
ABSTRACT
Respiratory Syncytial Virus (RSV) causes lower respiratory tract infections that can be severe and sometimes fatal. The risk for severe RSV infection is highest in infants and older adults. A safe and effective RSV vaccine for older adults represents a serious unmet medical need due to higher morbidity and mortality in this age group. In this randomized, partially double-blind, placebo-controlled, phase 1 dose-escalation study, we evaluated the safety, tolerability and immunogenicity of an investigational messenger ribonucleic acid (mRNA) vaccine encoding the RSV fusion protein (F) stabilized in the prefusion conformation. The study was conducted in healthy younger adults (ages ≥18 and ≤49 years) and healthy older adults (ages ≥60 and ≤79 years). Participants received mRNA-1777 (V171) or placebo as a single intramuscular dose. For each dose level, three sentinel participants were administered open-label mRNA-1777 (V171). Seventy-two younger adults were randomized and administered 25, 100, or 200 µg mRNA-1777 (V171) or placebo, and 107 older adults were randomized and administered 25, 100, 200 or 300 µg mRNA-1777 (V171) or placebo. Primary objectives were safety and tolerability and secondary objectives included humoral and cell-mediated immunogenicity. All dose levels of mRNA-1777 (V171) were generally well tolerated and no serious adverse events related to the vaccine were reported. Immunization with mRNA-1777 (V171) elicited a humoral immune response as measured by increases in RSV neutralizing antibody titers, serum antibody titers to RSV prefusion F protein, D25 competing antibody titers to RSV prefusion F protein, and cell-mediated immune responses to RSV-F peptides.
Introduction
Respiratory Syncytial Virus is a non-segmented RNA virus with two subtypes, RSV A and RSV B. The virus commonly infects most infants by 1 year of ageCitation1 and can cause reinfections in children and adults.Citation2 Infections due to RSV generally cause upper respiratory tract infection, but can progress to lower respiratory tract infection (LRTI), most often in vulnerable populations such as infants, the immunocompromised, and the elderly. Complications of RSV-related LRTI include respiratory distress, pneumonia, and bronchiolitis, which can be fatal. In infants, RSV infection has been identified as the leading cause of hospitalization in the first 6 months of life and the most common cause of LRTI-related deaths during the neonatal period.Citation3 Morbidity and mortality in older adults are extensive and generally increase with age.Citation4 The prevalence and burden of RSV in adults may be greatly underestimated because testing for RSV in adults is not routinely performed; however, adults with RSV infections have mortality and hospitalization rates comparable to that of seasonal influenza.Citation4–6 In the United States alone, studies have estimated that RSV infections in older adults cause over 175,000 hospitalizations and 14,000 deaths per yearCitation7. RSV treatment in adults is limited to supportive care and general symptom management. A licensed RSV vaccine is not available.
The majority of adults demonstrate some level of humoral immunity to RSV due to past infection; despite this pre-existing immunity, repeat infections still occur. Though immune correlates of protection against RSV-associated disease in adults have not yet been defined, studies of RSV specific-immune response in the older adults have suggested that lower antibody titers to the F and G proteins, decreased nasal IgA levels, and waning cellular immunity may be risk factors for RSV-associated disease.Citation8–13
A significant part of the current research and development efforts for adult RSV vaccines target the RSV fusion (F) protein which, along with the G protein, induces RSV-neutralizing antibody responses. Compared to the G protein, the F protein is more conserved across RSV types A and B.Citation14 The F protein exists in two conformations, a metastable prefusion conformation and a postfusion conformation. Studies of humoral immune response to natural RSV infection have shown that the majority of the human antibody response targets the prefusion conformation.Citation15,Citation16 The advent of structure-based design methods to stabilize the prefusion conformation of the F proteinCitation17 has revitalized RSV vaccine research in recent years with a large focus on prefusion F-based immunogens.Citation18 Current stabilized prefusion F-based development candidates include a number of protein subunit vaccines with or without adjuvants,Citation19–22 a recombinant adenoviral vector,Citation23 virus-like particlesCitation24 and nanoparticles.Citation25
We recently reported the pre-clinical evaluation of modified mRNA/lipid nanoparticle-based vaccine formulations expressing RSV prefusion F.Citation26 These mRNA vaccine candidates elicited RSV-neutralizing antibody responses as well as CD4 and CD8 T-cell response in mice and demonstrated protection against RSV A and B in cotton rat challenge models.
mRNA vaccine technology has the potential to offer rapid antigen design and scalable production.Citation27,Citation28 The nucleic acid approach eliminates the need to produce and purify proteins, a process which can be costly or challenging for unstable antigens. It also eliminates the need for egg or cell culture systems.Citation29 A number of phase I clinical trials assessing mRNA vaccines against infectious diseases were recently undertaken, including: rabies, chikungunya, cytomegalovirus, zika, influenza, human metapneumovirus, and parainfluenza type 3Citation27 and SARS-CoV-2Citation30. Although the majority of these studies are ongoing or unpublished to date, the phase I studies of H10N8 and H7N9 influenza investigational vaccines were reported to be well tolerated and to induce robust and durable antibody responses.Citation31 These data support the clinical evaluation of investigational mRNA vaccines for the prevention of RSV disease.
Here, we report a first-in-human phase 1 study in healthy younger and older adults to evaluate the safety and immunogenicity of mRNA-1777 (V171), an investigational prophylactic RSV vaccine encoding the full-length RSV F protein stabilized in the prefusion conformation.
Materials and methods
Study design
This was a phase 1, first-in-human, randomized, partially double-blind, placebo-controlled, dose-escalation study to evaluate the safety, tolerability, and immunogenicity of mRNA-1777 (V171) in healthy younger participants (ages ≥18 and ≤49 years, Part A) and healthy older participants (ages ≥60 and ≤79 years, Part B) (Protocol mRNA-1777-P101, V171). The study was conducted at three sites in Australia between November of 2016 and May of 2019.
The first three participants in each arm and at each dose level in Part A and Part B were enrolled as open-label sentinels. The purpose of the sentinel groups was to identify potential toxicities prior to enrollment of the randomized expansion cohorts. Participants assigned to the expansion cohorts were randomized to receive mRNA-1777 (V171) or placebo in a 3:1 ratio. Randomization in the expansion cohort was carried out using an interactive voice response system. All doses of mRNA-1777 (V171) and placebo were administered as an intramuscular (IM) injection (up to 0.4 mL) into the deltoid muscle as a single dose on Day 1.
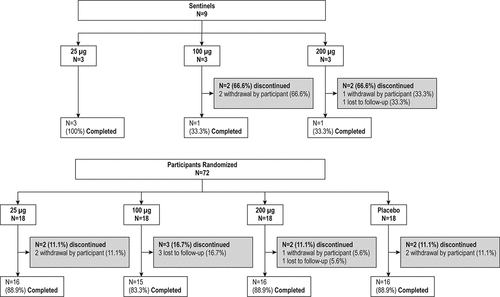
Part A included three sequential treatment arms of 25 µg, 100 µg and 200 µg doses. Each treatment arm consisted of three sentinel participants and 24 expansion cohort participants (18 active and 6 placebo) (). Safety data from the sentinel cohort within each treatment arm in Part A were reviewed by the investigator in consultation with the sponsor prior to dosing the expansion cohort. Once the final participant in a treatment arm was dosed and completed the Day 8 evaluation, the safety monitoring committee (SMC) reviewed the safety data from the sentinel and expansion cohorts prior to escalation to the next treatment arm.
Figure 1. Participant Disposition for Group A, Younger Adults
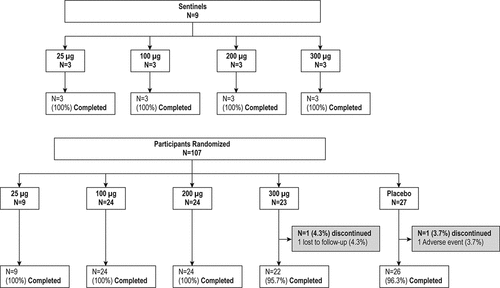
Part B included four sequential treatment arms of 25 µg, 100 µg, 200 µg and 300 µg doses. Each treatment arm consisted of three sentinel participants and expansion cohorts (). In part B, the safety data from the sentinel cohort within each treatment arm, in addition to the sentinel and blinded safety data in Part A at the same dose level, were reviewed by an independent SMC prior to dosing the expansion cohort. The 25 µg arm consisted of 12 participants (nine active and three placebo). The 100 µg and 200 µg arms each consisted of 24 active and eight placebo, and the 300 µg arm consisted of 23 active and eight placebo. The 300 µg arm was included as a conditional group to further characterize the relationship between dose, tolerability, and immunogenicity in healthy older adults, and was implemented by the sponsor with approval of the SMC based on the tolerability and immunogenicity observed in the lower dose groups.
Figure 2. Participant Disposition for Group B, Older Adults
Doses were selected based upon data from pre-clinical safety and immunogenicity studies as well as clinical studies with other Moderna, Inc. mRNA-based vaccines. The sample size of the study was set to provide 97.5% confidence that the true vaccine-related serious adverse event (SAE) rate was <2.9% in participants receiving mRNA-1777 (V171) if no vaccine-related SAE was reported in the participants receiving mRNA-1777 (V171) (dose groups 25 µg, 100 µg and 200 µg).
Study objectives
The primary objective of this study was to assess the safety and tolerability of mRNA-1777 (V171) versus placebo. The secondary objectives were to determine the immunogenicity of mRNA-1777 (V171) by serum neutralizing titers against RSV A and RSV B, serum antibody titers to RSV F protein, and serum palivizumab-and D25-competing antibody titers to RSV F protein. Cell-mediated immune responses to RSV F peptides were measured by IFN-γ enzyme-linked immunospot (ELISPOT) and flow cytometry using intracellular cytokine staining.
Participants
Males and females of non-childbearing potential in good general health between the ages of 18 and 49 (inclusive; Part A) or between 60 and 79 years of age (inclusive; Part B) were included. Good general health was determined by medical history, physical examination, vital sign measurements, laboratory tests, and an ECG performed at screening. Participants with a body mass index (BMI) ≥18 kg/m2 to ≤32 kg/m2 were included in the study. Key exclusion criteria included any ongoing, symptomatic acute or chronic illness requiring medical or surgical care, a history of diabetes, a history of active cancer within the last 10 years, significant psychological disorders, previous participation in studies with lipid nanoparticles (LNP), a history of severe allergies, or receipt of any other licensed vaccines within 4 weeks prior to dose administration. For a full list of inclusion and exclusion criteria see Appendices A and B, respectively. This study was conducted in accordance with principals of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies.
Vaccine description
The mRNA-1777 (V171) vaccine consisted of chemically modified mRNA encoding a full length (membrane-associated) RSV F protein stabilized in the prefusion conformation using the modification described by McLellan et al.Citation17(mDS-Cav1),Citation26 and an LNP delivery system. The LNP consisted of cholesterol, DSPC (1,2-Distearoyl-sn-glycero-3-phosphocholine), MC3 (also known as DLin-MC3-DMA), and PEG2000-DMG. The mRNA-1777 (V171) vaccine was diluted to the target dose using 0.9% Sodium Chloride Injection, USP or BP as the diluent. Placebo was a matched volume of 0.9% Sodium Chloride Injection, USP or BP. All products were prepared, packaged, and labeled according to Good Manufacturing Practice (GMP) guidelines from the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and applicable local laws and regulations. Drug supplies were shipped, stored, and distributed in accordance with the trial protocol.
Safety assessments
Physical examinations were conducted at screening and Days 1 (baseline), 8, 29, 180, and 366. Electrocardiograms were taken at screening and Day 1. Clinical laboratory assessments were taken at screening and Days 1, 2, 8, and 29. Safety was assessed by collecting solicited adverse events (AEs) (local and systemic reactogenicity events) for 7 days post-vaccination, unsolicited AEs for 90 days post-vaccination, and serious AEs, adverse events of special interest (AESI), medically attended AEs, and new onset of chronic medical illness (NOCIs) for 1 year following vaccination.
The intensity of AEs and laboratory abnormalities was categorized by the investigator as mild (Grade 1), moderate (Grade 2), severe (Grade 3), or possibly life-threatening (Grade 4) using the Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventative Vaccine Clinical Trials as a guideline.Citation32 All AEs were determined by the investigator to be related or not related to the vaccine. Furthermore, during the time of enrollment, rules were in place to pause the study if pre-specified criteria were met.
Immunogenicity assessments
Serum neutralizing antibody titers to RSV A (Long strain) and RSV B (Washington strain) were measured at baseline and Days 8, 29, 60, 90, 180, and 366 using a near infrared-dye (NIRDye) based In-Cell Western neutralization assay as previously described.Citation33 Briefly, serum samples are serially diluted and mixed with RSV virus for 1 hour before being added to HEp-2 cells and incubated for 3 days. The cells were washed, fixed and incubated with mouse monoclonal antibodies to RSV F and RSV N. The plates were washed and incubated with biotinylated anti-mouse IgG followed by infrared dye-streptavidin to detect the RSV specific signal. The stained plates were scanned using a Li-Cor Aerius (LI-COR Biosciences, Lincoln, Nebraska) infrared fluorescence scanner. Anti-RSV antibody levels in human serum were reported as the dilution of serum required to achieve 50% response in the assay (denoted NT50).
Serum antibody titers to RSV prefusion F protein were evaluated using an electrochemiluminescent (ECL) assay at baseline, Days 8, 29, 60, 90, 180, and 366. The ECL assay measures total IgG antibodies binding to prefusion F using the Meso Scale Discovery Sector Imager S600 Analyzer. Palivizumab- D25-competing antibody titers to RSV postfusion and prefusion proteins, respectively, were evaluated with competition AlphaLISA (PerkinElmer) at Baseline, Day 8, 29, 60, 90, 180, and 366 similarly to previously described.Citation26 Briefly, serially diluted serum samples are mixed with AlphaLISA acceptor beads conjugated to prefusion or postfusion F protein for 30 minutes. Biotinylated palivizumab or D25 antibody was added and incubated. Next, streptavidin-coated donor AlphaLISA beads were added to the mixture and incubated and plates were read for the AlhaLISA signal on a PHERAStar FS (BMG Labtech).
Cell-mediated immune (CMI) responses were analyzed using the interferon-γ ELISPOT assayCitation34 and the intracellular cytokine (IFN-γ, TNF-α and IL-2) staining flow cytometry assay at baseline and Days 15 and 60. For the intracellular cytokine staining assay, peripheral blood mononuclear cells (PBMC) were thawed, rested overnight in complete RPMI medium, and counted the following morning. Approximately 1 million cells were stimulated with an RSV F peptide pool and anti-CD28/CD49d antibodies for two hours at 37°C followed by four additional hours in the presence of GolgiPlug (BD Biosciences). The cells were stored 4°C overnight and stained the next day with LIVE/DEAD™ fixable stain (Invitrogen), washed, permeabilized (BD Cytofix/Cytoperm), and stained with cocktail of fluorescently labeled anti-CD3 (clone UCHT1, BD Pharmingen), CD4 (clone SK3, BD Biosciences), CD8 (clone SK1, BD Biosciences), IL-2 (clone 5344.111, BD Pharmingen), IFN-γ (Clone 4S.B3, BD Pharmingen), and TNF-α (clone Mab 11, BD Biosciences) mAbs. Data were acquired on the BD LSR Fortessa X-20.
For each of the CMI assays, PBMC were stimulated with a peptide pool based on an RSV B sequence spanning the RSV F protein (15mers, overlapping by 11 amino acids, JPT Peptide Technologies catalog PM-HRSVB-FGF0). Data are background subtracted based on the negative control stimulation, which was dimethyl sulfoxide, the diluent used for the peptide pool.
Data analysis
A formal hypothesis was not tested. The overall safety and tolerability of the tested vaccine were assessed and reported as a summary of serious and non-serious solicited and unsolicited adverse events. Clinical laboratory test results, vital sign measurements, electrocardiograms, and physical examination data were presented in data listings. The Safety Set included all participants in the randomized set who received vaccine or placebo.
Immunogenicity analyses were reported for the per-protocol (PP) set. The PP set was defined as all participants who received a dose of mRNA-1777 (V171) vaccine or placebo, had at least one immunogenicity result available, and had no major protocol violations that impacted immunogenicity response at the corresponding time point.
For the RSV A and B neutralization assay, the RSV prefusion F serum antibody titer assay and the palivizumab and D25 antibody competition assays, the data were natural log transformed for analysis. The means, the mean changes from baseline, and the corresponding 95% CIs were computed based on the T-distribution and were exponentiated to obtain geometric mean titers (GMTs), geometric mean fold increase (GMFI) from baseline, and the corresponding 95% CIs. For the ELISPOT and intracellular cytokine staining assays, original values were used to calculate the means and mean changes from baseline, and the corresponding 95% CIs were computed based on the T-distribution.
The GMTs and 95% CIs, as well as GMFI from baseline and 95% CIs for neutralizing antibody titers against RSV A and RSV B, absolute serum antibody titers to RSV F protein, and serum palivizumab-and D25-competing antibody titers to RSV F protein were plotted by part, treatment group, and time. Mean percent of positive cells and 95% CIs for intracellular cytokine staining were plotted by part, treatment group and time. The mean spot forming cells and 95% CIs for ELISPOT were plotted by part, treatment group, and time. All analyses were conducted using SAS® software (SAS Institute Inc., Cary, North Carolina) Version 9.3 or higher.
Results
Participants
Part A of the study enrolled three sentinel participants for each dose (25 μg, 100 μg, 200 μg) (nine total) and a total of 72 participants were randomized in the expansion cohorts: 18 each received either 25 μg, 100 μg, or 200 μg of mRNA-177 (V171) or placebo (). The study completion percentages for the randomized expansion cohorts were 88.9% (25 μg), 83.3% (100 μg), 88.9% (200 μg) and 88.9% (placebo) ().
Part B of the study also enrolled 3 sentinel participants for each dose (25 μg, 100 μg 200 μg, and 300 μg) (12 total), and a total of 107 participants were randomized in the expansion cohorts (). Nine participants received 25 μg, 24 participants received 100 μg, 24 participants received 200 μg, 23 participants received 300 μg of mRNA-177 (V171), and 27 participants received placebo. One hundred percent (100%) of the participants in the 25 μg, 100 μg, and 200 μg groups, 95.7% of the 300 μg group and 96.3% of the placebo group completed the study ().
The participant baseline characteristics for the randomized cohorts for Parts A and B are shown in . Baseline characteristics for the sentinel groups for Parts A and B are shown in Supplemental Table 1. The demographic characteristics were generally similar across treatment groups in Parts A and B. In Part A, the majority of randomized participants were male (98.6%) and white (87.5%), with a mean age of 28.7 years (range: 18 to 47 years) and a mean BMI of 24.5 kg/m2. In Part B, the majority of randomized participants were female (57.9%) and white (97.2%), with a mean age of 66.9 years (range: 60 to 79 years) and a mean BMI of 26.4 kg/m2. The mean values for age and BMI were generally similar across treatment groups in Parts A and B.
Table 1. Participant baseline characteristics: randomized cohorts, Part A and Part B
Safety
An overall summary of treatment emergent adverse events (TEAEs) for the safety sets is shown in (data for the sentinel sets are shown in Supplemental Table 2). Treatment-emergent AEs included both solicited and unsolicited AEs.
Table 2. Summary of treatment-emergent adverse events
Of the younger adult participants randomized to receive mRNA-1777 (V171), 52 (96.3%) reported TEAEs and 51 (94.4%) reported treatment-related TEAEs. Of the younger adult participants randomized to receive placebo, 15 (83.3%) reported TEAEs and 8 (44.4%) reported treatment-related TEAEs. Overall, 20 (37.0%) participants who received mRNA-1777 (V171), and 7 (38.9%) participants who received placebo reported medically attended AEs. There were no deaths, TEAEs leading to withdrawal, AESIs, NOCIs, or new onsets of autoimmune disorders. One (5.6%) younger adult participant randomized to receive 200 μg mRNA-1777 (V171) reported an SAE (ligament operation) that was unrelated to study vaccine.
Of the older adult participants randomized to receive mRNA-1777 (V171), all 80 (100.0%) reported TEAEs and 77 (96.3%) reported treatment-related TEAEs. Of the older adult participants randomized to receive placebo, 24 (88.9%) reported TEAEs and 10 (37.0%) reported treatment-related TEAEs. Overall, 38 (47.5%) participants who received mRNA-1777 (V171) and 17 (63.0%) participants who received placebo reported medically attended AEs. There were no AESIs, NOCI, or new onsets of autoimmune disorder in the older adult groups. Six (7.5%) older adult participants randomized to receive mRNA-1777 (V171) and 2 (7.4%) randomized to receive placebo reported SAEs; all were unrelated to study vaccine. There was one death due to cardiomyopathy in one participant randomized to receive placebo (not related to treatment) in an older adult participant.
Solicited injection site adverse events
A summary of participants with solicited injection-site AEs by maximum toxicity grade in the younger and older adult randomized safety sets is presented in and Supplemental Table 3.
Figure 3. Solicited Injection-Site Adverse Events
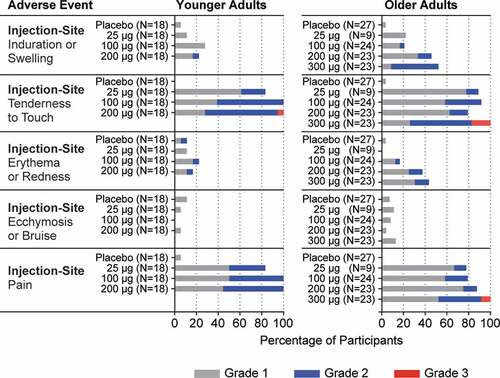
Overall, in the younger adult group, 52 (96.3%) participants randomized to receive mRNA-1777 (V171) and 4 (22.2%) participants randomized to receive placebo reported injection-site AEs. The number of participants who reported injection-site AEs was similar across mRNA-1777 (V171) treatment groups. Injection-site pain and tenderness to touch were the most common injection-site AEs, each reported by 51 (94.4%) participants randomized to receive mRNA-1777 (V171) (Supplemental Table 3). The majority of solicited injection-site AE were Grade 1 and Grade 2; one participant randomized to receive 200 μg mRNA-1777 (V171) reported a Grade 3 injection-site tenderness to touch. No Grade 4 injection-site AEs were reported.
Overall, in the older adult group, 79 (98.8%) participants were randomized to receive mRNA-1777 (V171) and 3 (11.1%) participants randomized to receive placebo reported injection-site AEs (Supplemental Table 3). The number of participants who reported injection-site AEs was similar across mRNA-1777 (V171) treatment groups. Injection-site pain and tenderness to touch were the most common injection-site AEs in participants randomized to receive mRNA-1777 (V171), reported in 70 (87.5%) and 72 (90.0%), respectively. The majority of solicited injection-site AEs were Grade 1 and Grade 2. Five participants reported Grade 3 injection-site AEs; all were in the 300 μg mRNA-1777 (V171) treatment group: 2 reported Grade 3 injection-site pain and 3 reported Grade 3 injection-site tenderness to touch. No Grade 4 injection-site AEs were reported.
Solicited systemic adverse events
A summary of randomized participants reporting solicited systemic AEs is presented in and Supplemental Table 4. In the younger adult group overall, the proportion of participants who reported solicited systemic AEs was higher in the 200 μg mRNA-1777 (V171) treatment group (94.4%) than in the 25 μg, 100 μg, and placebo treatment groups (66.7%, 72.2%, and 33.3%, respectively). The most common solicited systemic events across mRNA-1777 (V171) treatment groups were fatigue, generalized myalgia, headache, and malaise.
Figure 4. Solicited Systemic Adverse Events
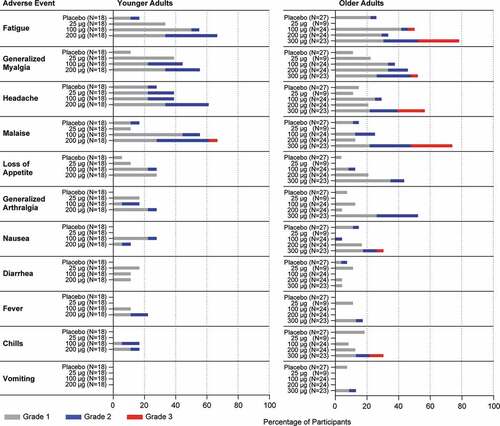
In the older adult group overall, the proportion of participants who reported solicited systemic AEs was higher in the 300 μg treatment group (91.3%) than in the 25 μg, 100 μg, 200 μg and placebo treatment groups (55.6%, 70.8%, 62.5%, and 37.0%, respectively). The most common solicited systemic events across mRNA-1777 (V171) treatment groups were fatigue, generalized myalgia, headache, and malaise.
In the younger adult group, all of the reported solicited systemic adverse events were Grade 1 or Grade 2 with the exception of 1 younger adult participant in the 200 μg treatment group reporting Grade 3 malaise (Supplemental Table 4). In the older adult group, all but one of the Grade 3 events were reported by participants in the 300 μg treatment group: fatigue in 6 (26.1%), myalgia in 1 (4.3%), headache in 4 (17.4%) and malaise in 6 (26.1%), nausea in 1 (4.3%), and chills in 2 (8.7%). One (4.3%) participant in the 100 μg mRNA-1777 (V171) treatment group reported Grade 3 fatigue. No Grade 4 solicited systemic adverse events were reported in the study (Supplemental Table 4).
Unsolicited treatment-emergent adverse events
Unsolicited TEAEs were collected from Days 1 to 90. In the younger adults, 66.7%, 33.3%, and 66.7% of participants, in the 25 μg, 100 μg, and 200 μg mRNA-1777 (V171) groups, respectively, and 61.1% in the placebo group reported an unsolicited treatment-emergent adverse event. Overall, in the younger adults, all of the reported unsolicited TEAEs from mRNA-immunized participants were of Grade 1 and Grade 2 toxicity.
In the older adults, 77.8%, 66.7%, 70.8%, and 60.9% in the 25 μg, 100 μg, 200 μg, and 300 μg mRNA-1777 (V171) groups, respectively, and 63.0% in the placebo group reported an unsolicited TEAE. Overall, most of the unsolicited TEAEs, regardless of relationship to treatment, were of Grade 1 or Grade 2 toxicity. Grade 3 unsolicited TEAEs were reported by 22.2%, 8.3%, 12.5%, and 8.7% in the 25 μg, 100 μg, 200 μg, and 300 μg mRNA-1777 (V171) groups, respectively, and 3.7% in the placebo group. Grade 4 unsolicited TEAEs were reported by 2 (2.5%) participants in the 200 μg mRNA-1777 (V171) group and included prothrombin time prolonged (treatment-related) and hyperbilirubinemia (not related to treatment).
In both the younger and older adult groups, there were no clinically relevant changes from baseline in vital sign measurements observed and no clinically relevant changes were generally observed for hematology and coagulation, serum chemistry, or urinalysis results. Dose-dependent increases from baseline in mean bilirubin, C-reactive protein, leukocyte, and neutrophil levels were observed on Day 2 in vaccinated subjects and generally returned to baseline by Day 8.
RSV A and B serum neutralization
RSV neutralization titers were evaluated for the two RSV subtypes A and B through one-year post-vaccination (). As the majority of the general adult population has some level of serum neutralization activity due to past infections, the baseline RSV neutralization titers were measured and the postvaccination GMFI from baseline were calculated ().
Figure 5. Serum Neutralization Titers against RSV A and RSV B
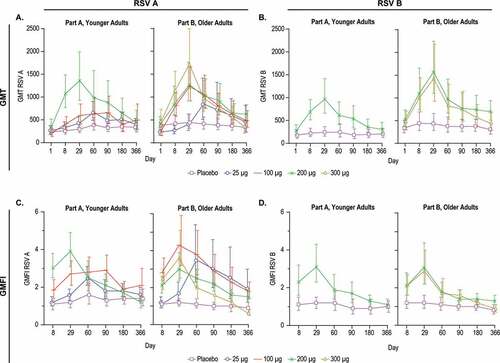
In the younger adults, a GMT dose response was observed for RSV A across the 25 μg, 100 μg, and 200 μg groups, with peak responses generally ranging from Days 29 to 90 (), Supplemental Table 5). Throughout this report, the term peak is defined as the highest observed value across the time points sampled.
In the older adults, an RSV A dose-response in GMT was observed across the 25 μg, 100 μg, 200 μg, and 300 μg groups, with peak responses generally ranging from Days 29 to 60 ), Supplemental Table 5). In contrast, the groups receiving placebo displayed minimal variation in RSV A GMT over the course of the year (), Supplemental Table 5).
A dose-response in the RSV A GMFI was observed in the younger adult cohort ()). The peak GMFI was 2.5-fold for the 25 μg dose, 2.9-fold for the 100 μg group and 3.9-fold for the 200 μg group (), Supplemental Table 6). In the older adults, peak GMFI for RSV A was 3.5-fold for the 25 μg dose group, 4.3-fold for the 100 μg, 3.0-fold for the 200 μg group and 3.6-fold for the 300 μg group (), Supplemental Table 6). In the placebo groups, GMFIs remained relatively constant for RSV A neutralization titers over the course of the year, ranging from 1.1 to 1.6-fold for the younger adults and 0.9 to 1.2-fold for the older adults.
Neutralization titers against RSV B were measured for all visits for the 200 μg and placebo groups in younger adults, and the 200 μg, 300 μg dose, and placebo groups in older adults (), and only at Days 1 and 29 for the 25 μg and 100 μg groups (Supplemental Tables 5,6). Vaccination with 200 μg mRNA-1777 (V171) resulted in increased RSV B neutralization GMT in the younger adults with a maximum observed GMFI of 3.1-fold (). In the older adults, vaccination with 200 μg or 300 μg of mRNA resulted in similar peak GMTs and GMFIs (). The younger and older adult groups receiving placebo displayed minimal variation in RSV B titers over all timepoints, with GMFI ranging from 0.9 to 1.2 and 0.8 to 1.2-fold, respectively (), Supp Table 6).
Overall, vaccination with mRNA-1777 (V171) increased RSV A and B serum neutralization GMTs at all doses tested in both the younger and older adults. Geometric mean titers and GMFIs generally declined over time after Day 60; but remained increased over baseline and placebo through at least Day 180 in all mRNA-vaccinated groups ().
RSV prefusion F serum antibody titers
Vaccination with mRNA-1777 (V171) increased prefusion F-specific serum antibody titers in both the younger and older adult groups (). A dose-response was observed across the 25 μg, 100 μg, and 200 μg dose groups in the GMTs over time for absolute serum antibody titers to RSV prefusion F protein for both the younger and older adult groups ()). In the older adult group, a higher GMT was not observed in the 300 μg dose group as compared to the 200 μg dose group. The highest observed serum titers for both GMTs and GMFIs ranged from Days 29 to Day 60 among all groups ().
Figure 6. Serum Antibody Titers to Prefusion F Protein
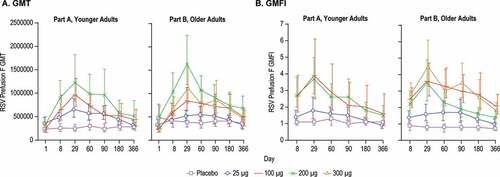
The highest observed GMFI for absolute serum antibody titers to RSV prefusion F in younger adults were 1.8, 3.9, and 3.7-fold for 25 μg, 100 μg, and 200 μg, respectively ()), all at day 29. In the older adult cohort, the highest GMFI were 1.7, 3.6, 3.5, and 4.5-fold for 25 μg, 100 μg, 200 μg and 300 μg, respectively ()). In contrast, the GMFIs for the placebo groups remained relatively flat over the year, with GMFIs ranging from 1.0 to 1.3-fold across all timepoints in the younger adults and 0.7 to 0.9-fold across all timepoints in the older adult cohort ()).
Palivizumab and D25 competing antibodies
The serum antibodies from immunized participants were evaluated for the ability to compete with the palivizumab or D25 antibody for binding to postfusion F or prefusion F proteins, respectively. Palivizumab binds to an epitope on site II which is present in both prefusion and postfusion forms of RSV F protein, and D25 specifically recognizes an epitope on site Ø, which is only present in the prefusion conformation of the protein.Citation17 An increase in D25 competing antibody (DCA) geometric mean concentration (GMC) was detected after immunization with mRNA-1777 (V171) at all doses tested in both the younger and older adults (Supplemental Figure 1A). DCA in the younger adults increased 2.2, 3.7, and 3.7-fold over Day 1 for the 25 μg, 100 μg, and 200 μg groups, respectively at the peak (Day 29) ()). In older adults, the DCA GMFIs were 2.4, 3.3, 3.4, and 4.1-fold for the 25 μg, 100 μg, 200 μg, and 300 μg groups, respectively at peak (Day 29). The placebo groups displayed minimal variation over all timepoints ranging from 0.8 to 1.1-fold in the younger adults and 0.7 to 1.0-fold in the older adults.
Figure 7. D25 and Palivizumab Competing Antibodies
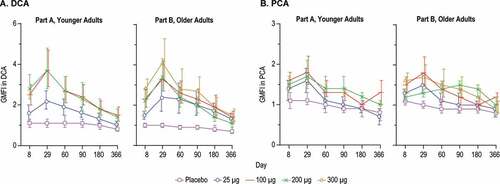
Palivizumab competing antibody (PCA) GMFIs and GMCs in the mRNA-vaccinated groups were numerically lower as compared to the DCA levels (, Supplemental Figure 1). Palivizumab competing antibody GMCs increased in all mRNA dose groups, generally peaking from Day 29 to Day 90 (Supplemental Figure 1). In the younger adults, PCA GMFIs were 1.6, 1.8, and 1.7-fold for the 25 μg, 100 μg, and 200 μg groups, respectively, at the peak (Day 29). In the older adults PCA GMFIs were 1.5, 1.8, 1.5, and 1.7-fold for the 25 μg, 100 μg, 200 μg, and 300 μg groups, respectively, at the peak time points ()). The placebo PCA GMFIs remained relatively flat over all time points tested, ranging from 0.8 to 1.1-fold in the younger adults and 0.8 to 1.1-fold in the older adult group.
Cell-mediated immunogenicity
T-cell responses to RSV Fusion protein were assessed using IFN-γ ELISPOT and intracellular cytokine staining assay ( and ). All dose groups in both the younger and older adults showed an increase in the mean ELISPOT on Day 15 compared to placebo with no apparent dose-response (). Mean ELISPOT responses remained increased at Day 60 in the 100, 200, and 300 μg dose groups.
Figure 8. IFN-γ ELISPOT
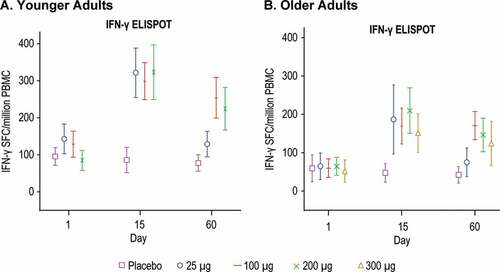
Figure 9. CD4 T cell-responses by Intracellular Cytokine Staining
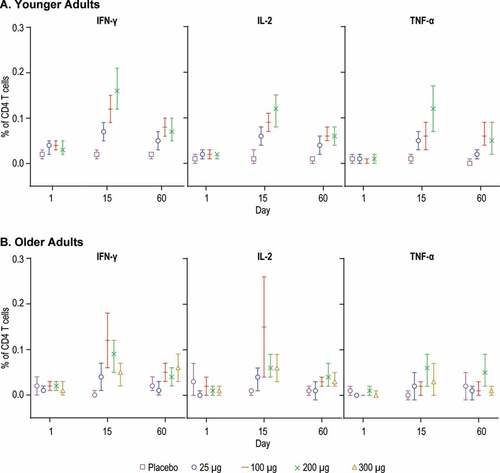
Results from the intracellular cytokine staining assay demonstrated a dose-response across the 25 μg, 100 μg, and 200 μg dose groups in the GM of % CD4 T cells positive for IFN-γ, IL-2 and TNF-α in the younger adult group at Day 15, and to a lesser extent at Day 60 ()). In the older adults, the mean % CD4 T cells expressing IFN-γ, IL-2 and TNF-α increased at Day 15; however, a dose-response was not apparent ()). CD8 T cell responses were not detected overall in any of the dose groups in either younger or older adults (Supplemental Figure 2).
Discussion
This first-in-human study of mRNA-17777 (V171) showed that at dose levels up to 300 μg, the mRNA-1777 (V171) vaccine was generally well tolerated. No SAEs related to study vaccine were reported. A few of the solicited injection-site AEs, such as tenderness to touch and injection site pain were more frequently reported in the mRNA treated groups versus placebo. Participants in the mRNA treated group also reported solicited systemic AEs more frequently as compared to placebo. The vast majority of all AEs from mRNA treated participants were transient Grade 1 or 2 events. These safety results are similar to findings from other reports of mRNA vaccines.Citation30,Citation31 The safety profile observed in the current study supports continuing the clinical evaluation of this or other RSV mRNA vaccines.
This study also demonstrates the ability of the investigational vaccine to increase humoral and cellular immunity against the RSV F protein in healthy younger and older adults. Although a correlate of protection against RSV disease in adults has not yet been established, it is accepted that a desirable vaccine candidate should increase RSV-neutralizing titers against both the RSV A and B subtypes.Citation18 In the current study, the highest observed GMFI in RSV A neutralization titers was at day 29 and ranged from 2.5 to 3.9-fold across the 25 μg, 100 μg, and 200 μg dose levels in the younger adults and 3.0 to 4.3-fold across the 25 μg, 100 μg, 200 μg and 300 μg dose levels in the older adults. For RSV B neutralizing antibody response, the highest observed GMFI was also at day 29 and was 3.1-fold (200 μg) in the younger adults and 3.1-fold (200 μg) and 2.9-fold (300 μg) in the older adults. Furthermore, neutralization antibody GMTs and GMFIs remained increased over baseline and placebo through at least Day 180 in all mRNA-1777 (V171) treatment groups.
Vaccination with mRNA-1777 (V171) also increased prefusion F-specific serum antibody titers and the levels of antibodies which compete with D25 for binding to prefusion F protein. While site Ø and site II are both expressed on the prefusion F protein, studies of adult antibody repertoire to natural infection have shown that antibodies targeting site Ø are more abundant and more potent overall as compared to those targeting site II.Citation35,Citation36 Therefore, it is not surprising that immunization with a prefusion encoding mRNA vaccine increased the competing antibodies to both sites, with numerically higher concentrations and GMFI in DCA as compared to PCA. Increased T-cell responses to RSV F were also detected in the mRNA-vaccinated groups relative to respective baseline as well as levels in the placebo group. Intracellular cytokine staining showed that the response was predominantly from the CD4 T cell subset, which differs from the preclinical results from similar vaccine candidates tested in mice, where potent CD4 and CD8 T cell responses were identified.Citation26
Although this study was not designed to compare immunogenicity responses between the older and younger adults, the humoral and cellular immunogenicity appeared to be generally similar between the two age groups. This is an important observation given that one of the target groups for this vaccine are older adults, a population that demonstrates reduced or poor immune responses to some vaccines.Citation37,Citation38 This phenomenon has been attributed to general immune function decline with age.Citation39 Here we are limited by the size of the study and larger evaluations would be required to fully characterize the immunogenicity in this age group, particularly those with comorbidities that might impact vaccine responses.
Recently there have been several other clinical trials with various investigational vaccines containing RSV prefusion-F-stabilized immunogens conducted in healthy adults. Direct comparison to these data is limited by differences in assays utilized and cohort demographics across studies. The WHO has recently established an international standard for antiserum to RSV;Citation40 however, the testing for the current study and most published RSV vaccine trials began prior to the integration of this standard into the assays. Taking these limitations into account, results can be generally compared by viewing fold changes in neutralization titers before and after vaccination. This method for comparison when two (or more) assays are involved makes three key assumptions: (1) that the within-participant increase (ratio) in assay value is an appropriate metric for their likely level of protection by the vaccine (or at least for the strength of their response), (2) that such ratios can be compared between participants (i.e. that a fourfold increase has a similar meaning in two participants),Citation41 and that a calibration curve for one assay against the other (i.e. the scatter of points obtained from repeatedly measuring in both assays samples with a wide range of titers) would be sufficiently close to linear.
There have been a few relevant studies in recent years evaluating RSV prefusion F vaccine candidates in adults with the goal of development for use in older adults or for maternal vaccination to protect infants. In a study reported by DeVincenzo et al., healthy adults aged 18–50 were immunized with a recombinant adenoviral vector containing prefusion F (Ad26.RSV.preF).Citation23 At day 28 post-vaccination, the RSV A IC50 GMT in the Ad26 vaccinated group was approximately 5-to 6-fold higher than baseline (267 versus 1589). This study preselected participants with the lowest 25% RSV titers at baseline as part of the design of the human challenge study, and the impact of such pre-selection on fold change in GMFI is not well understood. The Ad26.RSV.preF vaccine was also evaluated in a phase 1 study of adults ≥60 years old, and induced 2.3-fold (5 x 1010 viral particles) and 3.1-fold increase (1 x 10 11 viral particles) in RSV A neutralization GMTs from baseline to day 28 after a single dose.Citation42
Several recombinant protein-based prefusion F investigational vaccines have also been evaluated. Langley et al. reported an increase in RSV A neutralization titers ranging from 3.2 to 4.9-fold after vaccination with investigational prefusion F protein subunit vaccines (with or without alum adjuvant) in a phase I study in healthy adult men ages 18–44.Citation20 Additionally, Beran et al. described two phase II studies of RSV-prefusion F protein subunit vaccines (with and without alum adjuvant) in healthy adult women ages 18–45Citation21. Across both of those studies, a range of 3.1 to 3.9 fold ratio in GMT neutralizing titer from baseline to day 30 post-vaccination was reported.Citation21 More recently, an interim analysis was released from an ongoing study in healthy women aged 18–85 for three formulations of subunit prefusion RSV vaccines (with and without alum) demonstrating a geometric mean fold rise of 7.2 to 10.6 for RSV A neutralization and 10.4 to 19.8 for RSV B neutralization at day 30 post-vaccination.Citation22 Lastly, Crank, et al. reported an analysis of immunogenicity from VRC-317, a phase 1 study of a prefusion F subunit protein vaccine candidate in healthy adults 18–50 years of age. The results showed a greater than 10-fold change in RSV A neutralizing antibody levels (week 4 over baseline), and greater than ninefold for RSV B at the 150 μg dose levels. The VRC-317 vaccine candidate contains a soluble version of DS-Cav-1 prefusion F while the DS-Cav-1 in mRNA-1777 (V171) is membrane associated; it is currently unknown if, or to what extent, this contributes to the level of immunogenicity observed in the respective studies. The relative contribution to efficacy of any differences in the magnitude and durability of neutralizing antibodies, or other immune responses elicited, such as antigen-specific CD4 or CD8 T-cell responses across RSV F vaccine candidates remains to be determined.
There are a number of limitations to the current study. The safety assessments are primarily limited by study size and larger sample sizes will be needed to better understand the complete safety profile of this mRNA vaccine. The increased AEs observed in the vaccinated group as compared to placebo were primarily solicited reactogenicity events typical of vaccines. We are unable based on this study design to attribute them to the mRNA, lipid nanoparticle or the in-situ production of the RSV F protein. In terms of immunogenicity analysis, a precise correlate of protection for RSV in adults has not been established. Here, we have measured both humoral and T cell-mediated immunity, each of which may contribute to protection. The study enrolled a high percentage of males, particularly in the younger group due to the exclusion of women with childbearing potential. Additionally, there was a majority of white participants due to the demographics of the population where the study took place. It is unknown what the effect of ethnicity or gender is on the immunogenicity or safety due to the size of this study and thus larger and more diverse studies will be required to evaluate the impact of these factors.
Overall, this first-in-human study demonstrated that mRNA-V177 (V171) increased RSV F specific humoral and cellular immunity in younger and older healthy adults and was generally well tolerated. These results support additional studies with mRNA-based prefusion F vaccines optimized for immunogenicity for the prevention of RSV infection in vulnerable adult populations.
Disclosure of potential conflicts of interest
A.A., R.R., X.C., W.L., J.S., K.C., D.S, C.W., and E.L. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may hold stock in Merck & Co., Inc., Kenilworth, NJ, USA. A.E. is an inventor on a patent related to the vaccine and is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ USA, and may hold stock in Merck & Co., Inc., Kenilworth, NJ, USA. C.A.S. L.P and H.L. and T.Z. are employees of Moderna, Inc., Cambridge, MA 02139, USA, and may hold stock in Moderna, Inc., Cambridge, MA 02139, USA. I.S., is an employees of Moderna, Inc., Cambridge, MA 02139, USA. C.J.S., N.F. and P.G. have nothing to disclose.
Supplemental Material
Download MS Word (581.8 KB)Acknowledgments
The authors would like to thank the participants who enrolled in the study and the staff at PPD for contract research assistance for this study. We thank Karyn Davis, BA, Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, for editorial assistance. The authors would like to thank Melissa Whiteman, Ying Homan, Harilakshmi Kannan, Wen Feng, Reshma Panemangalore, Yu Cao, Sarah Carpenter, Lena Hofer, and Krystyn Delfino for their support in generation of immunogenicity data.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Glezen WP, Taber LH, Frank AL, Kasel JA. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child. 1986;140(6):543–46. doi:10.1001/archpedi.1986.02140200053026.
- Hall CB. Respiratory syncytial virus and parainfluenza virus. N Engl J Med. 2001;344(25):1917–28. doi:10.1056/nejm200106213442507.
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet. 2012;380(9859):2095–128. doi:10.1016/s0140-6736(12)61728-0.
- Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. Jama. 2003;289(2):179–86. doi:10.1001/jama.289.2.179.
- Shi T, Denouel A, Tietjen AK, Campbell I, Moran E, Li X, Campbell H, Demont C, Nyawanda BO, Chu HY, et al. Global disease burden estimates of respiratory syncytial virus-associated acute respiratory infection in older adults in 2015: A systematic review and meta-analysis. J Infect Dis. 2019. doi:10.1093/infdis/jiz059.
- Kwon YS, Park SH, Kim MA, Kim HJ, Park JS, Lee MY, Lee CW, Dauti S, Choi WI. Risk of mortality associated with respiratory syncytial virus and influenza infection in adults. BMC Infect Dis. 2017;17(1):785. doi:10.1186/s12879-017-2897-4.
- Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352(17):1749–59. doi:10.1056/NEJMoa043951.
- Cherukuri A, Patton K, Gasser RA, Zuo F, Woo J, Esser MT, Tang RS. Adults 65 years old and older have reduced numbers of functional memory T cells to respiratory syncytial virus fusion protein. J Clin Vacc Immunol. 2013;20(2):239–47. doi:10.1128/CVI.00580-12.
- Walsh E, Falsey AR. Humoral and mucosal immunity in protection from natural respiratory syncytial virus infection in adults. J of Infect Dis. 2004;190(2):373–78. doi:10.1086/421524.
- Duncan CB, Walsh EE, Peterson DR, Lee FE, Falsey AR. Risk factors for respiratory failure associated with respiratory syncytial virus infection in adults. J Infect Dis. 2009;200(8):1242–46. doi:10.1086/605948.
- Walsh EE, Peterson DR, Kalkanoglu AE, Lee FE, Falsey AR. Viral shedding and immune responses to respiratory syncytial virus infection in older adults. J Infect Dis. 2013;207(9):1424–32. doi:10.1093/infdis/jit038.
- Piedra PA, Jewell AM, Cron SG, Atmar RL, Glezen WP. Correlates of immunity to respiratory syncytial virus (rsv) associated-hospitalization: establishment of minimum protective threshold levels of serum neutralizing antibodies. Vaccine. 2003;21(24):3479–82. doi:10.1016/s0264-410x(03)00355-4.
- Luchsinger V, Piedra PA, Ruiz M, Zunino E, Martinez MA, Machado C, Fasce R, Ulloa MT, Fink MC, Lara P, et al. Role of neutralizing antibodies in adults with community-acquired pneumonia by respiratory syncytial virus. Clin Infect Dis. 2012;54(7):905–12. doi:10.1093/cid/cir955.
- Meng J, Stobart CC, Hotard AL, Moore ML. An overview of respiratory syncytial virus. PLoS Pathog. 2014;10(4):e1004016. doi:10.1371/journal.ppat.1004016.
- Magro M, Mas V, Chappell K, Vazquez M, Cano O, Luque D, Terron MC, Melero JA, Palomo C. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc Natl Acad Sci USA. 2012;109(8):3089–94. doi:10.1073/pnas.1115941109.
- Ngwuta JO, Chen M, Modjarrad K, Joyce MG, Kanekiyo M, Kumar A, Yassine HM, Moin SM, Killikelly AM, Chuang GY, et al. Prefusion f-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med. 2015;7(309):309ra162. doi:10.1126/scitranslmed.aac4241.
- McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GB, Yang Y, Zhang B, Chen L, Srivatsan S, Zheng A, et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science. 2013;342(6158):592–98. doi:10.1126/science.1243283.
- Graham BS. Immunological goals for respiratory syncytial virus vaccine development. Curr Opin Immunol. 2019:5957–64. doi:10.1016/j.coi.2019.03.005.
- Crank MC, Ruckwardt TJ, Chen M, Morabito KM, Phung E, Costner PJ, Holman LA, Hickman SP, Berkowitz NM, Gordon IJ, et al. A proof of concept for structure-based vaccine design targeting rsv in humans. J Science. 2019;365(6452):505–09. doi:10.1126/science.aav9033.
- Langley JM, Aggarwal N, Toma A, Halperin SA, McNeil SA, Fissette L, Dewe W, Leyssen M, Toussaint JF, Dieussaert I. A randomized, controlled, observer-blinded phase 1 study of the safety and immunogenicity of a respiratory syncytial virus vaccine with or without alum adjuvant. J Infect Dis. 2017;215(1):24–33. doi:10.1093/infdis/jiw453.
- Beran J, Lickliter JD, Schwarz TF, Johnson C, Chu L, Domachowske JB, Van Damme P, Withanage K, Fissette LA, David M-P, et al. Safety and immunogenicity of 3 formulations of an investigational respiratory syncytial virus vaccine in nonpregnant women: results from 2 phase 2 trials. J of Infect Dis. 2018;217(10):1616–25. doi:10.1093/infdis/jiy065.
- Schmoele-Thoma B, Falsey AR, Walsh EE, Swanson K, Zareba A, Cooper D, Gruber WC, Jansen KU, Radley D, Scott D, et al. 2755. Phase 1/2, first-in-human study of the safety, tolerability, and immunogenicity of an RSV prefusion F-based subunit vaccine candidate. Open Forum Infect Dis. 2019;6(Supplement_2):S970–S970. doi:10.1093/ofid/ofz360.2432.
- DeVincenzo J, Gymnopoulou E, De Paepe E, Murray B, Rosemary Bastian A, Haazen W, Noulin N, Comeaux C, Heijnen E, Eze K, et al. 902. A randomized, double-blind, placebo-controlled study to evaluate the efficacy of a single immunization of ad26.RSV.Pref against RSV infection in a viral challenge model in healthy adults. Open Forum Infect Dis. 2019;6(Suppl2):S27–S28. doi:10.1093/ofid/ofz359.061.
- Blanco JCG, Fernando LR, Zhang W, Kamali A, Boukhvalova MS, McGinnes-Cullen L, Morrison TG. Alternative virus-like particle-associated prefusion F proteins as maternal vaccines for respiratory syncytial virus. J Virol. 2019;93(23):e00914–19. doi:10.1128/jvi.00914-19.
- Marcandalli J, Fiala B, Ols S, Perotti M, de van der Schueren W, Snijder J, Hodge E, Benhaim M, Ravichandran R, Carter L, et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell. 2019;176(6):1420–1431.e1417. doi:10.1016/j.cell.2019.01.046.
- Espeseth AS, Cejas PJ, Citron MP, Wang D, DiStefano DJ, Callahan C, Donnell GO, Galli JD, Swoyer R, Touch S, et al. Modified MRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. NPJ Vaccines. 2020;516. doi:10.1038/s41541-020-0163-z.
- Jackson NAC, Kester KE, Casimiro D, Gurunathan S, DeRosa F. The promise of MRNA vaccines: A biotech and industrial perspective. NPJ Vaccines. 2020;5(1):11. doi:10.1038/s41541-020-0159-8.
- Mascola JR, Fauci AS. Novel vaccine technologies for the 21st century. Nat Rev Immunol. 2020;20(2):87–88. doi:10.1038/s41577-019-0243-3.
- Zhang C, Maruggi G, Shan H, Li J. Advances in MRNA vaccines for infectious diseases. Frontiers in Immunol. 2019;10:594. doi:10.3389/fimmu.2019.00594.
- Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, McCullough MP, Chappell JD, Denison MR, Stevens LJ, et al. An MRNA vaccine against SARS-COV-2 - preliminary report. N Engl J Med. 2020. doi:10.1056/NEJMoa2022483.
- Feldman RA, Fuhr R, Smolenov I, Mick Ribeiro A, Panther L, Watson M, Senn JJ, Smith M, Almarsson O, Pujar HS, et al. MRNA vaccines against H10nN and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine. 2019;37(25):3326–34. doi:10.1016/j.vaccine.2019.04.074.
- Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research (US). Guidance for industry: toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. Research CfBEa, ed. 2007.
- Citron MP, Patel M, Purcell M, Lin SA, Rubins DJ, McQuade P, Callahan C, Gleason A, Petrescu I, Knapp W, et al. A novel method for strict intranasal delivery of non-replicating RSV vaccines in cotton rats and non-human primates. Vaccine. 2018;36(20):2876–85. doi:10.1016/j.vaccine.2018.02.110.
- Dubey S, Clair J, Fu T-M, Guan L, Long R, Mogg R, Anderson K, Collins KB, Gaunt C, Fernandez VR, et al. Detection of HIV vaccine-induced cell-mediated immunity in HIV-seronegative clinical trial participants using an optimized and validated enzyme-linked immunospot assay. JAIDS. 2007;45(1):20–27. doi:10.1097/QAI.0b013e3180377b5b.
- Gilman MS, Castellanos CA, Chen M, Ngwuta JO, Goodwin E, Moin SM, Mas V, Melero JA, Wright PF, Graham BS, et al. Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci Immunol. 2016;1(6). doi:10.1126/sciimmunol.aaj1879.
- Graham BS. Vaccine development for respiratory syncytial virus. Curr Opin Virol. 2017;23:107–12. doi:10.1016/j.coviro.2017.03.012.
- Osterholm MT, Kelley NS, Sommer A, Belongia EA. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect Dis. 2012;12(1):36–44. doi:10.1016/s1473-3099(11)70295-x.
- Weinberger B. Vaccines for the elderly: current use and future challenges. Immun Ageing. 2018;15:3. doi:10.1186/s12979-017-0107-2.
- Ciabattini A, Nardini C, Santoro F, Garagnani P, Franceschi C, Medaglini D. Vaccination in the elderly: the challenge of immune changes with aging. Semin Immunol. 2018:4083–94. doi:10.1016/j.smim.2018.10.010.
- McDonald JU, Rigsby P, Dougall T, Engelhardt OG. Establishment of the first WHO international standard for antiserum to respiratory syncytial virus: report of an international collaborative study. Vaccine. 2018;36(50):7641–49. doi:10.1016/j.vaccine.2018.10.087.
- Guess HA, Broughton DD, Melton LJ 3rd, Kurland LT. Population-based studies of varicella complications. Pediatrics. 1986;78(4 Pt 2):723–27. PMID: 3763290. https://www.ncbi.nlm.nih.gov/pubmed/3763290.
- Williams K, Bastian AR, Feldman RA, Omoruyi E, de Paepe E, Hendriks J, van Zeeburg H, Godeaux O, Langedijk JPM, Schuitemaker H, et al. Phase 1 safety and immunogenicity study of a respiratory syncytial virus vaccine with an adenovirus 26 vector encoding pre-fusion f (ad26.RSV.Pref) in adults 60 years and older. J Infect Dis. 2020;222(6):979–88. doi:10.1093/infdis/jiaa193.
Appendix A.
Study Inclusion Criteria
Each participant must meet all of the following criteria to be enrolled in this study:
1. The subject is male or female (nonchildbearing potential) between 18 and 49 years of age (inclusive; Part A) or between 60 and 79 years of age (inclusive; Part B) at Screening.
2. Female participants of nonchildbearing potential are defined as: postmenopausal (defined as amenorrhea at least 12 consecutive months and documented plasma follicle-stimulating hormone [FSH] level at or above the postmenopausal level) or surgically sterile (i.e., hysterectomy, bilateral tubal ligation, or bilateral oophorectomy). NOTE: These procedures must be confirmed with medical records.
3. Male participants agree to use appropriate contraception for sexual intercourse up through1 month post vaccination.
4. The subject has a body mass index between 18 and 32 kg/m2, inclusive, at Screening. Body mass index will be calculated using the following formula: body weight (kg)/(height [m]2). The numerical result will be rounded to the nearest 0.1.
5. The subject is considered by the investigator to be in good general health as determined by medical history, clinical laboratory assessments, vital sign measurements, 12-leadelectrocardiograms (ECGs), and physical examination findings at Screening. With regard to liver function tests at Screening: ALT and AST must be equal to or below 1.5 × upper limit of normal (ULN). If ALT and AST levels are between 1.0 × and 1.5 × ULN, the blood glucose level must also be equal to or below the ULN. Alkaline phosphatase and direct bilirubin must be equal to or below the ULN. Total bilirubin may be up to 2 × ULN as long as direct bilirubin is equal to or below the ULN.
6. The subject agrees to comply with all protocol requirements.
7. The subject agrees to comply with the study restrictions.
8. The subject is a nonsmoker and/or has not used nicotine or nicotine-containing products (e.g., nicotine patch) for at least 4 weeks prior to Screening.
9. The subject understands the study procedures and agrees to participate by providing written informed consent (including Future Biomedical Research) before any study procedures, including Screening, are performed.
10. The subject has access to consistent and reliable means of telephone contact, which may be in the home, workplace, or by personal mobile electronic device.
11. The subject agrees to stay in contact with the study site for the duration of the study, to provide updated contact information as necessary, and has no current plans to move from the study area for the duration of the study.
Appendix B.
Study Exclusion Criteria
Subjects meeting any of the following criteria will be excluded from the study:
1. The subject is mentally or legally incapacitated, has significant psychological disorder at the time of screening or expected during the conduct of the study, or has a history of clinically significant psychiatric disorder within the last 5 years. Subjects who have had situational depression may be enrolled in the study at the discretion of the investigator.
2. The subject has any ongoing, symptomatic acute or chronic illness requiring medical or surgical care; asymptomatic conditions or findings or conditions (e.g., mild hypertension, dyslipidemia) that are not associated with evidence of end-organ are not exclusionary provided that they are being appropriately managed and are clinically stable (i.e., unlikely to result in symptomatic illness within the time-course of this study), in the opinion of the investigator. An exception is symptomatic osteoarthritis, which is permitted as long as surgery is not anticipated during the study. Note that illnesses or conditions may be exclusionary, even if otherwise stable, due to therapies used to treat them, at the discretion of the investigator.
3. The subject has a history of diabetes, including a fasting blood glucose greater than 125 mg/dL (7 mmol/L) at Screening.
4. The subject has a fasting blood glucose above the ULN but below 125 mg/dL (7 mmol/L) and a hemoglobin A1c above the ULN at Screening.
5. The subject has a history of active cancer (malignancy) in the last 10 years. Exception is subjects with adequately treated non-melanomatous skin carcinoma, who may participate in the study.
6. The subject has participated in another investigational study involving any investigational product (i.e., study drug, biologic, device) within 30 days, or 5 half-lives of the investigational product, whichever is longer, before dose administration.
7. The subject has previously participated in an investigational study involving LNPs.
8. The subject has a history of Guillain-Barré syndrome.
9. The subject has a history of narcolepsy.
10. The subject has any known or suspected autoimmune disease or immunosuppressive condition, acquired or congenital, as determined by medical history and/or physical examination.
11. The subject has had chronic (defined as more than 14 continuous days) or current administration of a systemic immunosuppressant or other immune-modifying drug, including any dose of oral corticosteroids, within 6 months prior to dose administration (Day 1). The use of topical, inhaled, and nasal glucocorticoids will be permitted.
12. The subject received immunoglobulins and/or any blood products within the 3 months preceding the administration of the study drug or at any time during the study.
13. The subject has any acute illness at the time of enrollment (defined as the presence of a moderate or severe illness with or without fever, or an oral temperature >38.0°C on the planned day of dose administration).
14. The subject has any significant disorder of coagulation requiring ongoing treatment, or a history of either more than 1 deep vein thrombosis or pulmonary embolism, or a single deep vein thrombosis or pulmonary embolism in the past 5 years. Subjects receiving prophylactic aspirin (≤150 mg/day), and without clinically apparent bleeding tendency, are eligible.
15. The subject has a history of asthma or allergic asthma in the past 5 years.
16. The subject has a history of multiple significant and/or severe allergies (e.g., food, drug, latex allergy), or has had an anaphylactic reaction or significant intolerability to prescription or nonprescription drugs, food, or vaccine.
17. The subject is expected to be receiving or is currently receiving antipyretic or analgesic medication on a daily or every other day basis from randomization through Day 8 (a daily dose of ≤150 mg of aspirin given under the guidance of a physician is not a contraindication to enrollment). For all analgesics and antipyretics, including a daily dose of aspirin ≤150 mg, a washout of 3 days will be required before dose administration.
18. The subject has a reported or documented history of alcohol abuse or drug addiction or nonmedicinal recreational drug use (excluding nonprescription health supplements and herbal remedies) within 1 year before the planned day of dose administration.
19. The subject has a positive test result for drugs of abuse, cotinine, or alcohol at Screening or before dose administration (Day 1).
20. The subject has any abnormality or permanent body art (e.g., tattoo) that would obstruct the ability to observe local reactions at the injection site (deltoid region).
21. The subject has any condition that, in the opinion of the investigator, would pose a health risk to the subject if enrolled or could interfere with evaluation of the study drug or interpretation of study results (including neurologic or psychiatric conditions deemed likely to impair the quality of safety reporting).
22. The subject received any other licensed vaccines (inactivated or live vaccines) within 4 weeks prior to dose administration or who are planning to receive any vaccine within 4 weeks from dose administration.
23. The subject has any unstable chronic medical condition, including one that has resulted in change in therapy (medication or other) in the 30 days prior to randomization or hospitalization in the previous year or might be predicted to result in hospitalization in the year after enrollment. Subjects with severe, untreated, or uncontrolled underlying medical disease that might either compromise subject safety or affect the ability to assess safety of the investigational product are excluded.
24. The subject has a positive test result for hepatitis B surface antigen, hepatitis C virus antibody, or human immunodeficiency virus types 1 or 2 antibodies at Screening.
25. The subject has donated blood or blood products >450 mL within 30 days of dosing.