
ABSTRACT
T cell immunity, such as CD4 and/or CD8 T cell responses, plays a vital role in controlling the virus infection and pathological damage. Several studies have reported SARS-CoV-2 proteins could serve as ideal vaccine candidates against SARS-CoV-2 infection by activating the T cell responses. In the current study, based on the SARS-CoV-2 sequence and distribution of host human leukocyte antigen (HLA), we predicted the possible epitopes for the vaccine against SARS-CoV-2 infections. Firstly, the current study retrieved the SARS-CoV-2 S and N protein sequences from the NCBI Database. Then, using the Immune Epitope Database Analysis Resource, we predicted the CTL epitopes of the SARS-CoV-2 S and N proteins according to worldwide frequency distributions of HLA-A, -B, and -C alleles (>1%). Our results predicted 90 and 106 epitopes of N and S proteins, respectively. Epitope cluster analysis showed 16 and 34 respective clusters of SARS-CoV-2 N and S proteins, which covered 95.91% and 96.14% of the global population, respectively. After epitope conservancy analysis, 8 N protein epitopes and 6 S protein epitopes showed conservancy within two SARS-CoV-2 types. Of these 14 epitopes, 13 could cover SARS coronavirus and Bat SARS-like coronavirus. The remaining epitope (KWPWYIWLGF1211-1220) could cover MERS coronavirus. Finally, the 14-epitope combination could vaccinate 89.60% of all individuals worldwide. Our results propose single or combined CTL epitopes predicted in the current study as candidates for vaccines to effectively control SARS-CoV-2 infection and development.
Introduction
Beginning December 2019, a cluster of acute respiratory disease, known as novel coronavirus-infected pneumonia (COVID-19), occurred in Wuhan, Hubei Province, China.Citation1–3
In January 2019, SARS-CoV-2 was identified and confirmed as the cause of COVID-19.Citation4 Then, the full genome sequences of SARS-CoV-2 (NC_045512.2) were published in the National Center for Biotechnology Information (NCBI) website. The full-genome sequence and phylogenetic analysis indicated that SARS-CoV-2 forms a clade that is distinct from the beta coronaviruses associated with human severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS).Citation3,Citation4 Phylogenetic analysis also revealed that the gene sequence of SARS-CoV-2 is about 88% identical to that of bat SARS-like coronavirus ZXC21 (bat-SL-CoVZXC21, accession no. MG772934.1) and ZC45 (MG772933.1),Citation5 which indicated SARS-CoV-2 has closer homology to bat SARS-like coronavirus and bats could be the primary source of SARS-CoV-2.Citation6,Citation7
The antigenicity of coronaviruses, such as SARS-CoV, seems to be largely dependent upon two viral proteins that comprise the nucleocapsid protein (N) and the spike protein (S). The N protein, which is a nucleocapsid phosphoprotein, has been demonstrated to be highly immunogenic and seems to be an important component of the humoral response to SARS-CoV.Citation8,Citation9 The S protein, which is a surface glycoprotein, is a large type-I transmembrane glycoprotein that is not only responsible for receptor binding and membrane fusion, but also serves as a potent immunogen that induces neutralizing antibodies.Citation8,Citation10 Whole-sequence alignment showed about 79% sequence identity between SARS-CoV-2 and SARS-CoV.Citation5 Thus, the SARS-CoV-2 S and N proteins could serve as target antigens for immune intervention protocols against coronaviruses.
T cell immunity, such as CD4 and/or CD8 T cell responses, plays a vital role in controlling the SARS-CoV infection and/or pathological damage after infection with MERS-CoV.Citation11,Citation12 T cell responses have been shown to provide long-term protection.Citation13–15 Several studies reported that T cells had the strongest immunogenicity to structural proteins in peripheral blood mononuclear cells of convalescent SARS-CoV patients.Citation16,Citation17 Based on the high avidity of cytotoxic T lymphocytes (CTLs), CTLs are considered the major eradicators of viral infections through adaptive immune response. Several studies showed that the presented CTL epitopes of S and N proteins in the context of human leukocyte antigen (HLA) alleles will aid in characterizing the virus control mechanisms and immunopathology of SARS-CoV infection and could provide a new strategy to develop an epitope-based vaccine for SARS.Citation18–22 For example, in 2007 and 2008, Cheung et al. predicted the SARS N protein peptide sequence for human MHC class I binding and screened for potential CTL epitopes to control the SARS-associated coronavirus infection in vitro and in vivo.Citation18,Citation23 In 2020, Ibrahim and Kafi used a computational approach for vaccine design to search for candidate epitopes to control MERS-CoV infections.Citation24 But the epitope vaccine method is only in the research stage, and there is no relevant clinical trial report for coronavirus. However, for other viruses, the peptides vaccines have entered clinical trial, such as HPV peptide vaccines.Citation25
When a virus infects cells, the viral antigens are presented to the host immune system through the antigen processing machine (APM). The APM is composed of a proteasome (where the antigens are degraded into peptides), transporters associated with antigen presentation (TAPs, which are responsible for translocating peptide precursors), endoplasmic reticulum aminopeptidases (ERAPs, which trim peptides to fit HLA molecules), and the major histocompatibility complex (HLA, which presents antigen peptides to the cell surface).Citation26,Citation27 The CTL epitopes bind to the cleft of various HLA-I molecules through features embedded in the peptide sequence and, more specifically, in anchor residues of HLA-I molecules.Citation28 Then, the HLA-I antigen processing system plays important roles in eliminating the infected cells. However, epitope-based vaccines are limited by HLA specificity, as HLA molecules are highly polymorphic. Therefore, it may be difficult to produce an epitope-based vaccine that is effective in patients with different HLA molecules, thus making it impractical for large-scale vaccination programs.Citation28
In the current study, based on the distribution characteristics of HLA alleles across all populations, we predicted putative CTL epitopes of the novel coronavirus (SARS-CoV-2) N and S proteins using immunoinformatic methods, which combine predictors of proteasomal processing, TAP transport, and MHC binding to produce an overall score indicating the intrinsic potential of each peptide as a T cell epitope. Our results provide likely candidate CTL epitopes or combinations thereof for vaccine development to effectively control SARS-CoV-2.
Methods
Sequence retrieval of SARS-CoV-2 N and S proteins
The ID of SARS-CoV-2 (NC_045512.2) was retrieved from NCBI (https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.2).
Epitope prediction
T cell CTL epitopes of the SARS-CoV-2 N and S proteins were predicted using the Proteasomal cleavage/TAP transport/MHC class I combined predictor, which combines predictors of proteasomal processing, TAP transport, and MHC binding to produce a total score indicating the intrinsic potential of each peptide as a T cell epitope. Based on HLA allele data from the Immune Epitope Database (IEDB) Analysis Resource (http://tools.iedb.org/processing/), only HLA alleles occurring in at least 1% of the human population or with an allele frequency of 1% or more were selected. This prediction method is recommended by IEDB. The total score combines the proteasomal cleavage, TAP transport, and MHC binding predictions, which predicts a quantity proportional to the amount of peptide presented by MHC molecules on the cell surface. As the prediction method recommended, high score equal to the high efficiency of the epitope presented by MHC molecules. The total score was used as the cut off value for epitope selection in the present study.
Epitope cluster analysis
Based on sequence identity, the predicted epitopes were grouped into clusters using Epitope Cluster Analysis (http://tools.iedb.org/cluster/). A cluster is defined as a group of sequences having a sequence similarity greater than the specified minimum sequence identity threshold. An identity set percentage means that any member of the cluster will be at least the set percentage identical to at least one member of the cluster.Citation29 In the current study, a cluster was defined as a group of sequences that have a sequence similarity >70% minimum sequence identity threshold, which is the default of the cluster analysis (http://tools.iedb.org/cluster/help/)
Population coverage
The population coverage method calculates the fraction of individuals predicted to respond to a given set of epitopes with known MHC restrictions (http://tools.iedb.org/population/). This calculation is based on HLA genotypic frequencies assuming non-linkage disequilibrium between HLA loci. According to the results, we selected >95% population coverage as the threshold value.
The blast analysis of the 13 coronavirus N and S proteins
Blast method was used to analyze identity proportion of the N and S protein sequences between NC_045512.2 (Wuhan seafood market pneumonia virus isolate Wuhan-Hu-1) and other 12 coronaviruses which are MT007544.1 (Severe acute respiratory syndrome coronavirus 2 isolate Australia/VIC01/2020), FJ882944.1 (SARS coronavirus ExoN1 isolate P3pp23), NC_004718.3 (SARS coronavirus Tor2), MG987420.1 (Middle East respiratory syndrome-related coronavirus isolate NL13892) and MG021451.1 (Middle East respiratory syndrome-related coronavirus isolate NL13845), NC_006213.1 (Human coronavirus OC43 strain ATCC VR-759), NC_006577.2 (Human coronavirus HKU1), KY983587.1 (Human coronavirus 229E strain HCoV_229E/Seattle/USA/SC3112/2015), NC_005831.2 (Human Coronavirus NL63) which contain all 7 known human coronaviruses, and MG772934.1 (Bat SARS-like coronavirus isolate bat-SL-CoVZXC21), KY417144.1 (Bat SARS-like coronavirus isolate Rs4084), KT444582.1 (SARS-like coronavirus WIV16) which contain none human coronaviruses.
Epitope conservancy analysis
Epitope conservancy analysis was used to calculate the degree of conservancy of an epitope within N and S protein sequence of 13 coronaviruses set at different degrees of sequence identity (http://tools.iedb.org/conservancy/). The degree of conservancy is defined as the fraction of protein sequences containing the epitope at a given identity level that the selected epitopes completely matched at least two human coronaviruses above.
Results
The flow chart of epitope prediction analysis and results is illustrated in .
Figure 1. The flow chart of epitope prediction analysis and results
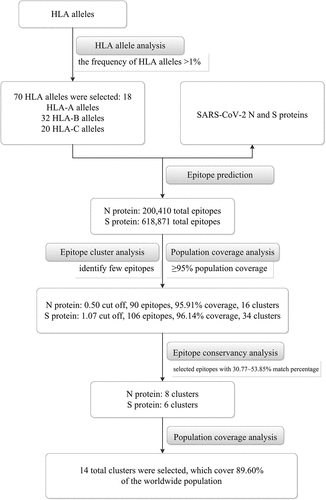
HLA allele analysis
The average frequency of HLA alleles (>1%) across all population samples was selected in the current study (http://tools.iedb.org/processing/help/). In total, 70 HLA alleles were selected for analysis according to the IEDB Analysis Resource (). There were 18 HLA-A alleles, 32 HLA-B alleles, and 20 HLA-C alleles.
Table 1. List of 70 HLA alleles (18 HLA-A alleles, 32 HLA-B alleles, 20 HLA-C alleles) selected occurring in at least 1% of the human population
Epitope prediction
A total of 200,410 epitopes of SARS-CoV-2 N protein were predicted against the 70 alleles of the HLA-A, -B, and -C loci. We selected 0.60, 0.55, 0.50, 0.47, 0.45, 0.40, and 0.30 as the cut off values of total scores for epitope selection, and 71, 82, 90, 99, 103, 110, and 130 epitopes were found within these cut offs. According to the epitope cluster analysis and population coverage results, we chose 0.50 as the cut off value for epitope selection (90 epitopes), as this value identified few epitopes while reaching more than 95% population coverage ().
Table 2. The epitopes number, cluster numbers, and population coverage results of the predicted SARS-CoV-2 N and S proteins
For the SARS-CoV-2 S protein, 618,871 total epitopes were predicted against the 70 alleles of the HLA-A, -B and -C loci. We selected 0.70, 0.80, 0.90, 0.95, 1.00, 1.05, 1.07, 1.10, and 1.15 as cut off values for epitope selection, and 80, 95, 106, 113, 133, 158, 179, 247, and 313 epitopes were found. According to the epitope cluster analysis and population coverage results, 1.07 was chosen as the cut off value for epitope selection (106 epitopes), as this value indicated few epitopes reaching more than 95% population coverage ().
Epitope cluster analysis
Using the selected epitopes (90 epitopes in N protein and 106 epitopes in S protein), the predicted epitopes were grouped into 16 and 34 clusters, respectively. Among the 16 N protein clusters, cluster 1 contained the most epitopes (11 epitopes), which was represented by HLA-A*30:02 (8/11), A*29:02 (8/11), A*30:01 (1/11), A*31:01 (1/11), B*15:25 (1/11), B*15:01 (1/11), A*03:01 (1/11), B*07:02 (1/11), and B*08:01 (1/11). Clusters 9 to 16 contained only one epitope each (). Among the 34 S protein clusters, clusters 1 and 2 contained the most epitopes (5 epitopes each). Cluster 1 was represented by HLA-B*15:25 (2/5), B*15:01 (1/5), A*03:01 (1/5), A*23:01 (1/5), A*24:02 (1/5), and A*30:02 (1/5). Cluster 2 included HLA- A*23:01 (2/5), A*29:02 (1/5) and A*02:01 (1/5). Clusters 16 to 34 contained only one epitope each ().
Table 3. The epitopes sequences, amino acid position and its presented HLA alleles of SARS-CoV-2 N protein by cluster analysis
Table 4. The epitopes sequences, amino acid position and its presented HLA alleles of SARS-CoV-2 S protein by cluster analysis
Population coverage
The HLA allele frequencies and associated data for different individual populations from worldwide studies were provided by the allelefrequencies.net database (http://www.allelefrequencies.net/), which covered 10,656,469 individuals from 1,270 populations in 16 different geographical areas including 115 countries and 21 different ethnicity groups.
The worldwide population coverage of the 90 and 106 epitopes of N and S proteins were 95.91% and 96.14%, respectively (). For the 90 N protein epitopes, the average number of epitope hits/HLA combinations recognized by the population was 5.57, and the minimum number recognized by 90% of the population was 1.51. For the 106 S protein epitopes, the average number of epitope hits/HLA combinations recognized by the population was 7.08, and the minimum number recognized by 90% of the population was 1.56.
The blast analysis of the 13 coronavirus N and S proteins
The BLAST results of N and S proteins between NC_045512.2 and 12 other sequences (MT007544.1, MG772934.1, KY417144.1, KT444582.1, FJ882944.1, NC_004718.3, MG987420.1, MG021451.1, NC_006213.1, NC_006577.2, KY983587.1, and NC_005831.2) selected in the current study are shown in . The percent identities of other human coronaviruses except SARS-CoV-2 and SARS were relatively low (29.43%~50.97% for N protein; 30.78%~37.63% for S protein).
Table 5. The results of comparing N and S protein of SARS-CoV-2 with Bat coronavirus and other human coronaviruses (SARS, MERS, HCoV-OC43, HCoV-HKU1, HCoV-229E and HCoV-NL63)
Epitope conservancy analysis
The identity proportion results of 16 epitopes within 13 N protein sequences above are shown in . The identity is 80%~100% for 16 epitopes within 7 N protein sequences belong to the same subgenus (Sarbecovirus, including SARS-CoV-2). The identity is 23.53%~80% for 16 epitopes within other 6 N protein sequences belong to the different genus (Alphacoronavirus) or subgenus (Merbecovirus). The identity proportion results of 34 epitopes within 13 S protein sequences above are shown in . The identity is higher (60%~100%) for NP13 (KRSFIEDLLF814-823), NP17 (KWPWYIWLGF1211-1220) and NP28 (YEQYIKWPWY1214-1223) within 13 S protein sequences. For the remaining 31 epitopes, the identity is 33.33%~100% for the epitopes within 7 S protein sequences belong to the same subgenus (Sarbecovirus, including SARS-CoV-2); the identity is 28.57%~55.56% for the epitopes within other 6 N protein sequences belong to different genus (Alphacoronavirus) or subgenus (Merbecovirus).
Table 6. The epitope conservancy analysis results for N protein among the 13 coronaviruses
Table 7. The epitope conservancy analysis results for S protein among the 13 coronaviruses
Among the 16 SARS-CoV-2 N protein epitopes, 7 epitopes showed 53.85% (7/13) match between 13 protein sequences (). For NP 3 (MSRIGMEVTPSGTWLTY317-333), this match was 46.15% (6/13). The above 8 epitopes had 23 alleles (). HLA-B*35:01 (3/8) was most frequent in the epitopes, followed by B*15:01 (2/8), B*15:02 (2/8), B*15:25 (2/8), A*30:02 (2/8), and B*53:01 (2/8). The remaining 17 HLA alleles were present in only one epitope. No epitope was found with the N protein of other human coronaviruses except SARS-CoV-2 and SARS. Among the 34 SARS-CoV-2 S protein epitopes, 3 epitopes showed 53.85% (7/13) match between 13 protein sequences (). Two epitopes had a 46.15% (6/13) match. The NP17 epitope (KWPWYIWLGF1211-1220) was found in the S protein of two SARS-CoV-2 and two MERS-related coronaviruses. The above 6 epitopes had 13 alleles (). HLA-A*23:01, B*35:01, B*58:01, C*03:04, and C*03:03 were present in 2 epitopes. The remaining 8 HLA alleles were present in only one epitope.
Table 8. The epitope sequences and its presented HLA alleles of the 14 combination epitopes
Based on the epitope conservancy analysis results, we selected epitopes with 30.77%~53.85% match percentage as epitope candidates, which means the selected epitopes completely matched at least two human coronaviruses. Finally, 14 total epitopes (8 epitopes for N protein, 6 epitopes for S protein) were selected, which cover 89.60% of the worldwide population (). The average number of epitope hits/HLA combinations recognized by the population was 3.22, and the minimum number recognized by 90% of the population was 0.96. For SARS-CoV (FJ882944.1) and SARS-like coronavirus (KT444582.1), 13 epitopes (8 clusters for N protein, 5 clusters for S protein) were covered, which occurred 84.94% of the worldwide population. For Bat SARS-like coronavirus (KY417144.1), 12 epitopes (7 clusters for N protein, 5 clusters for S protein) were covered, which occurred 81.91% of the worldwide population. For Bat SARS-like coronavirus (MG772934.1), 11 epitopes (8 clusters for N protein, 3 clusters for S protein) were covered, which occurred 84.94% of the worldwide population. For MERS (MG987420.1 and MG021451.1), only one epitope (S protein) was covered, which occurred 26.18% of the worldwide population ().
Table 9. The population coverage results (%) of the 14 combination epitopes
Discussion
Modern immunoinformatic methodologies provide new strategies for the design and synthesis of antigen-specific epitope-based vaccines against viral or pathogenic infections. In the current study, according to the SARS-CoV-2 S and N protein sequences, we predicted putative HLA-restricted CTL epitopes using immunoinformatic methods. We found 14-epitope combinations that have 30.77%~53.85% match percentage among the coronavirus sequences covering SARS-CoV-2, other 6 human coronaviruses and other coronavirus species (NC_045512.2, MT007544.1, MG772934.1, KY417144.1, KT444582.1, FJ882944.1, NC_004718.3, MG987420.1, MG021451.1, NC_006213.1, NC_006577.2, KY983587.1, and NC_005831.2). The worldwide population coverage is 89.60%, which indicates that these epitopes could serve as candidate epitopes for vaccines of SARS-CoV-2 among most of the global population.
Based on the antigenicity of the SARS-CoV-2 S and N proteins, they could be major targets for preventing and treating SARS-CoV-2 infection. In 2007 and 2008, Cheung et al. predicted the DNA vaccines encoding the N-protein peptides LLLDRLNQL223-231 and LALLLLDRL220-228 presented by HLA-A*02:01 could trigger the highest T-cell cytotoxicity toward N protein-expressing cells, which indicated that these two N-protein peptides could be valuable peptide candidates for SARS vaccine.Citation18,Citation23 In the current study, we also predicted that LLLDRLNQL223-231 could be presented by HLA-A*02:01 and HLA-A*02:06, which covered 39.08% and 1.95% individuals worldwide, respectively. Moreover, the sequence of the epitope is identical to those of SARS coronavirus (KT444582.1 and FJ882944.1) and nucleocapsid protein Bat SARS-like coronavirus (MG772934.1 and KY417144.1), but not MERS (MG987420.1 and MG021451.1), which indicated that LLLDRLNQL223-231 could also be a valuable vaccine candidate peptide for SARS and Bat SARS-like coronavirus. In 2020, Ahmed et al. revisited previously tested and functional HLA-restricted SARS coronavirus epitopes and found many epitopes were also conserved in SARS-CoV-2, such as epitope LLLDRLNQL223-231 presented by HLA-A*02:01 allele.Citation30 Thus, epitope LLLDRLNQL223-231 could potentially offer protection against these two viruses. In addition, in 2020, Austin Nguyen et al. analyzed viral peptide-MHC class I binding affinity across HLA-A, -B, and -C genotypes for all SARS-CoV-2 peptides in silico, and found that different HLA alleles showed various capacities to present highly conserved SARS-CoV-2 peptides.Citation31 Among the conserved sequences, PRWYFYYLGTGP106-117 in N protein was highly conserved. In the current study, we also predicted that FYYLGTGPEAGLPY110-123 was presented by HLA-A*29:02 and HLA-C*14:02 alleles. In 2006, Zhou et al. predicted the SARS S protein epitopes and identified KLPDDFMGCV411–420 as a novel HLA-A*02:01-restricted S protein epitope.Citation11 Their results indicated that the epitope could be a novel SARS-associated coronavirus-specific CTL epitope and a potential target for characterizing virus control mechanisms and evaluating candidate SARS vaccines. Then, Liu et al. also predicted a SARS-CoV N protein-derived CTL epitope and identified QFKDNVILL346–354, which was restricted by HLA-A*24:02, by a series of in vitro studies.Citation19 In the current study, we did not predict these two epitopes in the SARS-CoV-2 N and S proteins. After aligning SARS-CoV-2 and SARS-CoV, we found only one mutation in the S protein epitope KLPDDFMGCV411–420 and two different mutations in QFKDNVILL346–354 between SARS-CoV-2 and SARS-CoV. Then, we replaced these mutations in SARS-CoV-2 and reconducted the predictions; KLPDDFMGCV411–420 and QFKDNVILL346–354 could be predicted, which indicated that these mutations could change the affinity between epitopes and HLA molecules.
Evasion of the host CTL response through the mutation of key epitopes is a major challenge to achieving natural or therapeutic vaccine-induced immune control of SARS-CoV-2. Therefore, we used two SARS-CoV-2 sequences (NC_045512.2 and MT007544.1) retrieved from NCBI to predict the epitopes and found that 14 epitopes shared identity with these two SARS-CoV-2 sequences, which include two major types of SARS-CoV-2 (Designated L and S). Thus, the 14-epitope combination could be feasible in vaccines for SARS-CoV-2, including SARS-CoV-2 types L and S.
Moreover, 13 of the 14 epitopes we selected in the current study could also serve as candidate epitopes for the SARS coronavirus and Bat SARS-like coronavirus (MG772934.1, KT444582.1, KT444582.1, and FJ882944.1) due to identical sequences between the epitopes and coronaviruses; additionally, these 13 epitopes could cover 84.94% of individuals worldwide. However, the 13 combination epitopes did not cover MERS (MG987420.1 and MG021451.1) due to the different subgenus (MERS belongs to Merbecovirus and SARS-CoV-2 belongs to Sarbecovirus) with significant differences in the sequences between SARS-CoV-2 (NC_045512.2) and MERS (MG987420.1 and MG021451.1). The identity of the N and S proteins was 50.0% and 35.0%, respectively, between SARS-CoV-2 (NC_045512.2) and MERS (MG987420.1 and MG021451.1). However, it was interesting that one epitope (KWPWYIWLGF1211-1220) had the same sequences between SARS-CoV-2 and MERS (NC_045512.2, MT007544.1, and MG987420.1, MG021451.1). This epitope (KWPWYIWLGF1211-1220) presented by HLA-A*23:01 and HLA-A*24:02 could cover 26.18% of individuals globally. In 2020, Ibrahim and Kafi also predicted the epitope KWPWYIWLGF1211-1220 because of the high scores that indicate high efficiency due to the prediction of a quantity proportional to the amount of peptide presented by MHC molecules on the cell surface.Citation24 No epitope completely matched with other four human coronaviruses (HCoV-OC43, HCoV-HKU1, HCoV-229E, and HCoV-NL63), because they belong to the different subgenus or different genus from SARS-CoV-2. HCoV-OC43 (NC_006213.1) and HCoV-HKU1 (NC_006577.2) belong to Embecovirus. HCoV-229E (KY983587.1) and HCoV-NL63 (NC_005831.2) belong to Alphacoronavirus. Thus, the 14-epitope combination could serve as vaccine candidate epitopes for SARS-CoV-2, Bat SARS-like coronavirus, SARS-like coronavirus, and MERS.
Our results indicated the possibility of using candidate CTL epitopes to produce vaccines to effectively control SARS-CoV-2 infections and development. In the current study, we obtained the 14-epitope combination based on the distribution of HLA-A, -B, and -C could cover 89.60% of individuals globally and overcome the limitation of HLA specificity. However, the epitopes we selected were only predicted with respect to binding ability between epitopes and specific MHC molecules based on the total scores as the cut off value for epitope selection. Thus, there were epitopes being ignored because the total scores were below the cut off value (0.5 for N protein, 1.07 for S protein). For example, there are no HLA-A*30:01 predicted epitopes in the entire sequence of the SARS-CoV-2 S protein in our results because all total scores were ≤0.6 with all 8,841 peptides for HLA-A*30:01. Moreover, in 2004, Wang et al. demonstrated that SARS-CoV protein-derived peptide-1 (RLNEVAKNL1167-1175) could induce peptide-specific CTLs both in vivo and in vitro.Citation21 In the current study, we have predicted this epitope using epitope prediction analysis; however, its total score was ≤-0.54. So, this epitope was not selected in the current study. Thus, the candidate epitopes which were predicted to reduce infection effects in the current study should also be demonstrated using peptide-sensitized peripheral blood mononuclear cells or isolated CD8+ CTL responses in vivo or in vitro level and animal model in the future. Our results will aid in exploring the possible use of these epitopes for the vaccine against SARS-CoV-2 infections.
Author contributions
Conceived and designed the experiments: Li Shi and Yufeng Yao. Performed the HLA data analysis: Sun Ming, Shuying Dai, Le Sun. Performed the immunoinformatic analysis: Yina Cun, Chuanyin Li, Lei Shi. Wrote the paper: Li Shi and Yufeng Yao.
Additional information
Funding
References
- Hui DS, Madani EIA, Ntoumi TA, Kock F, Dar RO, et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264–66.
- Lu H, Stratton CW, Tang YW. Outbreak of pneumonia of unknown etiology in Wuhan, China: the mystery and the miracle. J Med Virol. 2020;92(4):401–02. doi:10.1002/jmv.25678.
- Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, et al. Clinical Characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061. doi:10.1001/jama.2020.1585.
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–33. doi:10.1056/NEJMoa2001017.
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–74. doi:10.1016/S0140-6736(20)30251-8.
- Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, Yuen K-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–36. doi:10.1080/22221751.2020.1719902.
- Jiang S, Du L, Shi Z. An emerging coronavirus causing pneumonia outbreak in Wuhan, China: calling for developing therapeutic and prophylactic strategies. Emerg Microbes Infect. 2020;9(1):275–77. doi:10.1080/22221751.2020.1723441.
- Leung DT, Tam FC, Ma CH, Chan PK, Cheung JL, Niu H, et al. Antibody response of patients with severe acute respiratory syndrome (SARS) targets the viral nucleocapsid. J Infect Dis. 2004;190:379–86.
- Shi Y, Wan Z, Li L, Li P, Li C, Ma Q, et al. Antibody responses against SARS-coronavirus and its nucleocaspid in SARS patients. J Clin Virol. 2004;31(1):66–68. doi:10.1016/j.jcv.2004.05.006.
- He Y, Zhou Y, Wu H, Luo B, Chen J, Li W, et al. Identification of immunodominant sites on the spike protein of severe acute respiratory syndrome (SARS) coronavirus: implication for developing SARS diagnostics and vaccines. J Immunol. 2004;173:4050–57.
- Zhou M, Xu D, Li X, Li H, Shan M, Tang J, Wang M, Wang F-S, Zhu X, Tao H, et al. Screening and identification of severe acute respiratory syndrome-associated coronavirus-specific CTL epitopes. J Immunol. 2006;177(4):2138–45. doi:10.4049/jimmunol.177.4.2138.
- Coleman CM, Sisk JM, Halasz G, Zhong J, Beck SE, Matthews KL, et al. CD8+ T cells and macrophages regulate pathogenesis in a mouse model of middle east respiratory syndrome. J Virol. 2017;91.
- Tang F, Quan Y, Xin ZT, Wrammert J, Ma MJ, Lv H, Wang T-B, Yang H, Richardus JH, Liu W, et al. Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: a six-year follow-up study. J Immunol. 2011;186(12):7264–68. doi:10.4049/jimmunol.0903490.
- Peng H, Yang LT, Wang LY, Li J, Huang J, Lu ZQ, Koup RA, Bailer RT, Wu C-Y. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology. 2006;351(2):466–75. doi:10.1016/j.virol.2006.03.036.
- Fan -Y-Y, Huang Z-T, Li L, Wu M-H, Yu T, Koup RA, Bailer RT, Wu C-Y. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch Virol. 2009;154(7):1093–99. doi:10.1007/s00705-009-0409-6.
- Li CK, Wu H, Yan H, Ma S, Wang L, Zhang M, Tang X, Temperton NJ, Weiss RA, Brenchley JM, et al. T cell responses to whole SARS coronavirus in humans. J Immunol. 2008;181(8):5490–500. doi:10.4049/jimmunol.181.8.5490.
- Ng O-W, Chia A, Tan AT, Jadi RS, Leong HN, Bertoletti A, Tan Y-J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine. 2016;34(17):2008–14. doi:10.1016/j.vaccine.2016.02.063.
- Cheung YK, Cheng SC, Sin FW, Chan KT, Xie Y. Investigation of immunogenic T-cell epitopes in SARS virus nucleocapsid protein and their role in the prevention and treatment of SARS infection. Hong Kong Med J. 2008;14:27–30.
- Liu J, Wu P, Gao F, Qi J, Kawana-Tachikawa A, Xie J, Vavricka CJ, Iwamoto A, Li T, Gao GF, et al. Novel immunodominant peptide presentation strategy: a featured HLA-A*2402-restricted cytotoxic T-lymphocyte epitope stabilized by intrachain hydrogen bonds from severe acute respiratory syndrome coronavirus nucleocapsid protein. J Virol. 2010;84(22):11849–57. doi:10.1128/JVI.01464-10.
- Tsao YP, Lin JY, Jan JT, Leng CH, Chu CC, Yang YC, et al. HLA-A*0201 T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus nucleocapsid and spike proteins. Biochem Biophys Res Commun. 2006;344:63–71.
- Wang B, Chen H, Jiang X, Zhang M, Wan T, Li N, Zhou X, Wu Y, Yang F, Yu Y, et al. Identification of an HLA-A*0201-restricted CD8+ T-cell epitope SSp-1 of SARS-CoV spike protein. Blood. 2004;104(1):200–06. doi:10.1182/blood-2003-11-4072.
- Wang YD, Sin WY, Xu GB, Yang HH, Wong TY, Pang XW, He X-Y, Zhang H-G, Ng JNL, Cheng CSS, et al. T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus spike protein elicit a specific T-cell immune response in patients who recover from SARS. J Virol. 2004;78(11):5612–18. doi:10.1128/JVI.78.11.5612-5618.2004.
- Cheung YK, Cheng SC, Sin FW, Chan KT, Xie Y. Induction of T-cell response by a DNA vaccine encoding a novel HLA-A*0201 severe acute respiratory syndrome coronavirus epitope. Vaccine. 2007;25(32):6070–77. doi:10.1016/j.vaccine.2007.05.025.
- Ibrahim HS, Kafi SK. A computational vaccine designing approach for MERS-CoV infections. Methods Mol Biol. 2020;2131:39–145.
- Bezu L, Kepp O, Cerrato G, Pol J, Fucikova J, Spisek R, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: peptide-based vaccines in anticancer therapy. Oncoimmunology. 2018;7(12):e1511506. doi:10.1080/2162402X.2018.1511506.
- Germain RN, Margulies DH. The biochemistry and cell biology of antigen processing and presentation. Annu Rev Immunol. 1993;11(1):403–50. doi:10.1146/annurev.iy.11.040193.002155.
- Heemels MT, Ploegh H. Generation, translocation, and presentation of MHC class I-restricted peptides. Annu Rev Biochem. 1995;64:463–91.
- Hung CF, Ma B, Monie A, Tsen SW, Wu TC. Therapeutic human papillomavirus vaccines: current clinical trials and future directions. Expert Opin Biol Ther. 2008;8(4):421–39. doi:10.1517/14712598.8.4.421.
- Dhanda SK, Vaughan K, Schulten V, Grifoni A, Weiskopf D, Sidney J, et al. Development of a novel clustering tool for linear peptide sequences. Immunology. 2018;155(3):331–45. doi:10.1111/imm.12984.
- Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12(3):254. doi:10.3390/v12030254.
- Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A, et al. Human leukocyte antigen susceptibility map for severe acute respiratory syndrome coronavirus 2. J Virol. 2020;94.