 years - United States, 2017. Mmwr-morbid Mortal W. 2018;67:909–17. doi:10.15585/mmwr.mm6733a1.
- Walker TY, Elam-Evans LD, Yankey D, Markowitz LE, Williams CL, Fredua B, Singleton JA, Stokley S, et al. National, Regional, State, and selected local area vaccination coverage among adolescents aged 13–17 years - United States, 2018. Mmwr-morbid Mortal W. 2019;68:718–23. doi:10.15585/mmwr.mm6833a2.
- Wright TC, Stoler MH, Behrens CM, Sharma A, Zhang G, Wright TL. Primary cervical cancer screening with human papillomavirus: end of study results from the ATHENA study using HPV as the first-line screening test. Gynecol Oncol. 2015;136:189–97. doi:10.1016/j.ygyno.2014.11.076.
- Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, Edwards L, Parker RL, Denny L, Giffear M, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386:2078–88. doi:10.1016/S0140-6736(15)00239-1.