
ABSTRACT
Staphylococci are the main cause of nosocomial infections globally. The exotoxin staphylococcal enterotoxin B (SEB) produced by methicillin-resistant Staphylococcus aureus is a major cause of pathology after a staphylococcal infection. We previously isolated an anti-SEB human monoclonal antibody designated as M0313. Here we further characterize this antibody in vitro and in vivo. Immunoblotting analysis and ELISA results indicated that M0313 accurately recognized and bound to SEB. Its binding affinity to native SEB was measured at the low nM level. M0313 effectively inhibited SEB from inducing mouse splenic lymphocyte and human peripheral blood mononuclear cell proliferation and cytokine release in cell culture. M0313 also neutralized SEB toxicity in BALB/c female mice. Most importantly, M0313 promoted the survival of mice treated with SEB-expressing bacteria. In-vivo imaging revealed that M0313 treatment significantly reduced the replication of SEB-expressing bacteria in mice. The neutralization capacity of M0313 correlated with its ability to block SEB from binding to major histocompatibility complex II and T-cell receptor by binding to the SEB residues 85–102 and 90–92. Thus, the monoclonal antibody M0313 may be developed into a therapeutic agent.
Introduction
Staphylococcus aureus infections account for a significant increase in morbidity, mortality, the length of hospital stays, and medical costs.Citation1 Staphylococcal enterotoxin B (SEB) is one of the main pathogens involved in immune escape, toxic shock syndrome (TSS), and food poisoning in S. aureus infection.Citation2 Acting as a superantigen, it binds to class II molecules of the major histocompatibility complex (MHC II) and to specific V beta regions of the T-cell receptor (TCR), which results in the activation of monocytes/macrophages and T lymphocytes.Citation3 SEB induces the production of high levels of proinflammatory cytokines and chemokines, including interleukin-2 (IL-2), interleukin-6 (IL-6), interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and monocyte chemotactic protein-1 (MCP-1).Citation4-Citation7 In S. aureus-infected patients, high levels of systemic inflammatory cytokines may lead to endothelial cell injury, acute lung injury, acute respiratory distress syndrome, and vascular collapse (shock).Citation8-Citation10 In addition, SEB is often associated with food poisoning with symptoms of vomiting, abdominal pain, and diarrhea. SEB is considered a lethal and disabling agent with aerosol stability, and hence, it has been classified as a select agent by the Centers for Disease Control and Prevention and as a category B priority pathogen by the US National Institutes of Health.Citation11,Citation12 Concerns about SEB are compounded by the lack of an effective medical countermeasure for the large-scale treatment of the affected population.Citation13,Citation14
Currently, no vaccines or specific drug therapies have been approved for the treatment of SEB-induced illnesses. Current studies have suggested the use of various therapies including small molecule therapy (such as rapamycin, dexamethasone, pentoxifylline, pirfenidone, mimetic peptides of CD28 dimer interface, and mimetic peptides of the BB loop of MyD88), vaccines against SEB, and SEB-targeting monoclonal antibodies (mAbs; including mouse, lama, human-mouse chimera, and human mAbs).Citation8 High-affinity antibodies that at immediately are good partners for antibiotics and vaccines; thus, these combinations may represent the best approach to treat infections that are associated with emergency intervention, and the best options for immunocompromised patients who cannot be properly vaccinated.Citation15
Our laboratory has produced an anti-SEB human mAb named M0313 based on the blood samples of the volunteers who have enrolled in the phase 1 clinical trial of the recombinant five-antigen Staphylococcus aureus vaccine (rFSAV) by the high-throughput isolation of immunoglobulin genes from single human B cells and their expression as mAbs.Citation16,Citation17 Our present study aimed to detect the binding activity of M0313 to SEB, its ability to inhibit cell proliferation and cytokine release, its neutralization activity against SEB-induced TSS, and its protective activity against S. aureus-induced sepsis via in vitro and in vivo assays, as well as a preliminary study of the neutralization mechanism of M0313.
Materials and methods
Generation and purification of M0313, mutant SEB, and SEB
Mutant SEB (mSEB), in which three amino acids (L45 R, Y89A, and Y94A) of SEB were mutated, lost the ability to bond with MHC II without undergoing significant conformational changes; this protein was expressed in Escherichia coli and purified as a C-terminal six-histidine-tagged (6× His) fusion protein in our previous study.Citation18 SEB was also produced and purified in our previous study in the same way.
rFSAV contains five antigens: alpha-hemolysin (Hla), iron-regulated surface determinant B N2 domain (IsdB-N2), S. aureus protein A (SpA), mSEB, and manganese transport protein C (MntC). A total of 144 healthy adults aged between 18 and 65 y who participated in our phase 1 clinical trial were randomly assigned to different dose groups (low dose, middle dose, high dose, or placebo) in a ratio of 1:1:1:1 to evaluate the safety, tolerability, and preliminary immunogenicity of rFSAV. A total of 36 participants per group received four intramuscular shots of the vaccines or placebos on Days 0, 3, 7, and 14. Serum was collected and peripheral blood mononuclear cells (PBMCs) were isolated on Day 7 after the last injection was administered. This study has been registered at ClinicalTrials.gov under registration no. NCT02804711. M0313 was obtained based on the single B-cell technology by high-throughput isolation of immunoglobulin genes from the PBMCs of the high-dose group that received the experimental vaccine dose of 60 μg/single-protein.Citation16
The plasmid DNAs of M0313 heavy and light chains were extracted by Pure Yield Plasmid Maxiprep System (Promega, A2393). The plasmid DNAs (250 μg) of the M0313 heavy and light chains were added to 30 mL of Rockwell Park Memorium Institute (RPMI) 1640 medium, after which 1.50 mg of filter-sterilized polyetherimide was added to the RPMI/DNA solution, and the solution was vortexed vigorously for 3 s. The mixture was incubated at 20°C to 25°C for 15 min. HEK293 F suspension-adapted cells (30 mL at a concentration of 1.3 × 107 cells/mL) were added to the solution. Six hours after transfection, the HEK293 F suspension-adapted cells were fed with fresh HEK293 expression medium (OPM Bioscience, 81075–001) and incubated in an orbital shaker incubator for 5 d at 37°C, 125 rpm, and 5% CO2. Cellular supernatant was harvested by centrifuging the cells at 3,000 g for 30 min and mixed with protein A agarose (Beyotime, P2015) overnight at 4°C. Protein A agarose combined with M0313 was collected in an affinity column (Beyotime, FCL60). M0313 was eluted after washing with binding buffer (Thermo Fisher Scientific, 21001) and elution buffer (Thermo Fisher Scientific, 21004). The purity of acquired the antibody was determined by Coomassie blue staining of SDS-PAGE gels.
Western blotting
SEB and mSEB were dissolved in protein loading buffer and boiled for 5 min. The proteins were resolved on 10% SDS-polyacrylamide gels and the fractionated proteins were transferred from the gel onto polyvinylidene fluoride membranes (Millipore, IPVH00010) in a semi-dry transblot apparatus. The membranes were blocked in 5% skimmed milk for 2 h. The blots were washed and incubated with 1 μg/mL M0313 for 1 h. They were washed again and then incubated for 45 min with anti-human HRP-IgG (1:5000) (Promega, W4031). After subsequent washing, the membranes were developed using a chemiluminescence method according to manufacturer’s instructions (Thermo Fisher Scientific, 32106).
ELISA
To determine the titers of M0313, microtiter plates were coated with 1 μg/mL of SEB or mSEB at a volume of 100 μL overnight at 4°C. Nonspecific binding was blocked with 1% bovine serum albumin (BSA) for 2 h after the wells were washed three times. Serial dilutions of M0313 (0.000125 to 0.250 μg/mL) were incubated for 1 h at 37°C and then captured with anti-human HRP-IgG (Promega, W4031). Next, 100 μL of 1, 2, 4-trimethylbenzene (1, 2, 4-TMB) solution was added to each well and the plate was incubated in the dark at 37°C for 5 min. 2 M sulfuric acid (50 μL) was added and then the absorbance of wells was detected at 450 nm.
Biolayer interferometry
Biolayer interferometry (BLI) was performed using an Octet RED96 system (ForteBio, Fremont, USA) to measure the kinetic analysis of M0313 binding to SEB or mSEB. M0313 (50 nM) was immobilized onto an anti-human IgG capture biosensor (AHC), and two continuous diluents of SEB and mSEB at concentrations ranging from 3.12 to 50 nM were loaded in accordance with the preset sequence. Binding and dissociation of the antigens and M0313 were detected according to the detection procedure. Specific binding and dissociation curves, and dissociation constant (KD) values were determined.
Cell proliferation and cytokine release assays
The ability of M0313 to inhibit cell proliferation and cytokine release was tested using both mouse splenic lymphocytes and human PBMCs. Splenic lymphocytes from BALB/c female mice were isolated by Ficoll-Paque Plus (Dakewe Biotech Co., 7211011) gradient centrifugation, washed with RPMI 1640 medium, and resuspended in growth medium containing 45% RPMI 1640, 45% DMEM, 10% fetal bovine serum (FBS), 55 μM 2-mercaptoethanol, and penicillin-streptomycin solution (1×). Serially diluted M0313 samples were incubated with SEB for 30 min. Antigen-antibody complexes, SEB only (positive control) and RPMI 1640 medium (negative control) were added to a 96-well plate containing 5 × 105 lymphocytes per well. Human PBMCs were isolated by Ficoll-Paque Plus (TBD, LTS1077-1) gradient centrifugation of heparinized blood from normal human donors. PBMCs were washed with RPMI 1640 medium and resuspended in RPMI 1640 medium containing 10% FBS. Serially diluted M0313 samples were incubated with SEB for 30 min. Antigen-antibody complexes, SEB only (positive control) and RPMI 1640 medium (negative control) were added to a 96-well plate with 1 × 105 PMBC cells per well at a volume of 100 µL for cell proliferation assay, and to a 48-well plate with 6 × 105 PMBC cells per well at a volume of 600 µL for cytokine release assay. Cell proliferation was measured using CellTiter-Glo 2.0 Assay (Promega, G9241) after 48 h of incubation according to the manufacturer’s instructions. IL-2 (Dakewe Biotech Co., 1210202 & 1110202), IL-6 (Dakewe Biotech Co., 1210602 & 1110602), IFN-γ (Dakewe Biotech Co., 1210002 & 1110002), and TNF-α (Dakewe Biotech Co., 1217202 &1117202) were detected in the cellular supernatants using Bio-plex® cytokine ELISA kits after 24 h. Experiments were performed in triplicates. The inhibition rates of cell proliferation = (relative light unit [RLU] of positive control group − RLU of the mAb group)/(RLU of positive control group − RLU of negative control group). The inhibition rates of cytokines were calculated in the same way.
Animal experiments
All the animal experiments were approved by the Animal Ethical and Experimental Committee of the Third Military Medical University (Chongqing; permit number 2018–07). Six- to eight-week-old SPF female BALB/c mice were purchased from Beijing Vital River Lab Animal Technology Co., Ltd., maintained under pathogen-free conditions, and fed laboratory chow and water ad libitum. The SEB-expressing strain MRSA252 was obtained from the American Type Culture Collection. The bioluminescent S. aureus Xen29 strain was purchased from Xenogen Corp. (Alameda, CA, USA). Two clinical MRSA strains identified the type of enterotoxin were obtained from the General Hospital of Jinan Military Region.
To test the protective efficacy of M0313 against SEB toxic shock, the mice were intraperitoneally injected once with 20 μg of M0313 or normal saline (NS) 24 h prior to the intravenous administration of 20 μg SEB and 10 mg D-galactosamine. The mice were monitored for morbidity and mortality for 7 d. In another experiment, blood, lung, and spleen tissues were collected 5 h after the challenge. Blood was centrifuged for 20 min at 20°C to 25°C, 2000 rpm, and serum was collected to detect IL-2, IL-6, IFN-γ, and MCP-1 (Dakewe Biotech Co., 1217392) concentrations by ELISA. The lung and spleen tissues were fixed with 4% paraformaldehyde and embedded in paraffin. Four-micrometer thick sections were prepared and stained with hematoxylin and eosin for microscopic examination. Each lung and spleen section was rated on a score of 0 to 3 (from no abnormalities to severe) by the criteria based on edema, hyperemia, neutrophil infiltration, and structural damage.
To test the protective efficacy of M0313 against sepsis caused by different species of S. aureus, M0313 was administered intravenously to the mice, after which they were monitored for morbidity and mortality for 7 d. The mice were injected with 400 μg of M0313 or NS 24 h before or 1 h after the intravenous administration of 9 × 108 CFUs of MRSA252. The human clinical isolates JN028 and JN064 are strains obtained from the General Hospital of Jinan Military Region and were isolated and identified their enterotoxin type as follows: JN028 was obtained from the blood of a patient and identified to express enterotoxin A, C, D, and E; JN064 was obtained from the puncture fluid of a patient and identified to express enterotoxin B. The mice were intraperitoneally injected with 400 μg M0313 or NS 24 h before administering 8 × 108 CFUs of JN028 or 7 × 108 CFUs of JN064.
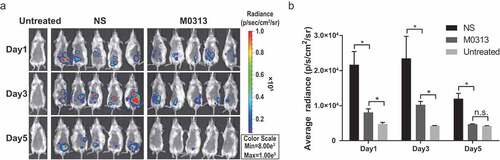
The tail vein of each mouse was injected with 400 μg of M0313 or NS and the abdominal hair was removed 24 h before the intraperitoneal injection of 6 × 108 CFUs of Xen29. Two untreated mice were used as the negative control group. The mice of the untreated group, M0313 group and NS group were anesthetized with isoflurane gas on Days 1, 3, and 5 after the challenge and monitored using an in vivo imaging system (IVIS) (Xenogen, Alameda, CA). The live images of mice and radiance data were processed in Living Image Version 4.4.
Inhibition of SEB binding to MHC II and TCR
The B-lymphocyte cell line “Raji” was used to detect antibodies that inhibited SEB-MHC II (HLA-DR, -DP, -DQ) interactions. FITC-conjugated SEB was prepared using HOOK Dye Labeling Kit (FITC) (Sangon Biotech, C006141). Next, Raji cells were incubated in 400 μL of RPMI 1640 medium containing 10% FBS with SEB (20 µg) at 37°C for 1 h either alone (positive control) or in combination with serial dilutions of M0313 (15.6 to 500 µg/mL) at 37°C for 2 h. After extensive washing, the cells were resuspended and the fluorescence intensity was analyzed by flow cytometry. Mouse splenic T lymphocytes were used to detect the inhibitory effect of M0313 on TCR interactions. Mouse splenic T lymphocytes were incubated in 400 μL of RPMI 1640 medium with 10% FBS with SEB (30 µg) incubated at 37°C for 30 min either alone (positive control) or in combination with two continuous diluents of M0313 (23.4 to 750 µg/mL) at 37°C for 1 h. After extensive washing with Hank’s balanced salt solution (1×), the cells were resuspended and the fluorescence intensity was analyzed by flow cytometry. The inhibition rates of SEB binding to MHC II = (relative fluorescence intensity [RFI] of positive control group − RFI of the mAb group)/RFI of positive control group. The inhibition rates of SEB binding to TCR were calculated in the same way.
Epitope mapping
Based on SEB sequence available in the MRSA COL (GI number: 57650036) genome, 20 overlapping peptides that spanned the entire length of SEB and consisted of 18 amino acid residues, three truncated peptides of SEB85-102, and six mutant peptides of SEB85-102 were synthesized by GenScript Biotech Corp. (Nanjing, China). The purity of all the above peptides was expected to be 75% or higher. To locate the core sequence of SEB that is recognized by M0313, ELISA plates were coated with coating buffer containing 2 μg/mL streptavidin (SA), SEB, or BSA overnight at 4°C. After three washes with 0.5% PBST, 1% BSA was added and the plates were further incubated for 2 h. The peptides were diluted to 4 μg/mL and linked to SA for 1 h. Then, the plates were incubated with M0313 (2 μg/mL) for 1 h and anti-human HRP-IgG for 45 min. The plates were further incubated with a TMB substrate, followed by 2 M sulfuric acid, and the absorbance was determined at 450 nm by microplate reader (Bio-Rad, Hercules, CA, USA). The values that were above the treble absorbance value of BSA were considered positive. Each experiment was performed in triplicates. The immunodominant epitope and key amino acids in the three-dimensional crystal structure of SEB were mapped using the PyMOL 1.1 program. SEB sequences from different S. aureus strains were retrieved from the GenBank database and compared using Jalview 2.10.5 software.
Statistical analyses
All the data were analyzed and graphs were plotted using the Prism software (GraphPad Prism 6, San Diego, USA). The data were presented as the mean ± standard error of mean. Intergroup difference was compared using an unpaired t-test and Kaplan–Meier test. A P value < .05 was considered statistically significant.
Results
Binding activity of M0313 to SEB
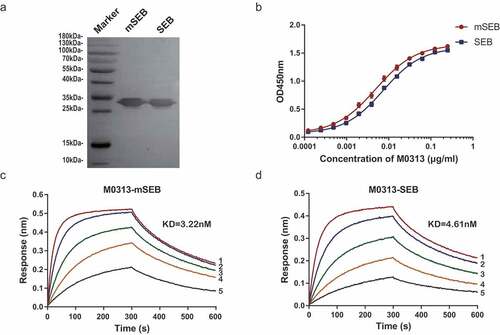
The purity of the produced and purified mAbs was over 95% as determined by SDS-PAGE. A 30 kDa band identified by western blotting indicated that M0313 recognized the linear epitope of mSEB and SEB (). ELISA demonstrated that M0313 bound to mSEB and SEB with an EC50 of 4.93 ng/mL and 7.37 ng/mL, respectively (). In addition, BLI performed to determine the binding kinetics of M0313 showed that M0313 bound to mSEB and SEB with equilibrium KD values of 3.22 nM and 4.61 nM ( and ); these results indicated that the binding affinity of M0313 to mSEB and the native toxin were comparable.
Figure 1. Binding activity of M0313 to SEB. (a) Combination of M0313 with linear epitope of SEB and mSEB detected by western blotting. (b) Binding activities of M0313 determined by ELISA. The absorbance of different concentrations of M0313 (0.000125 to 0.25 μg/mL) at 450 nm was shown. Kinetic analysis of the binding of M0313 to mSEB (c) and SEB (d) measured by BLI with a 300-s association step followed by a 300-s dissociation step. The Numbers 1 to 5 represent 50, 25, 12.5, 6.25, and 3.12 nM SEB were analyzed for binding to immobilized M0313, respectively.
Inhibition of cell proliferation and cytokine release
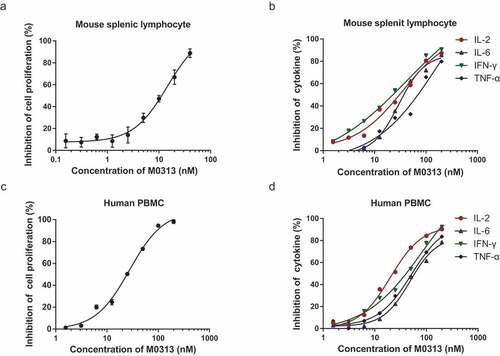
To determine the neutralizing effect of M0313 in vivo, the effect of SEB with and without the presence of M0313 was measured by cell proliferation and cytokine release assays using both mouse splenic lymphocytes and human PBMCs. The cell proliferation results were converted into RLUs by determining cellular ATP level by chemiluminescence. The inhibition rate of cell proliferation and cytokine release in both mouse splenic lymphocytes and human PBMCs positively correlated with M0313 concentration. As shown in , when the concentration of M0313 was 40 nM, the inhibition rate of mouse splenic lymphocyte proliferation reached 88.8%. When the concentration of M0313 was 200 nM, the inhibition rates of IL-2, IL-6, IFN-γ, and TNF-α were 87.3%, 85.7%, 90.6%, and 79.8%, respectively (). M0313 also showed inhibitory effect on the SEB-induced human PBMC proliferation and cytokine release. When the concentration of M0313 was 200 nM, the inhibition rate of human PBMC proliferation was 98.2%, and of IL-2, IL-6, IFN-γ, and TNF-α release were 90.2%, 78.5%, 92.3%, and 83.6%, respectively ( and ).
Figure 2. Inhibition of cell proliferation and cytokine release. (a) Inhibition of SEB-induced mouse splenic lymphocyte proliferation. Mouse splenic lymphocytes (5 × 105 cells) in the mAb group were incubated with 1 nM SEB and two continuous diluents of M0313 at concentrations ranging from 0.156 to 40 nM. (b) Inhibition of SEB-induced IL-2, IL-6, IFN- γ, and TNF-α release in the supernatant of mouse splenic lymphocytes. Mouse splenic lymphocytes (5 × 105 cells) in the mAb group were incubated with 2 nM SEB and two continuous diluents of M0313 at concentrations ranging from 1.56 to 200 nM. (c) Inhibition of SEB-induced human PBMC proliferation. Human PBMCs (1 × 105 cells) in the mAb group were incubated with 2 nM SEB and two continuous diluents of M0313 at concentrations ranging from 1.56 to 200 nM. (d) Inhibition of SEB-induced IL-2, IL-6, IFN-γ, and TNF-α release in the supernatant of human PBMCs. Human PBMCs (6 × 105 cells) in the mAb group were incubated with 2 nM SEB and two continuous diluents of M0313 at concentrations ranging from 1.56 to 200 nM.
Toxin neutralization in vivo
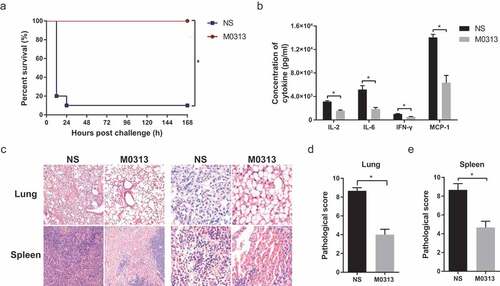
As SEB binds to MHC II with low affinity in mice, D-galactosamine must be added as a potentiating agent in toxic shock models. As shown in , 20 μg of SEB and 10 mg of D-galactosamine caused toxic shock that killed 90% of the mice within 7 d. At the same time, all the mice injected with 20 μg M0313 survived. Serum levels of inflammatory cytokines including IL-2, IL-6, IFN-γ, and MCP-1 were significantly lower (P < .05) in mice administered M0313 than in those that were not (). Furthermore, histological analysis showed that the lungs of mice in the NS group exhibited excessive cellular infiltration in the alveolar and interstitial spaces of the tissue. In contrast, mice in the M0313 treatment group showed a marked decrease in the SEB-driven cellular infiltration in the lungs and complete alveolar structure. The spleens of mice in the control group exhibited noticeable disruption of the spleen structure and many signs of pathological alterations. Both the lung and spleen of the control mice showed more infiltration of inflammatory cells, bleeding, and tissue damage compared to those in the M0313 group (). Consistent with the microscopic observations, both the pathological scores of the lungs and spleens in the M0313 group were significantly lower than those of the NS group ( and ) (P < .05).
Figure 3. Toxin neutralization in vivo. (a) Survival of mice in the M0313 and NS groups 7 d after the challenge (n = 10). (b) Concentrations of serum cytokines in both the groups 5 h after the challenge (n = 5). The histogram represents the concentrations of IL-2, IL-6, IFN-γ, and MCP-1. (c) Microscopic examination of the pathological sections of the lung and spleen tissues that were fixed 5 h after the challenge. Images were captured at 100 × and 400 × magnification. (d) Pathological score of the mouse lung. (e) Pathological score of the mouse spleen. “*” indicates P < .05.
Protection against S. aureus-induced sepsis
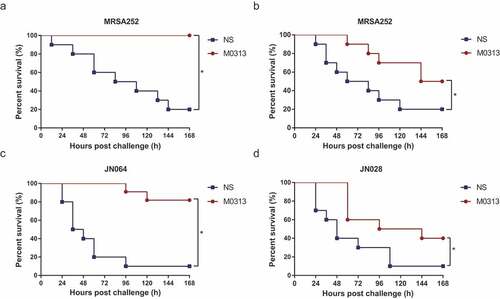
The effectiveness of the protective effect of M0313 against sepsis caused by different types of enterotoxin-expressing S. aureus was tested in the mouse model. As shown in , 9 × 108 CFUs of MRSA252 caused sepsis that killed 80% of the mice within 7 d, while all the mice injected with M0313 24 h before the challenge survived. Administration of M0313 1 h after the challenge slowed the mortality rate of the challenged mice (). The mice injected with JN028 and JN064 produced similar results ( and ). However, M0313 protected against JN064 better than it did against JN028, based on the type specificity of M0313 to SEB; 77.8% of the mice injected with M0313 survived JN064-induced sepsis and 33.3% survived JN028-induced sepsis.
Figure 4. Protection against S. aureus-induced sepsis. (a) Survival of mice administrated M0313 24 h before MRSA252 challenge (n = 10). (b) Survival of mice administrated M0313 1 h after MRSA252 challenge (n = 10). (c) Survival of mice administrated M0313 24 h before SEB-expressing JN064 challenge (n = 10). (d) Survival of mice administrated M0313 24 h before non-SEB-expressing JN028 challenge (n = 10). The abscissa represents the number of hours after the challenge, and the ordinate represents the per-day survival rate. “*” indicates significant difference (P < .05).
The mice protected by M0313 were challenged with the bioluminescence stain Xen29. In vivo bioluminescence results of the three groups are shown in . Bioluminescence observed in the NS group mice was significantly higher than that observed in the M0313 group on Days 1, 3, and 5 after the challenge (P < .05). On the fifth day after the challenge, in contrast to the obvious bioluminescence in the NS group mice, no significant difference in bioluminescence values was detected in the M0313-treated mice compared to that in the untreated mice (P ≥ 0.05). The quantitative analysis results of the bioluminescence signals are shown in . There were significant differences in the average radiance between the NS and M0313 groups for each day of living imaging (P < .05).
Figure 5. Live imaging of mice after intraperitoneal injection of Xen29. (a) In vivo bioluminescence images of untreated, NS, and M0313-treated mice (n = 2, 10, 10, respectively). Three groups of mice were monitored using the in vivo imaging system on Days 1, 3, and 5 after the challenge. (b) Quantitative analysis results of the bioluminescence signals. The histogram represents the average bioluminescence radiance. “*” indicates significant difference (P < .05) while “n.s.” indicates no significant difference (P ≥ 0.05).
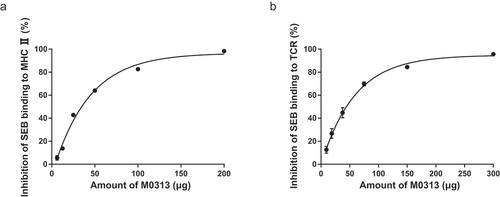
Inhibition of SEB binding to MHC II and TCR
In order to verify the mechanism underlying the toxin-neutralizing capacity of M0313, FITC-SEB was added to cells alone or together with M0313 and incubated. The inhibition rate of SEB binding to MHC II and TCR positively correlated with M0313 concentration. As shown in , when the amount of M0313 was 200 µg, the inhibition rate of SEB binding to MHC II was 98.3%. As shown in , when the amount of M0313 was 300 µg, the inhibition rate of SEB binding to TCR was 95.6%.
Figure 6. Inhibition of SEB binding to MHC II and TCR. (a) Inhibition of SEB binding to MHC II. Raji cells were incubated with 20 µg SEB and two continuous diluents of M0313 at amounts ranging from 6.25 to 200 µg. (b) Inhibition of SEB binding to TCR. Mouse splenic lymphocytes (5 × 105 cells) were incubated with 30 µg SEB and two continuous diluents of M0313 at amounts ranging from 9.38 to 300 µg.
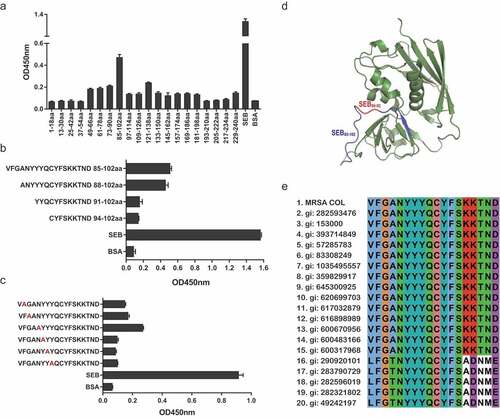
Epitope mapping of M0313 with SEB
To determine the immunodominant peptides of SEB binding to M0313, the entire length of the SEB amino acid sequence was truncated to synthetize twenty 18-mer peptides with six amino acids between the overlapped two adjacent peptides. Linear B-cell epitope mapping of SEB was determined with an overlapping 18-mer peptide by ELISA. The result shown in indicated that the immunodominant peptide was concentrated on SEB85-102 (VFGANYYYQCYFSKKTND). SEB85-102 was truncated three times to give three peptides: SEB88-102, SEB91-102, and SEB94-102. Using these peptides and SEB85-102, the binding sites were identified by ELISA (), and we found that SEB85-102 and SEB88-102 had stronger binding capacities to SEB. The second to seventh nonalanine amino acids of SEB88-102 were sequentially mutated into alanine (A), which then provided six new polypeptides that were used for detecting the binding activity with M0313. As shown in , the absorbance values of the three new polypeptides that contained any one amino acid mutation of SEB90-92 were negative. The mapping results of the immunodominant epitopes and key amino acids in the three-dimensional crystal structure of SEB shown in indicated that SEB85-102 and SEB90-92 were present in the exterior loop region of SEB. The conservation results of immunodominant epitope SEB85-102 revealed that sequences of the immunodominant epitope were highly conserved among the S. aureus strains, with over 90% amino acid identity match. The amino acid sequences of SEB90-92 were also highly conserved ().
Figure 7. Epitope mapping results of M0313 with SEB measured by ELISA. (a) Binding of M0313 to 20 overlapping peptides that spanned the entire length of SEB. (b) Binding of M0313 to three truncated peptides of SEB85-102. The ordinate represents the amino acid sequence and position of the polypeptides. (c) Binding condition of M0313 to six mutant peptides of SEB85-102. Red indicates the mutated amino acid. (d) The position of immunodominant peptides SEB85-102 (blue) and key amino acids SEB90-92 (red) in the three-dimensional structure of SEB. (e) Sequence alignment of SEB85-102 in SEB selected from MRSA COL and other 19 S. aureus strains.
Discussion
SEB is a potential biowarfare agent due to its inherent stability, high morbidity rate, high toxicity, and ease of dissemination.Citation19 If SEB is atomized, more than 80% of the people under attack may become seriously ill and develop symptoms of fever, myalgia, and shivering chills after 8 to 20 h. After prolonged paroxysms of coughing, dry cough, and inspiratory rales with dyspnea and vomiting are manifested. Heavy exposure can lead to more severe symptoms such as high fever, pulmonary edema, possible acute respiratory distress syndrome, or fatal septic shock.Citation12 In addition to inhalation, purified SEB can be ingested through contaminated water or food systems to affect many people. Clinical data have suggested that immunoglobulins can alleviate SEB-induced diseases.Citation20,Citation21 The use of IVIG has also proven useful in humans after the onset of SEB-induced toxic shock.Citation21,Citation22 Passive immunization using anti-SEB antibodies had a protective effect on rhesus monkeys 4 h before or after exposure to SEB aerosol.Citation23 The key to improve the treatment success rate for SEB-induced illness is to develop high-affinity and -specificity antibodies with high throughput that can effectively and quickly neutralize SEB. Therefore, mAb M0313 exhibited these characteristics in our study, we believe that it may be a good candidate acting against SEB.
In our study, M0313 demonstrated low nanomolar affinity for SEB; reduced cell proliferation and cytokine release caused by SEB; neutralizing activity against SEB-induced TSS, and protective action against SEB-expressing S. aureus-induced sepsis. By blocking the superantigen SEB in vivo, M0313 improved the survival rate, decreased serum inflammatory factor levels, and reduced pathological inflammation and replication of S. aureus in BALB/c mice. The protective action of M0313 against S. aureus clinical strains proved that M0313 protected one-third of non-SEB-expressing JN028, and protected more of SEB-expressing JN064. This may be due to the type specificity and cross-protection between different staphylococcal enterotoxins of M0313.Citation24 Flow cytometry results proved that the main mechanism of the intracellular neutralization of SEB by M0313 involved the inhibition of SEB binding to MHC II and TCR. These results were further verified by epitope mapping results that proved that SEB85-102 was the immunodominant epitope binding to M0313 and the amino acids present in the epitope SEB90-92 were the key ones. The sequence of the B-cell epitope SEB85-102 (VFGANYYYQCYFSKKTND) was highly consistent with that of the epitope SEB84-93 (DVFGANYYYQ) that was previously reported.Citation25 It is interesting that the residues Y89, Y91, Y92, and Y94 of SEB fall within the binding epitope of M0313, because Y89 and Y94 fall within the binding site of MHC II, and the aromatic pair of residues Y91 and Y92 are crucial for TCR binding to take place.Citation26,Citation27
There were three major limitations in our study. First, the efficacy of toxin-directed mAbs is naturally limited to disease types whose pathogenesis relies on these specific toxins. SEB is one of the 25 superantigens that S. aureus is capable of producing, so only a small percentage of S. aureus strains express SEB.Citation28,Citation29 Prophylactic M0313 administration protected more effective than post-infection passive immunotherapy with the same strain of MRSA, and the post-infection antibody treatment strategy only protected 37.5% of MRSA252. Considering that diverse toxins may be playing a role in disease pathogenesis depending on the clinical isolate involved, the utility of M0313 to prevent S. aureus infections may be limited. We do not expect that an anti-SEB mAb can be the single component of an immunotherapy that designs to combat against MRSA infections in all conditions. M0313 use is hence recommended to deter SEB biowarfare alone. Second, mice are poor responders to SEB due to the low affinity of SEB to mouse MHC compared with that for human MHC.Citation30 However, adding a potentiating agent such as D-galactosamine, actinomycin D, lipopolysaccharides, or viruses to amplify the responses to SEB is a crucial variable that has helped in deciphering derived data.Citation31-Citation33 The use of better animal models such as transgenic mice expressing HLA-DR or two low doses of SEB without using the abovementioned confounding potentiating agents is suggested for murine models of SEB-induced toxic shock.Citation34,Citation35 Third, local mutations of full-length SEB were not used to verify the immunodominant peptide SEB85-102 as an epitope. However, the most reliable method for identifying a B-cell epitope is X-ray crystallography. By more accurate epitope mapping using the crystal structure of SEB-neutralizing M0313, the binding sites of TCR, MHC II, and M0313 can be determined more accurately, on the basis of which the protection mechanism of M0313 can be clarified.Citation36
Identification and production of agents with protective action against SEB have garnered attention because of the biowarfare potential of SEB, and hence, several potential, neutralizing, anti-SEB mAbs have recently been reported such as mouse mAb 20B1, humanized mouse mAb Hu-1.6/1.1, Ch 82 M, and Ch63, mAb GC132, and other multiple mAbs composed of human Fabs and mAbs using phage-display technology. These mAbs have exhibited proven protective effect on SEB-induced lethal shock and SEB-expressing S. aureus-induced infections.Citation37-Citation44 Some of them significantly improved the survival of animals and reduced the dissemination of the bacteria in different infection mouse models, including S. aureus-induced bacteremia, superficial skin infection, deep tissue implant infection, and lethal pneumonia. M0313 derived from human sequences through various techniques can effectively locate novel therapeutic targets and may be less immunogenic than that derived from rodent sequences.Citation45 Single B-cell technology is based on the direct amplification of heavy (VH) and light (VL) chain variable region-encoding genes from single human B cells and their subsequent expression in cell culture systems.Citation46 It reserves natural VH and VL antibody pairings as it does not rely on random combinations compared to phage display.Citation47 In addition, because our blood samples were obtained from volunteers immunized with rFSAV, our antibodies had stronger affinity to the antigen after a series of affinity maturation processes. The vaccine immunization program triggered a stronger immune response to antigens and produced antibodies that recognized more dispersed epitopes compared to natural infection. Our study has determined the immunological characteristics of the anti-SEB human mAb M0313 derived using the single human B-cell antibody technique and proved its certain protective effect in the treatment of SEB-induced toxic shock and resistance to SEB-expressing S. aureus infection. M0313 has the potential to develop into a therapeutic agent against SEB biological warfare. More treatment strategies of multivalent mAb cocktails and combination of antibiotic and immunomodulator therapy should be explored for treating S. aureus infection in follow-up studies, because of the limited protection effect of a single anti-SEB mAb against MRSA-induced sepsis.Citation48-Citation50
Disclosure of potential conflicts of interest
The authors declare that the research was designed and conducted without any conflict of interest in terms of commercial or financial relationships.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 31370932) and the key project of innovative drug development (grant number 2015ZX09101033 and 2016ZX09J16102-002). None of the funding agencies played a role in study design, data collection or interpretation, or the decision to submit the work for publication.
Additional information
Funding
References
- Chambers HF, Deleo FR. Waves of resistance: staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. 2009;7:629–41. PMID:19680247. doi:10.1038/nrmicro2200.
- Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34. PMID:10627489. doi:10.1128/cmr.13.1.16-34.2000.
- Li H, Llera A, Tsuchiya D, Leder L, Ysern X, Schlievert PM, Karjalainen K, Mariuzza RA. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity. 1998;9:807–16. PMID:9881971. doi:10.1016/s1074-7613(00)80646-9.
- Krakauer T. Staphylococcal superantigens: pyrogenic toxins induce toxic shock. Toxins (Basel). 2019;11:178. PMID:30909619. doi:10.3390/toxins11030178.
- Hudson Reichenberg LC, Garg R, Fernalld R, Bost KL, Piller KJ. Systemic cytokine and chemokine responses in immunized mice challenged with staphylococcal enterotoxin B. Toxicon. 2017;133:82–90. PMID:28478060. doi:10.1016/j.toxicon.2017.05.005.
- Rajagopalan G, Sen MM, Singh M, Murali NS, Nath KA, Iijima K. Intranasal exposure to staphylococcal enterotoxin B elicits an acute systemic inflammatory response. Shock. 2006;25:647–56. PMID:16721274. doi:10.1097/01.shk.0000209565.92445.7d.
- Krakauer T, Buckley MJ, Fisher D. Proinflammatory mediators of toxic shock and their correlation to lethality. Mediators Inflamm. 2010;2010:517594. PMID:20634937. doi:10.1155/2010/517594.
- Krakauer T, Stiles BG. The staphylococcal enterotoxin (SE) family: SEB and siblings. Virulence. 2013;4:759–73. PMID:23959032. doi:10.4161/viru.23905.
- Raj HD, Bergdoll MS. Effect of enterotoxin B on human volunteers. J Bacteriol. 1969;98:833–34. PMID:4977485. doi:10.1128/JB.98.2.833-834.1969.
- McKallip RJ, Fisher M, Do Y, Szakal AK, Gunthert U, Nagarkatti PS, Nagarkatti M. Targeted deletion of CD44v7 exon leads to decreased endothelial cell injury but not tumor cell killing mediated by interleukin-2-activated cytolytic lymphocytes. J Biol Chem. 2003;278:43818–30. PMID:12904302. doi:10.1074/jbc.M304467200.
- Ahanotu E, Alvelo-Ceron D, Ravita T. Staphylococcal enterotoxin B as a BIOLOGICAL WEAPon: recognition, management, and surveillance of staphylococcal enterotoxin. Appl Biosaf. 2006;11:120–26. doi:10.1177/153567600601100303.
- Balali-Mood M, Moshiri M, Etemad L. Medical aspects of bio-terrorism. Toxicon. 2013;69:131–42. PMID:23339855. doi:10.1016/j.toxicon.2013.01.005.
- Lindsay CD, Griffiths GD. Addressing bioterrorism concerns: options for investigating the mechanism of action of Staphylococcus aureus enterotoxin B. Hum Exp Toxicol. 2013;32:606–19. PMID:23023027. doi:10.1177/0960327112458941.
- Henghold WB 2nd., Other biologic toxin bioweapons: ricin, staphylococcal enterotoxin B, and trichothecene mycotoxins. Dermatol Clin. 2004;22:257–62. PMID:15207307. DOI:10.1016/j.det.2004.03.004.
- Bagnoli F, Staphylococcus aureus toxin antibodies: good companions of antibiotics and vaccines. Virulence. 2017;8:1037–42. PMID:28267417. DOI:10.1080/21505594.2017.1295205.
- Song Z. Sorting of MRSA human monoclonal antibodies based on single B cell technology. Immunol J. 2019;35:254–61. doi:10.13431/j.cnki.immunol.j.20190041.
- Liao HX, Levesque MC, Nagel A, Dixon A, Zhang R, Walter E, Parks R, Whitesides J, Marshall DJ, Hwang -K-K. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J Virol Methods. 2009;158:171–79. PMID:19428587. doi:10.1016/j.jviromet.2009.02.014.
- Inskeep TK, Stahl C, Odle J, Oakes J, Hudson L, Bost KL, Piller KJ. Oral vaccine formulations stimulate mucosal and systemic antibody responses against staphylococcal enterotoxin B in a piglet model. Clin Vaccine Immunol. 2010;17:1163–69. PMID:20554806. doi:10.1128/cvi.00078-10.
- Pinchuk IV, Beswick EJ, Reyes VE. Staphylococcal enterotoxins. Toxins (Basel). 2010;2:2177–97. PMID:22069679. doi:10.3390/toxins2082177.
- Baudet V, Hurez V, Lapeyre C, Kaveri SV, Kazatchkine MD. Intravenous immunoglobulin (IVIg) modulates the expansion of V beta 3+ and V beta 17+ T cells induced by staphylococcal enterotoxin B superantigen in vitro. Scand J Immunol. 1996;43:277–82. PMID:8602461. doi:10.1046/j.1365-3083.1996.d01-36.x.
- Darenberg J, Soderquist B, Normark BH, Norrby-Teglund A. Differences in potency of intravenous polyspecific immunoglobulin G against streptococcal and staphylococcal superantigens: implications for therapy of toxic shock syndrome. Clin Infect Dis. 2004;38:836–42. PMID:14999628. doi:10.1086/381979.
- Schlievert PM. Use of intravenous immunoglobulin in the treatment of staphylococcal and streptococcal toxic shock syndromes and related illnesses. J Allergy Clin Immunol. 2001;108:S107–10. PMID:11586276. doi:10.1067/mai.2001.117820.
- LeClaire RD, Hunt RE, Bavari S, Protection against bacterial superantigen staphylococcal enterotoxin B by passive vaccination. Infect Immun. 2002;70:2278–81. PMID:11953360. DOI:10.1128/iai.70.5.2278-2281.2002.
- Johnson HM, Bukovic JA, Kauffman PE. Antigenic cross-reactivity of staphylococcal enterotoxins. Infect Immun. 1972;5:645–47. PMID:4674001. doi:10.1128/iai.5.5.645-647.1972.
- Pang LT, Kum WW, Chow AW. Inhibition of staphylococcal enterotoxin B-induced lymphocyte proliferation and tumor necrosis factor alpha secretion by MAb5, an anti-toxic shock syndrome toxin 1 monoclonal antibody. Infect Immun. 2000;68:3261–68. PMID:10816471. doi:10.1128/iai.68.6.3261-3268.2000.
- Rodstrom KEJ, Regenthal P, Bahl C, Ford A, Baker D, Lindkvist-Petersson K. Two common structural motifs for TCR recognition by staphylococcal enterotoxins. Sci Rep. 2016;6:25796. PMID:27180909. doi:10.1038/srep25796.
- Papageorgiou AC, Tranter HS, Acharya KR. Crystal structure of microbial superantigen staphylococcal enterotoxin B at 1.5 A resolution: implications for superantigen recognition by MHC class II molecules and T-cell receptors. J Mol Biol. 1998;277:61–79. PMID:9514739. doi:10.1006/jmbi.1997.1577.
- Holeckova B, Holoda E, Fotta M, Kalinacova V, Gondol J, Grolmus J. Occurrence of enterotoxigenic Staphylococcus aureus in food. Ann Agric Environ Med. 2002;9:179–82. PMID:12498587.
- Spaulding AR, Salgado-Pabon W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev. 2013;26:422–47. PMID:23824366. doi:10.1128/cmr.00104-12.
- Mollick JA, Chintagumpala M, Cook RG, Rich RR. Staphylococcal exotoxin activation of T cells. Role of exotoxin-MHC class II binding affinity and class II isotype. J Immunol. 1991;146:463–68. PMID:1987273.
- Nagaki M, Muto Y, Ohnishi H, Yasuda S, Sano K, Naito T, Maeda T, Yamada T, Moriwaki H. Hepatic injury and lethal shock in galactosamine-sensitized mice induced by the superantigen staphylococcal enterotoxin B. Gastroenterology. 1994;106:450–58. PMID:8299910. doi:10.1016/0016-5085(94)90604-1.
- Chen JY, Qiao Y, Komisar JL, Baze WB, Hsu IC, Tseng J. Increased susceptibility to staphylococcal enterotoxin B intoxication in mice primed with actinomycin D. Infect Immun. 1994;62:4626–31. PMID:7927730. doi:10.1128/IAI.62.10.4626-4631.1994.
- Sarawar SR, Blackman MA, Doherty PC. Superantigen shock in mice with an inapparent viral infection. J Infect Dis. 1994;170:1189–94. PMID:7963712. doi:10.1093/infdis/170.5.1189.
- Roy CJ, Warfield KL, Welcher BC, Gonzales RF, Larsen T, Hanson J, David CS, Krakauer T, Bavari S. Human leukocyte antigen-DQ8 transgenic mice: a model to examine the toxicity of aerosolized staphylococcal enterotoxin B. Infect Immun. 2005;73:2452–60. PMID:15784591. doi:10.1128/iai.73.4.2452-2460.2005.
- Huzella LM, Buckley MJ, Alves DA, Stiles BG, Krakauer T. Central roles for IL-2 and MCP-1 following intranasal exposure to SEB: a new mouse model. Res Vet Sci. 2009;86:241–47. PMID:18793785. doi:10.1016/j.rvsc.2008.07.020.
- Dutta K, Varshney AK, Franklin MC, Goger M, Wang X, Fries BC. Mechanisms mediating enhanced neutralization efficacy of staphylococcal enterotoxin B by combinations of monoclonal antibodies. J Biol Chem. 2015;290:6715–30. PMID:25572397. doi:10.1074/jbc.M114.630715.
- Chen G, Karauzum H, Long H, Carranza D, Holtsberg FW, Howell KA, Abaandou L, Zhang B, Jarvik N, Ye W. Potent neutralization of staphylococcal enterotoxin B in vivo by antibodies that block binding to the T-cell receptor. J Mol Biol. 2019;431:4354–67. PMID:30928493. doi:10.1016/j.jmb.2019.03.017.
- Karau MJ, Tilahun ME, Krogman A, Osborne BA, Goldsby RA, David CS, Mandrekar JN, Patel R, Rajagopalan G. Passive therapy with humanized anti-staphylococcal enterotoxin B antibodies attenuates systemic inflammatory response and protects from lethal pneumonia caused by staphylococcal enterotoxin B-producing Staphylococcus aureus. Virulence. 2017;8(7):1148–59. PMID:27925510. doi:10.1080/21505594.2016.1267894.
- Karauzum H, Chen G, Abaandou L, Mahmoudieh M, Boroun AR, Shulenin S, Devi VS, Stavale E, Warfield KL, Zeitlin L. Synthetic human monoclonal antibodies toward staphylococcal enterotoxin B (SEB) protective against toxic shock syndrome. J Biol Chem. 2012;287:25203–15. PMID:22645125. doi:10.1074/jbc.M112.364075.
- Larkin EA, Stiles BG, Ulrich RG. Inhibition of toxic shock by human monoclonal antibodies against staphylococcal enterotoxin B. PLoS One. 2010;5:e13253. PMID:20949003. doi:10.1371/journal.pone.0013253.
- Varshney AK, Wang X, Aguilar JL, Scharff MD, Fries BC. Isotype switching increases efficacy of antibody protection against staphylococcal enterotoxin B-induced lethal shock and Staphylococcus aureus sepsis in mice. mBio. 2014;5:e01007–14. PMID:24917594. doi:10.1128/mBio.01007-14.
- Varshney AK, Wang X, Scharff MD, MacIntyre J, Zollner RS, Kovalenko OV, Martinez LR, Byrne FR, Fries BC. Staphylococcal Enterotoxin B-specific monoclonal antibody 20B1 successfully treats diverse Staphylococcus aureus infections. J Infect Dis. 2013;208:2058–66. PMID:23922375. doi:10.1093/infdis/jit421.
- Varshney AK, Wang X, Cook E, Dutta K, Scharff MD, Goger MJ, Fries BC. Generation, characterization, and epitope mapping of neutralizing and protective monoclonal antibodies against staphylococcal enterotoxin B-induced lethal shock. J Biol Chem. 2011;286:9737–47. PMID:21233204. doi:10.1074/jbc.M110.212407.
- Tilahun ME, Kwan A, Natarajan K, Quinn M, Tilahun AY, Xie C, Margulies DH, Osborne BA, Goldsby RA, Rajagopalan G. Chimeric anti-staphylococcal enterotoxin B antibodies and lovastatin act synergistically to provide in vivo protection against lethal doses of SEB. PLoS One. 2011;6:e27203. PMID:22102880. doi:10.1371/journal.pone.0027203.
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. PMID:20811384. doi:10.1038/nrd3229.
- Guthmiller JJ, Dugan HL, Neu KE, Lan LY, Wilson PC. An efficient method to generate monoclonal antibodies from human B cells. Methods Mol Biol. 2019;1904:109–45. PMID:30539468. doi:10.1007/978-1-4939-8958-4_5.
- Tiller T. Single B cell antibody technologies. N Biotechnol. 2011;28:453–57. PMID:21473940. doi:10.1016/j.nbt.2011.03.014.
- Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Hiroshi Morisaki J, et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature. 2015;527:323–28. PMID:26536114. doi:10.1038/nature16057.
- Mariathasan S, Tan MW. Antibody-antibiotic conjugates: a novel therapeutic platform against bacterial infections. Trends Mol Med. 2017;23:135–49. PMID:28126271. doi:10.1016/j.molmed.2016.12.008.
- Tkaczyk C, Hamilton MM, Sadowska A, Shi Y, Chang CS, Chowdhury P, Buonapane R, Xiao X, Warrener P, Mediavilla J, et al. Targeting alpha toxin and ClfA with a multimechanistic monoclonal-antibody-based approach for prophylaxis of serious staphylococcus aureus disease. mBio. 2016;7. PMID:27353753. doi:10.1128/mBio.00528-16.