
ABSTRACT
Triple-negative breast cancer (TNBC) afflicts women at a younger age than other breast cancers and is associated with a worse clinical outcome. This poor clinical outcome is attributed to a lack of defined targets and patient-to-patient heterogeneity in target antigens and immune responses. To address such heterogeneity, we tested the efficacy of a personalized vaccination approach for the treatment of TNBC using the 4T1 murine TNBC model. We isolated tumor membrane vesicles (TMVs) from homogenized 4T1 tumor tissue and incorporated glycosyl phosphatidylinositol (GPI)-anchored forms of the immunostimulatory B7-1 (CD80) and IL-12 molecules onto these TMVs to make a TMV vaccine. Tumor-bearing mice were then administered with the TMV vaccine either alone or in combination with immune checkpoint inhibitors. We show that TMV-based vaccine immunotherapy in combination with anti-CTLA-4 mAb treatment upregulated immunomodulatory cytokines in the plasma, significantly improved survival, and reduced pulmonary metastasis in mice compared to either therapy alone. The depletion of CD8+ T cells, but not CD4+ T cells, resulted in the loss of efficacy. This suggests that the vaccine acts via tumor-specific CD8+ T cell immunity. These results suggest TMV vaccine immunotherapy as a potential enhancer of immune checkpoint inhibitor therapies for metastatic triple-negative breast cancer.
Introduction
Breast cancer patients are sub-typed based on the expression of receptors for estrogen (ER), progesterone (PR), or human epidermal growth factor receptor 2 (HER-2). Patients expressing one or more of these proteins are treated with existing therapies to control tumor growth and disease progression. However, 10–20% of patients fall into the triple-negative breast cancer (TNBC) designation, due to the absence of the ER, PR, and HER-2. TNBC exhibits an aggressive phenotype with a poor overall prognosis and occurs at a higher incidence in African-American women and more in premenopausal women than postmenopausal women.Citation1,Citation2 The current standard of care for TNBC has been surgery and chemotherapy. Despite aggressive treatment, recurrence is common, often resulting in metastasis to the vital organs within 18–24 months of detection.Citation3 For this reason, new therapies that induce lasting protection against relapse are needed.
Recently, the first immunotherapy targeting TNBC, the immune checkpoint inhibitor (ICI) anti-PD-L1 antibody atezolizumab used in combination with nanoparticle albumin-bound paclitaxel, was approved by the FDA for the treatment of adults with PD-L1-positive, unresectable, locally advanced or metastatic TNBC.Citation4 This therapy was shown to improve the median progression-free survival (PFS) from 5.5 months to 7.2 months, and complete response was observed in 10% of patients compared to 1% with chemotherapy alone.Citation4 Despite these promising results, a majority (>80%) of ICI-treated patients do not respond, and the mechanisms behind this lack of response are not well understood in breast and other cancers. However, because ICI has been shown to function through enhancement of the endogenous anti-tumor T cell response, lack of preexisting T cell immunity may drive ICI therapy resistance. For this reason, we hypothesize that induction of de novo tumor-specific T cells by vaccination will consequently render these tumors responsive to ICI therapy.
The major challenges in developing a vaccine immunotherapy for TNBC are the lack of known targets, intratumoral heterogeneity, and patient-to-patient variation in tumor antigens.Citation5 These limitations identify the clinical need for a personalized immunotherapy approach. However, individualized autologous immunotherapy failed in the clinic due to difficulty in growing tumor cell lines from patient tumor tissue, use of cell lines that do not recapitulate the patients’ tumor antigenicity, and suboptimal adjuvants in vaccines.Citation6,Citation7 To address these problems, we developed a personalized tumor membrane-based vaccine immunotherapy to treat metastatic TNBC that can be prepared from frozen or fresh tumor tissue without a need to establish cell lines and adjuvanted with naturally occurring immunostimulatory molecules. In human breast cancer, many tumor-associated antigens are expressed as plasma membrane-bound surface antigens, such as MUC-1, TROP-2, the mucin antigens TF, Tn, STn, and the glycolipid globo H, making it ideal to develop a membrane-based vaccine immunotherapy.Citation8–10 In addition, plasma membranes contain MHC molecules that complex with peptides derived from cytosolic tumor antigens. The novelty of this vaccine immunotherapy is the incorporation of glycolipid-anchored forms of immunostimulatory molecules (GPI-ISMs), such as B7-1 and IL-12, onto tumor membrane vesicles (TMV) derived from tumor tissue using a simple protein transfer method. These TMV-attached GPI-ISMs will simultaneously deliver tumor-specific antigens and stimulate immune cells to promote an effective anti-tumor immune response.Citation7,Citation11-14 The physical linkage of antigens and immunostimulatory adjuvant molecules has shown to be more efficient in inducing antigen-specific immune responses than unconjugated antigens and adjuvants in a vaccine formulation.Citation15–18
We tested vaccine efficacy using the murine 4T1 mammary carcinoma tumor model. 4T1 is a well-established murine TNBC model derived from a spontaneous tumor in BALB/c mouse that shares many characteristics with human breast cancer.Citation19,Citation20 4T1 tumor cells metastasize to organs, such as the lungs, and surgical resection of the primary tumor does not affect the growth of distal metastases, as pulmonary metastasis is the primary cause of death in the 4T1 model.Citation19 Monotherapy with ICI such as anti-CTLA-4 mAb or anti-PD-1 mAb is not effective in this model.Citation21 Consequently, the 4T1 model provides an opportunity to test the efficacy of new immunotherapies in combination with ICIs against metastatic disease with the goal of enhancing the efficacy of these FDA-approved ICIs. In this study we investigated whether personalized TMV vaccine therapy could be used to boost the efficacy of ICI therapy for indications such as TNBC, in which tumor antigens are not well defined. We evaluated vaccine efficacy by examining survival, metastasis, and anti-tumor immunity in the 4T1 TNBC model. Our results suggest that the combination of TMV vaccine immunotherapy and anti-CTLA-4 antibody synergistically reduces metastasis and increases the survival of the mice challenged with 4T1 tumor cells better than either monotherapy.
Materials and methods
Antibodies
Purified hamster anti-mouse CD80 (Clone 16-10A1), rat-anti-mouse CD80 (Clone 1G10) and rat anti-mouse IL-12 p40 (Clone C17.8) antibodies used for affinity chromatography and anti-mouse CTLA-4 antibody (Clone 9D9), and anti-mouse PD-L1 antibody (Clone 10 F.9G2), anti-mouse CD4 (Clone GK1.5) and mouse CD8 (Clone YTS 169.4) antibodies for administration into mice were purchased from Bio X Cell (West Lebanon, NH). Fluorochrome-conjugated anti-mouse monoclonal antibody specific for murine CD80 (Clone 16-10A1) was purchased from BD Biosciences (San Diego, CA) and anti-human CD80 (Clone 2D10) was purchased from BioLegend (San Diego, CA). Antibody specific for murine IL-12 p40 (Clone C17.8) was purchased from eBioscience (San Diego, CA) and anti-human IL-12 p40-specific antibody (Clone C11.5) was purchased from BD Biosciences. Anti-human CD3 antibody (Clone OKT3) used for T cell activation was obtained from BD Biosciences. All coating and biotin-conjugated detection antibodies used for IL-2 and IFN-γELISA assays were purchased from BD Biosciences.
Cell lines
Human MDA-MB-231, MDA-MB-453, BT-549, HCC-1187, Jurkat E6.1, NK-92, NK-92MI, and murine 4T1 cells were purchased from ATCC (Manassas, VA). CHO-K1 cell transfectants expressing GPI-ISMs were established in our laboratory as described before.Citation14,Citation22
Production and purification of GPI-proteins
GPI-B7-1 and GPI-IL-12 were expressed in CHO-K1 cells and immunoaffinity purified as previously described.Citation14,Citation22
Mice
Female BALB/cJ mice (6-8-week-old) were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained in accordance with Emory University IACUC approved institutional guidelines and protocols.
Murine TMV vaccine preparation and characterization
4T1 TMVs were prepared and incorporated with GPI-ISMs as previously described.Citation11 Briefly, tumors were grown in the mammary fat pads or hind flanks, excised upon reaching 10 mm in diameter and frozen at −80°C. Tumors were then minced and homogenized using an Omni TH tissue homogenizer along with disposable hard tissue rotor-stator generator probe tips from Omni International (Kennesaw, GA) and centrifuged over a 41% sucrose gradient at 100,000 g. TMVs were collected from the interface, washed, and resuspended in phosphate-buffered saline (PBS). TMV concentration was then determined using a micro BCA assay kit from Thermo Scientific (Rockford, IL). TMVs were then incorporated with GPI-proteins at 2.5 μg of each molecule/100 μg TMV for 3 h at 37°C with gentle rotation. The incorporated TMVs were centrifuged to remove unincorporated GPI-proteins and the pelleted TMVs were resuspended in PBS. Incorporation of GPI-mB7-1 (anti-mouse CD80-APC, Clone 16-10A1) and GPI-mIL-12 (anti-mouse IL-12 p40-PE, Clone C17.8) was evaluated using flow cytometry. FlowJo version 9.7.6 software was used for flow cytometry data analysis from TreeStar (Ashland, OR). The GPI-ISM incorporated TMVs (TMV vaccine) were stored at −80°C until use. TMVs were analyzed for size distribution/particle diameter using a Zetasizer Nano ZS from Malvern Panalytical (Westborough, MA). Scanning electron microscopy was performed by the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University.
Human TMV vaccine preparation
Human TNBC cell lines were grown in bulk and TMVs were prepared from the cell pellets by homogenization and ultracentrifugation as described for murine tumor cell lines and tumor tissue.Citation14 De-identified human breast cancer tumor tissues were obtained from Emory pathology tumor tissue bank and processed as described above. Human GPI-IL-12 and GPI-B7-1 were purified from CHO-K1 cells and incorporated into human TMVs prepared from human TNBC cell lines and breast cancer tumor tissues as described above. Incorporation of GPI-IL-12 and GPI-B7-1 was confirmed by flow cytometry analysis using fluorochrome-conjugated antibodies (data not shown).
Luminex assay for serum cytokines/chemokines
Mice were immunized with the TMV vaccine as described (). Mice were bled on d 5 after the final immunization and serum was collected. Samples were pooled from each group and an eBioscience 36-plex Luminex assay was performed by Charles River Laboratories (Wilmington, MA). Samples were run in duplicate and cytokine/chemokine levels were extrapolated using a standard curve for each protein.
Tumor challenge studies
4T1 cells (2 x 104) were suspended in 100 μl of PBS and injected into the second mammary fat pad. Tumors were resected on d 9–10 upon reaching 3–4 mm in size. Mice were then treated with immunotherapy and followed to assess survival or metastasis. Mice were euthanized when they reached IACUC endpoint for survival studies.
Immunotherapy studies
For survival and metastasis studies, 100 μg of 4T1 TMV vaccine was given s.c. to some groups 2 d after surgical removal of the primary tumor and a booster dose given 9 d after surgery (). For immune response studies (), mice were given three injections of TMV vaccine spaced at 2-week intervals. Immune responses were then evaluated 5 d after the final dose by Luminex assay of the serum.
For survival and metastasis studies, anti-CTLA-4 mAb (Clone 9D9) was given intraperitoneally (i.p.) 1 d following TMV vaccine administration and subsequently every 3 d for a total of 4 doses: dose 1 (200 μg), 2 (100 μg), 3 (100 μg), and 4 (100 μg) in PBS. This dosing schedule was adapted from a study published by Allison and coworkers.Citation23 For immune response studies (), anti-CTLA-4 mAb was given twice after each TMV vaccine administration: on d1 (200 μg) and d4 (100 μg) after the first dose of vaccine and then 100 μg doses following the second and third administration of the vaccine for a total of 6 injections of anti-CTLA-4 mAb. Anti-PD-L1 mAb (Clone 10 F.9G2) was given i.p. 1 d following TMV vaccine and subsequently every 3 d for a total of 4 doses (200 μg/dose in PBS).Citation23
Clonogenic assay
For the clonogenic assay, the TMV vaccine and anti-CTLA-4 mAb therapies were given as described above. A clonogenic assay was performed as previously described.Citation24 Briefly, mice were sacrificed on d 28–35 post tumor challenge, lungs were digested and processed to a single cell suspension using collagenase type IV (Sigma-Aldrich) in a 2-h incubation at 37°C and then passed through a 70 μm cell strainer. Cells were washed and suspended in DMEM with 10% FBS (Hyclone) containing 6-thioguanine (Sigma-Aldrich). Serial dilutions were made and cultures grown for 7 d at 37°C, 5% CO2. 4T1 cells are resistant to 6-thioguanine, but normal lung cells and fibroblasts are sensitive and fail to survive, leading to the formation of distinct 4T1 colonies. Colonies were visualized after staining with 0.5% crystal violet in ethanol and counted.Citation25
In vitro immune response assays
Human GPI-anchored B7-1 and IL-12 incorporated into human TMVs were used for stimulating IL-2 and IFN-γ secretion by Jurkat E6.1 and NK-92 cells. Human NK-92 cells (2 x 105/ml) were stimulated with TMVs containing incorporated GPI-IL-12 (40 μg/ml TMV) from various human TNBC cell lines or de-identified patient-derived breast cancer tumor tissue for 48 h. Supernatant was collected and IFN-γ was measured by ELISA. Human Jurkat E6.1 cells (6 x 105/ml) were stimulated with TMVs with/without GPI-B7-1 (40 μg/ml TMV) from various human TNBC cell lines or patient-derived breast cancer tumor tissue in combination with anti-CD3 (Clone OKT3, 1 μg/ml) for 24 h. Supernatant was collected and IL-2 was measured by ELISA.
Statistical analysis
A log-rank (Mantel-Cox) test was used to evaluate Kaplan-Meier survival curves. A Student’s t test (unpaired, 2-tailed) or Mann–Whitney U test was used to compare control and experimental groups. P values <.05 were deemed statistically significant (*p < .05, **p < .01). GraphPad Prism 6 software was used to analyze data.
Results
TMV vaccine preparation and characterization
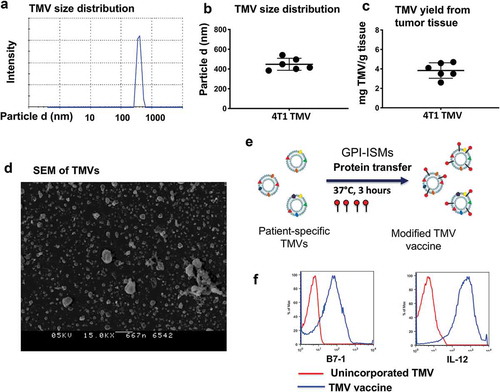
TMVs were prepared from surgically resected tumors from syngeneic mice, characterized using physicochemical analytical methods for yield and size, and then incorporated with purified GPI-B7-1 and GPI-IL-12 by protein transfer. The data suggest that TMVs are uniform in size ranging from 300 to 500 nm () and the yield ranged from 3 to 5 mg per gram of starting tissue (). Electron microscopy analysis also confirmed the particulate nature of the TMVs (). Efficient incorporation of GPI-B7-1 and GPI-IL-12 by protein transfer onto membranes () was confirmed via flow cytometry ().
Figure 1. TMV vaccine preparation and characterization from 4T1 tumor tissue. (a) Zetasizer analysis of TMVs, (b) Size of 4T1 TNBC TMVs, (c) Yield of TMV, (d) Scanning electron microscopy (SEM) of TMV from 4T1 breast cancer tumor tissue, (e) Schematics of incorporating GPI-ISMs onto TMVs by protein transfer approach and (f) Flow cytometry analysis of TMV vaccine showing protein transfer-mediated incorporation of GPI-B7-1 and GPI-IL-12 onto 4T1 TMV
TMV vaccine and anti-CTLA-4 mAb combination immunotherapy induces a cytokine response
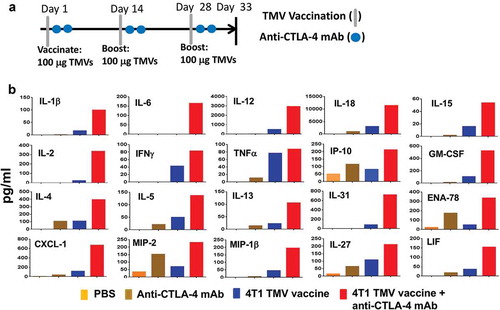
Serum cytokine levels in response to vaccination are used as a biomarker of cellular immune response and toxicity of a vaccine. In order to determine whether TMV vaccine immunotherapy with and without anti-CTLA-4 mAb induces cellular immune responses, we collected serum 5 d after the last booster dose and analyzed serum cytokines and chemokines using a 36-plex Luminex assay (eBioscience) at Charles River Labs (). TMV vaccination induced both Th1 and Th2 cytokines, such as TNF-α, IFN-γ, IL-2, IL-4, IL-12, IL-18, and IL-31 in the serum. In addition, combination with anti-CTLA-4 mAb increased serum levels of these proteins. Almost no measurable response was observed in mice receiving PBS or anti-CTLA-4 mAb alone. Additionally, combination therapy induced a greater increase of the chemokines ENA-78, CXCL-1, MIP-2, MIP-1β, and LIF than TMV immunotherapy alone (data not shown). No changes were observed for IL-1α, G-CSF, M-CSF, IFN-α, IL-3, IL-10, IL-9, Eotaxin, MCP-3, MIP-1α, RANTES, or MCP-1 (data not shown). However, these cytokine and chemokine levels are far below what would be considered a “cytokine storm”Citation26 suggesting that immune pathology would not be induced by TMV vaccine treatment or TMV/anti-CTLA-4 mAb combination therapy. The lack of robust increase in cytokine/chemokines in the TMV vaccine and the combination groups may be due to the membrane-anchored nature of GPI-ISMs, which might have restricted systemic activation and release of cytokine/chemokine in the circulation. No significant anti-4T1 antibody titers were detectable following immunotherapy, even when TMV vaccination was combined with anti-CTLA-4 mAb (data not shown), suggesting that either TMV therapy or combination therapy does not enhance B cell responses against 4T1 cells or 4T1 cells do not have a cell surface B cell antigen.
Figure 2. Co-administration of anti-CTLA-4 mAb with immunotherapy increases key immune mediators in serum. (a) Immunotherapy protocol design. BALB/c mice (n = 5) were immunized three times 14 d apart with PBS, anti-CTLA-4 mAb, TMV vaccine, or TMV vaccine + anti-CTLA-4 mAb. Serum was collected and pooled on d 5 after the final immunization. (b) eBioscience 36-Plex Luminex assay was performed by Charles River Laboratories. The values were an average of duplicate readings
TMV vaccine immunotherapy in combination with anti-CTLA-4 mAb increases the survival of mice after resection of primary tumor
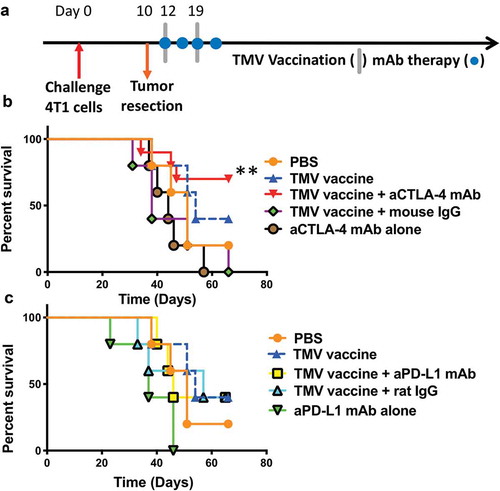
Next, we determined whether the increased immune response induced by TMV vaccine immunotherapy conferred protection against metastatic disease. Mice were inoculated with 4T1 cells orthotopically and the primary tumors were resected on d 10 (), by which time spontaneous metastasis had occurred.Citation19 These mice were then treated with the TMV vaccine immunotherapy in the absence or presence of anti-CTLA-4 mAb or anti-PD-L1 mAb. Our results show that only 20% of mice survived in the control groups by d 60. Anti-CTLA-4 mAb therapy alone provided no significant increase in survival, as no mice survived by d 60. However, in the group receiving TMV vaccine and anti-CTLA-4 antibody combination therapy, 70% of the mice (7/10) survived for more than 66 d (). A log-rank analysis (Mantel–Cox test) demonstrated that a significant increase in survival was observed in the combination group including TMV immunotherapy and anti-CTLA-4 mAb treatment in comparison to anti-CTLA-4 mAb therapy or TMV immunotherapy alone. Interestingly, combination therapy with anti-PD-L1 mAb did not increase survival (), despite the upregulated expression of PD-L1 on 4T1 cells following exposure to IFN-γ in vitro (data not shown). Together, this data shows that TMV immunotherapy in combination with anti-CTLA-4 antibody therapy is more effective at preventing 4T1 metastasis than either monotherapy.
Figure 3. A combination of TMV vaccine and anti-CTLA-4 mAb enhances the survival of mice after resection of primary tumor. (a) Tumor challenge/resection and immunotherapy protocol design. (b) Kaplan-Meier survival curves of control and anti-CTLA-4 mAb treated groups. Comparison of TMV vaccine + anti-CTLA-4 mAb and anti-CTLA-4 mAb alone yields p = .0074, n = 5–10. Comparison of TMV vaccine + anti-CTLA-4 mAb and TMV vaccine + Mouse IgG yields p = .0105, n = 5–10. Log-rank (Mantel-Cox) test used for comparison analysis. (c) Kaplan-Meier survival curves of control and anti-PD-L1 mAb treated groups. **p ≤ 0.01.
TMV vaccine immunotherapy in combination with anti-CTLA-4 antibody post tumor challenge and surgical resection of the primary tumor reduces lung metastasis
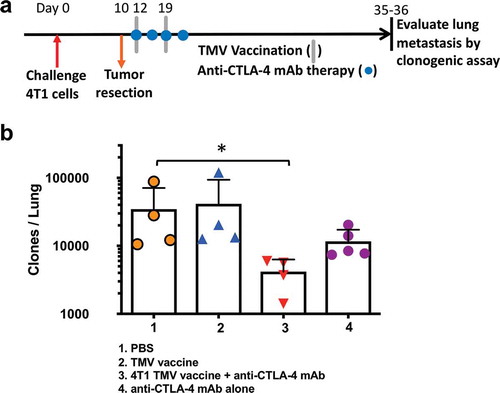
The primary cause of death in the 4T1 model is pulmonary metastasis.Citation19 Therefore, we hypothesized that the increased survival in mice co-administered with the TMV vaccine immunotherapy and anti-CTLA-4 antibody therapy may be due to a reduction in lung metastasis. To test this, we challenged BALB/c mice orthotopically with 4T1 cells and then resected the primary tumor on d10. TMV vaccine and anti-CTLA-4 mAb were administered post resection as described in . After 35–36 d, the mice were sacrificed, and a clonogenic assay was performed as described in Methods to assess the extent of lung metastasis. Only the group receiving the combination therapy demonstrated a significant reduction in the number of metastatic tumor cells present in the lung (), exhibiting a ten-fold reduction in the number of metastatic cells detected compared to PBS-treated controls (~20,000 colonies in control PBS group versus to ~2,500 colonies in combination therapy group).
Figure 4. Reduction in metastasis to the lung in mice receiving a combination of TMV vaccine and anti-CTLA-4 mAb. (a) Tumor challenge, resection, and immunotherapy protocol design. (b) Metastasis evaluated using a clonogenic assay at d 35–36 post orthotopic challenge. Statistical differences were calculated using Mann–Whitney U test *p < .05, n = 4–5.
CD8+ T cells are critical for the control of metastasis in the 4T1 breast cancer model
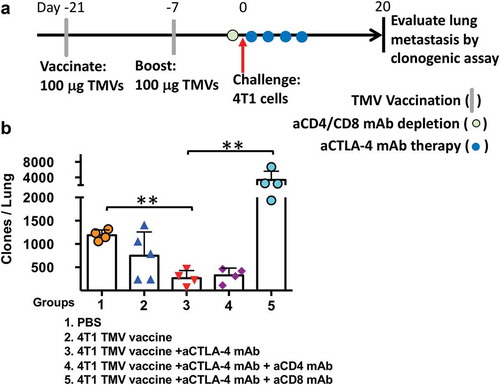
To determine the role of CD8+ and CD4+ T cells in reducing 4T1 lung metastasis in the TMV vaccine and anti-CTLA-4 mAb combination treatment, T cell depletion experiments were carried out (). The depletion of CD4+ T cells in the vaccinated mice prior to tumor challenge yielded no difference in control of metastasis. However, CD8+ T cell depletion resulted in the loss of 4T1 metastasis control, with a 10-fold increase in comparison to mice treated with the combination therapy. Interestingly, the metastasis in the CD8+ T cell-depleted mice was ~4-fold higher than the control mice (), suggesting that CD8+ T cells primed by the tumor may also play a role in controlling the 4T1 metastasis in the absence of vaccination.
Figure 5. The depletion of CD8 T cells abrogates the impact of immunotherapy on 4T1 metastasis. (a) Immunotherapy, cell depletion, and tumor challenge protocol design. (b) Metastasis evaluated using a clonogenic assay at d 20 post orthotopic challenge with 4T1 cells. Statistical differences were calculated using t-test. **p < .01, n = 4–5.
Human GPI-ISMs incorporated onto TMVs prepared from human TNBC cell lines and human breast cancer tissue retains functional activity
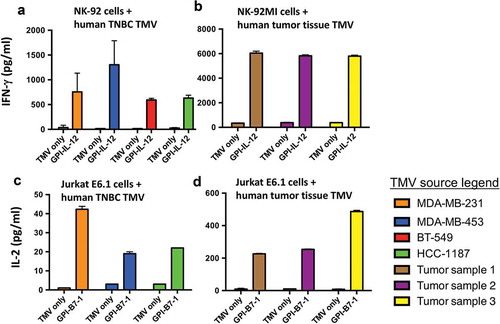
In order to address whether human GPI-ISMs retained their activity in human TNBC TMVs, we prepared TMVs from established human TNBC cell lines and human breast cancer tumor tissues. Human GPI-ISMs incorporated into these TMVs stimulated human NK-92 cells and Jurkat E6.1 cells effectively compared to unincorporated TMVs (). TMVs incorporated with human GPI-IL-12 induced IFN-γ secretion by NK-92 cells (). To confirm whether GPI-ISMs incorporated onto TMVs prepared from patient-derived breast cancer tissue can also stimulate human immune cells, we prepared TMVs from three tumor samples and incorporated with GPI-ISMs. Human breast cancer TMVs incorporated with human GPI-IL-12 also stimulated IFN-γ secretion by NK-92MI cells (). Stimulation of Jurkat E6.1 with unmodified TMV did not result in the production of IL-2, even in the presence of anti-CD3 mAb; however, incorporation of GPI-B7-1 effectively stimulated IL-2 production in the presence of anti-CD3 (). Human breast cancer TMVs incorporated with human GPI-B7-1 also stimulated IL-2 secretion by Jurkat E6.1 cells (). These results demonstrate that GPI-ISMs incorporated into human breast cancer TMVs retain their functional activity and suggest that TMV vaccines can be developed from human breast cancer tumor tissue for translational studies.
Figure 6. Human GPI-ISMs incorporated into TMVs prepared from human TNBC cell lines or breast cancer tumor tissues are able to stimulate PBMC, NK-92, and Jurkat E6.1 cells. (a) Human NK-92 cells and (b) human NK-92MI cells (2 x 105/ml) were stimulated with TMVs containing incorporated human GPI-IL-12 (40 μg/ml TMV) from various human TNBC cell lines (a) or patient-derived breast cancer tumor tissue (b) for 48 h. Supernatant was collected and IFN-γ was measured by ELISA. Human Jurkat E6.1 cells (6 x 105/ml) were stimulated with TMVs with/without GPI-B7-1 (40 μg/ml TMV) from various human TNBC cell lines (c) or patient-derived breast cancer tumor tissue (d) in combination with anti-human CD3 mAb for 24 h. Supernatant was collected and IL-2 was measured by ELISA. IL-2 levels were undetectable in Jurkat E6.1 with anti-CD3 alone. Values were average of triplicates and represented as Mean ± SD
Discussion
Previous studies by our group have shown that TMV vaccine-induced anti-tumor immunity is capable of inhibiting tumor growth in an antigen-dependent manner and inducing immune memory formation in a HER2+ breast cancer model.Citation14 Further, irradiated tumor cell vaccines containing a combination of membrane-anchored B7-1 and IL-12 induced stronger anti-tumor immunity compared to the vaccine containing either one of the molecules in murine breast cancer models.Citation7,Citation27,Citation28 In this study we have shown that the TMV vaccine immunotherapy can be used to treat poorly immunogenic tumors with undefined tumor-associated antigens when used in combination with anti-CTLA-4 blockade therapy. In the 4T1 TNBC breast cancer model, TMV vaccine/anti-CTLA-4 mAb combination therapy resulted in increased overall survival, reduced lung metastasis, as well as enhanced CD8 T cell responses. Together, the data suggest that TMV tumor tissue-derived vaccination as a potential strategy for treating cancers that exhibit a high degree of heterogeneity and lack well-defined tumor antigens, such as TNBC. It is not clear why the combination of TMV vaccine and mouse IgG further reduces survival in comparison to TMV vaccine alone or TMV vaccine in combination with rat IgG. This observation warrants further investigation with larger cohorts. In contrast to our observations in other murine tumor models,Citation11,Citation14 the TMV vaccine therapy alone did not protect mice against 4T1 tumor challenge. This may be due to the low level of poorly immunogenic antigens, which could be overcome through a different dosing schedule, dose of TMV vaccine, or combination therapy with an immune checkpoint inhibitor. Further studies are being conducted to investigate these possibilities.
There are many cancer vaccine immunotherapy strategies being tested in preclinical models. However, most rely on the presence of a known antigen, determination of mutated neoepitopes in individual patients that can elicit an immune response,Citation29 or development of cell lines from each patient. Unfortunately, these remain labor-intensive and time-consuming. Unlike these approaches, our TMV vaccine can be prepared from frozen tumor tissue without antigen identification or cell line establishment, facilitating vaccine development time and reducing labor, thereby simplifying clinical application. The use of frozen tumor tissue and TMV vaccines makes the logistics of preparing and transporting vaccines simpler in the clinical setting. Human GPI-ISMs not only remain at the surface of patient-derived TMV after protein transfer, but they also retain their functional activity, which can be measured by functional readouts such as IFN-γ and IL-2 secretions by PBMCs or lymphocyte cell lines. This further confirms the applicability of the approach for the clinical development of the TMV vaccine.
The present study also found that, while the TMV vaccine synergized with CTLA-4 blockade, it did not synergize with anti-PD-L1 mAb treatment. It is known that these two checkpoint blockade therapies target distinct immune pathways.Citation30 Recent studies have shown that blocking CTLA-4 signaling can boost the CD4+ T cell effector population, reduce regulatory T cells, and engage several subsets of CD8+ T cells,Citation30 while PD-1 signaling blockade predominantly induces the expansion of exhausted CD8+ T cells by targeting “stem” like progenitors.Citation31 Another study using dual checkpoint blockade observed that anti-CTLA-4 and anti-PD-1 antibodies expand and activate unique T cell repertoires in a murine TNBC model.Citation32 This study also postulated that anti-CTLA-4 antibody primarily functions to prime naïve T cells in the lymph node and reduce Treg numbers in the tumor, while anti-PD-1 antibody enhances the anti-tumor T cell functions.Citation32 The 4T1 model is highly immunosuppressive, which significantly dampens initial T cell priming and activation.Citation19 Therefore, it is possible that anti-CTLA-4 antibody blocks the negative signaling during vaccine-induced T cell priming. This may explain why we only observed a synergistic effect with anti-CTLA-4 antibody and not anti-PD-L1 antibody. Although PD-L1 expression on tumor cells often correlates with response rate to anti-PD-L1 in human TNBC, not all patients whose tumors are PD-L1+ve respond to ICI therapy, suggesting that tumors may be using other immunosuppressive mechanisms.Citation4 It is also possible that in vivo expression of PD-L1 was diminished or lost on 4T1 tumor cells, making them non-responsive to anti-PD-L1 therapy.
The anti-CTLA-4 mAb Yervoy, approved for metastatic melanoma, enhances the overall response rate in 10.9% of patients.Citation33 However, it is not known why a majority of patients fail to respond to treatment. It has been shown that vaccination with tumor-derived antigens enhances the efficacy of anti-CTLA-4 mAb therapy in animal models, suggesting that a larger percentage of patients would benefit from Yervoy treatment if additional stimulation of the anti-tumor immune response could be elicited.Citation21,Citation34,Citation35 Thus, our vaccination approach provides an opportunity to expand the use of anti-CTLA-4 mAb in patients who are resistant to the therapy.
In summary, the preclinical data in this study suggest that a combination of our TMV-vaccine immunotherapy along with anti-CTLA-4 mAb therapy work together to suppress metastasis, stimulate CD8 T cell immunity, and enhance overall survival in the highly aggressive, poorly immunogenic 4T1 murine breast cancer model. In addition, this protection is dependent upon CD8+ T cell immunity, not anti-tumor antibody responses.
TNBC is a highly heterogeneous disease; therefore, the 4T1 model may only represent a subset of human TNBC cases. The TMV vaccine approach used in our studies employs plasma membrane vesicles derived from tumor cells which may not fully capture the mutant tumor antigens present in the cytoplasm. However, we expect these mutant proteins are processed and presented on membrane-associated MHC class I molecules. Our novel combinatorial approach may prove to be a promising treatment option for TNBC patients that critically need personalized approaches to help combat the heterogeneous nature of their disease and prevent relapse.
Disclosure of Potential Conflicts of Interest
The corresponding author (PS) holds shares in Metaclipse Therapeutics Corporation, a company that is planning to use GPI-anchored molecules to develop membrane-based breast cancer vaccines in the future as suggested in the current manuscript.
Acknowledgments
This work was supported by NCI/NIH SBIR Phase I and Phase II grants (CA176920 and CA221559 to C.P. and P.S), NCI/NIH R01 grant (R01 CA202763 to P.S. and C.P.), a diversity supplemental award to LEM (R01 CA202763-S), and a seed grant from the Georgia Research Alliance based in Atlanta, Georgia. The authors thank Dr. D’Souza laboratory at the Mercer University, Atlanta, GA for allowing us to use their Malvern Zetasizer. We also gratefully acknowledge the services of Emory University Robert P. Apkarian Integrated Electron Microscopy Core facility for the SEM analysis of TMVs and critical reading of the manuscript by Dr. Kristen Jacobsen, Metaclipse Therapeutics Inc.
Additional information
Funding
References
- Boyle P. Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol. 2012;23(Suppl 6):vi7–12. doi:https://doi.org/10.1093/annonc/mds187.
- Dietze EC, Sistrunk C, Miranda-Carboni G, O’Regan R, Seewaldt VL. Triple-negative breast cancer in African-American women: disparities versus biology. Nat Rev Cancer. 2015;15:248–54. doi:https://doi.org/10.1038/nrc3896.
- Dawson SJ, Provenzano E, Caldas C. Triple negative breast cancers: clinical and prognostic implications. Eur J Cancer. 2009;45(Suppl 1):27–40. doi:https://doi.org/10.1016/S0959-8049(09)70013-9.
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im S-A, Shaw Wright G, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–21. doi:https://doi.org/10.1056/NEJMoa1809615.
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–99. doi:https://doi.org/10.1038/nature10933.
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi:https://doi.org/10.1056/NEJMoa1113205.
- Selvaraj P, Yerra A, Tein L, Shashidharamurthy R. Custom designing therapeutic cancer vaccines: delivery of immunostimulatory molecule adjuvants by protein transfer. Hum Vaccin. 2008;5:1–5.
- Croce MV, Segal-Eiras A. The use of carbohydrate antigens for the preparation of vaccines for therapy in breast cancer. Drugs Today (Barc). 2002;38:759–68. doi:https://doi.org/10.1358/dot.2002.38.11.820135.
- Croce MV, Rabassa ME, Pereyra A, Segal-Eiras A. Differential expression of MUC1 and carbohydrate antigens in primary and secondary head and neck squamous cell carcinoma. Head Neck. 2008;30:647–57. doi:https://doi.org/10.1002/hed.20756.
- Livingston PO, Ragupathi G, Musselli C. Autoimmune and antitumor consequences of antibodies against antigens shared by normal and malignant tissues. J Clin Immunol. 2000;20:85–93. doi:https://doi.org/10.1023/A:1006697728855.
- McHugh RS, Nagarajan S, Wang YC, Sell KW, Selvaraj P. Protein transfer of glycosylphosphatidylinositol-B7-1 into tumor cell membranes: A novel approach to tumor immunotherapy. Cancer.Res. 1999;59:2433–37.
- Poloso NJ, Nagarajan S, Bumgarner GW, Selvaraj P. Development of therapeutic vaccines by direct modification of cell membranes from surgically removed human tumor tissue with immunostimulatory molecules. Vaccine. 2001;19:[pii] 2029–2038. doi:https://doi.org/S0264-410X(00)00424-2.
- Nagarajan S, Selvaraj P. Human tumor membrane vesicles modified to express glycolipid-anchored IL-12 by protein transfer induce T cell proliferation in vitro: A potential approach for local delivery of cytokines during vaccination. Vaccine. 2006;24:2264–74. doi:https://doi.org/10.1016/j.vaccine.2005.11.045.
- Patel JM, Vartabedian VF, Bozeman EN, Caoyonan BE, Srivatsan S, Pack CD, Dey P, D'Souza MJ, Yang L, Selvaraj P. Plasma membrane vesicles decorated with glycolipid-anchored antigens and adjuvants via protein transfer as an antigen delivery platform for inhibition of tumor growth. Biomaterials. 2015;74:231–44. doi:https://doi.org/10.1016/j.biomaterials.2015.09.031.
- Liu G, Tarbet B, Song L, Reiserova L, Weaver B, Chen Y, Li H, Hou F, Liu X, Parent J, et al. Immunogenicity and efficacy of flagellin-fused vaccine candidates targeting 2009 pandemic H1N1 influenza in mice. PLoS One. 2011;6(6):e20928. doi:https://doi.org/10.1371/journal.pone.0020928.
- Zhu M, Ding X, Zhao R, Liu X, Shen H, Cai C, Ferrari M, Wang HY, Wang R-F. Co-delivery of tumor antigen and dual toll-like receptor ligands into dendritic cell by silicon microparticle enables efficient immunotherapy against melanoma. J Controlled Release. 2018;272(72–82):72–82. doi:https://doi.org/10.1016/j.jconrel.2018.01.004.
- Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440(808–812):808–12. doi:https://doi.org/10.1038/nature04596.
- Bates JT, Graff AH, Phipps JP, Grayson JM, Mizel SB. Enhanced antigen processing of flagellin fusion proteins promotes the antigen-specific CD8+ T cell response independently of TLR5 and MyD88. J Immunol. 2011;186:6255–62. doi:https://doi.org/10.4049/jimmunol.1001855.
- Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol. 2001;20:20.2. doi:https://doi.org/10.1002/0471142735.im2002s39.
- Kaur P, Nagaraja GM, Zheng H, Gizachew D, Galukande M, Krishnan S, Asea A. A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC Cancer. 2012;12:120. doi:https://doi.org/10.1186/1471-2407-12-120.
- Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun. 2013;13:5.
- Patel JM, Kim MC, Vartabedian VF, Lee YN, He S, Song JM, Choi HJ, Yamanaka S, Amaram N, Lukacher A, et al. Protein transfer-mediated surface engineering to adjuvantate virus-like nanoparticles for enhanced anti-viral immune responses. Nanomed. 2015;11(1097–1107). doi:https://doi.org/10.1016/j.nano.2015.02.008
- Waitz R, Solomon SB, Petre EN, Trumble AE, Fasso M, Norton L, Allison JP. Potent induction of tumor immunity by combining tumor cryoablation with anti-CTLA-4 therapy. Cancer Res. 2012;72:430–39. doi:https://doi.org/10.1158/0008-5472.CAN-11-1782.
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(2315–2319). doi:https://doi.org/10.1038/nprot.2006.339.
- Song Y, Xue L, Du S, Sun M, Hu J, Hao L, Gong L, Yeh D, Xiong H, Shao S, et al. Caveolin-1 knockdown is associated with the metastasis and proliferation of human lung cancer cell line NCI-H460. Biomed Pharmacother. 2012;66(6):439–47. doi:https://doi.org/10.1016/j.biopha.2012.03.001.
- Yiu HH, Graham AL, Stengel RF. Dynamics of a cytokine storm. PLoS One. 2012;7:e45027. doi:https://doi.org/10.1371/journal.pone.0045027.
- Bozeman EN, Cimino-Mathews A, Machiah DK, Patel JM, Krishnamoorthy A, Tien L, Shashidharamurthy R, Selvaraj P. Expression of membrane anchored cytokines and B7-1 alters tumor microenvironment and induces protective antitumor immunity in a murine breast cancer model. Vaccine. 2013;31:2449–56. doi:https://doi.org/10.1016/j.vaccine.2013.03.028.
- Cimino AM, Palaniswami P, Kim AC, Selvaraj P. Cancer vaccine development: protein transfer of membrane-anchored cytokines and immunostimulatory molecules. Immunol.Res. 2004;29:231–40. doi:https://doi.org/10.1385/IR:29:1-3:231.
- Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–39. doi:https://doi.org/10.1038/s41586-018-0792-9.
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. Distinct cellular mechanisms underlie Anti-CTLA-4 and Anti-PD-1 checkpoint blockade. Cell. 2017;170(6):1120–1133.e1117. doi:https://doi.org/10.1016/j.cell.2017.07.024.
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–21. doi:https://doi.org/10.1038/nature19330.
- Crosby EJ, Wei J, Yang XY, Lei G, Wang T, Liu C-X, Agarwal P, Korman AJ, Morse MA, Gouin K, et al. Complimentary mechanisms of dual checkpoint blockade expand unique T-cell repertoires and activate adaptive anti-tumor immunity in triple-negative breast tumors. Oncoimmunology. 2018;7(5):e1421891. doi:https://doi.org/10.1080/2162402x.2017.1421891.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi:https://doi.org/10.1056/NEJMoa1003466.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–36. doi:https://doi.org/10.1126/science.271.5256.1734.
- Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011;241:104–18. doi:https://doi.org/10.1111/j.1600-065X.2011.01007.x.