
ABSTRACT
Our previous research verified that HSP (heat shock protein) 110 could enhance the anti-tumor effect of HPV16 E749-57 epitope. In this study, to optimize the immunotherapy of this vaccine type, we developed and evaluated the anti-tumor immunity of a nanoparticle vaccine format assembling with E749-57-HSP110 fusion expression plasmid and RGD-GGG-K18 polypeptide. The nanoparticle vaccine was self-assembled from positively charged RGD-GGG-K18 polypeptide and negatively charged fusion expression plasmid pIRES2-3× E7-HSP110-EGFP. The particle size, stability, expression of E749-57-HSP110 fusion protein and the target ability of nanoparticle were determined, respectively. Specific CTL responses were determined by E7 tetramer staining and cytotoxicity assay in TC-1 tumor-bearing mice (CD4/CD8 knockout). The preventive and therapeutic experiments of nanoparticle vaccine were investigated in TC-1 tumor-bearing mice. Results showed that the RGD-GGG-K18 polypeptide and pIRES2-3× E7-HSP110-EGFP plasmid self-assembled nanoparticles about 100 nanometers in diameter when the charge ratios of peptide/plasmid were 2. The nanoparticles effectively entered TC-1 cells directed by RGD target-peptide, and correctly expressed the E7-HSP110 fusion protein. The HSP110 effectively facilitated nanoparticles activating CD8+T cells than nanoparticles without HSP110, including the CD8+ T cell number and the IFN-γ level; in contrast, the CD4+T cells immune response remained indiscriminate among the mice groups. This nanoparticle formulation inhibited tumor growth and prolonged the survival duration in the prophylactic and therapeutic mouse models. Therefore, the RGD-based tumor-targeting nanoparticle expressing E749-57-HSP110 fusion protein can efficiently evoke anti-tumor activity and thus suggests it might be a favorable candidate for cervical cancer immunotherapy.
1. Introduction
It is well established that human papillomavirus (HPV) is the etiologic agent of cervical cancer.Citation1,Citation2 HPV16 and 18 are the main subtypes of inducing cervical cancer, and HPV16 infection accounts for more than half of cervical cancer cases.Citation3,Citation4 The HPV16 E7 oncoproteins are constitutively expressed in the infected lesions and are responsible for maintenance of cellular transformation and tumorigenic property. Therefore, E7 oncoprotein is an attractive target for designing vaccine and immunotherapeutic strategies against HPV16-associated cervical cancer.Citation1 The available prophylactic vaccines for HPV can effectively prevent high-risk HPV infection, but they are incapable in the treatment for preexisted cervical cancer. Thus, it is critical to develop therapeutic vaccine against the tumor lesions. Recently, a number of alternative vaccine approaches have been developed targeting E7 oncoprotein, and met with varying degrees of success in preclinical and clinical trials.Citation5 These documented vaccine strategies have included vaccines based on DNA, vectors, dendritic cells, peptides or proteins, and combination approaches. Among these, the peptide-based vaccines comprising cytotoxic T lymphocyte (CTL) epitope have shown particularly promising method in virtue of efficacy and safety, and are easy to prepare, as CTL epitope plays a pivotal role to induce cellular immune response against tumor via binding MHC (major histocompatibility complex) molecules. To date, HPV16 E749-57 polypeptide as an immuno-dominant CTL epitope has been extensively used in therapeutic vaccine for cervical cancer.Citation6–10 However, the epitope peptide is of small molecular size, degradable, and has relatively low affinity for MHC molecules, resulting in weak immunogenicity and moderate efficacy. Therefore, epitope-based vaccines require optimizing strategies to prime more powerful immunogenic performance.
One of such potential strategies is the utilization of chaperone property of HSP110 to enhance the polypeptide epitope immunogenicity. It is well known that T cell immunity plays a key role in anti-tumor process. HSP110 has strong peptide-binding ability,Citation11 efficiently presents the peptide to APCs (antigen-presenting cells), and accordingly accelerates peptide-MHC recognition, for augmenting the specific T cell immune response. APCs capture HSP-antigen complex by receptor-mediated endocytosis, which is essential for HSP to promote antigen cross presentation.Citation12 The relevance of the peptides associated with HSPs for inducing specific immune responses is demonstrated by several studies, and HSP110 purified from diverse tumors and functioning as tumor vaccines have shown to cause tumor regression in animal models,Citation13 of which the key factor is the successful activation of CD8+ T cell-dependent tumor immunity.Citation14 Wang et al. reported that tumor-derived HSP110 elicits a more potent antitumor response on a molar basis than HSP70.Citation15 The enhanced immunogenicity has been attributed to HSP110’s more efficient chaperoning capability.Citation16 Kim et al. suggested that the affinity with which the chaperone binds antigen significantly contributes to its ability to generate a cytotoxic T-lymphocyte (CTL) response.Citation17 Our previous study has also indicated that HSP110 can effectively bind polypeptides under heat shock and produce good adjuvant effects.Citation8,Citation18
Tumor-targeting is another pivotal strategy for tumor immunotherapy. RGD (Arginine-Glycine-Aspartate containing peptide: ACRGDMFFCA) is a target sequence of αvβ3 integrin that specifically expresses on tumor cells and has been widely used in the research of anti-tumor targeted therapy.Citation19,Citation20 The combination of RGD peptide and chemotherapy drugs has attracted special attention and has been studied in depth. These delivery systems are able to transfer drugs to the endothelial cells and cancer cells of the blood vessels through the combination of RGD peptides and the expression of ανβ3 integrin in tumor cells.Citation21 The tumor targeting strategy based on this theory is of great concern because the phagocytosis of integrin receptor ligands has been proved to be accomplished through the receptor mediated endocytosis. Therefore, in the process of internalization, RGD binding drugs or DNA plasmid could facilitate them to enter cancer cells.Citation21–23
It also has been documented that nanoparticles can condense plasmid DNA into small particles by means of electrostatic interaction between positively charged peptide segments and negatively charged DNA skeleton.Citation24 And recent researches have shown that these systems are effective in delivering cationic peptide-related antigens and transferring genes to DC.Citation25,Citation26 However, the application of RGD peptide in self-assembled nanoparticles for cervical cancer has not been proposed. In our previous results, we demonstrate that HSP110 as biological adjuvant can enhance the anti-tumor therapy of cytotoxic T lymphocyte epitope HPV16 E749-57 in mice.Citation8 In this study we further used the RGD-GGG-K18 cationic peptide to encase the plasmid E7-HSP110 expressing E749-57-HSP110 protein into nanoparticles, which demonstrated the effective anti-tumor capacity as evidence by the potent anti-tumor immune response and therapeutic efficiency ex vivo and in vivo.
2. Materials and methods
2.1. Plasmids
The E7 CTL epitope E749-57 (RAHYNIVTF) fragment was amplified by PCR from the plasmid BmtI-3XE7-BglII in pUC19-Amp (Sino-Biological, Beijing, China), and the restriction enzyme sites Bmt I and Bgl II (New England Biolabs, USA) were introduced into the upstream and downstream of the epitope, and the fragment was inserted between the corresponding enzyme sites of the pIRES2-EGFP vector (Sino-Biological). Then, plasmid pGEM-HSPH1 (Sino-Biological) was used as template and PCR method was used to clone HSP110, and BglII and Sal II (New England Biolabs) were introduced upstream and downstream of fragment HSP110 respectively. The fragment was inserted between Bgl II and Sal II restriction enzyme sites of the pIRES2-EGFP vector and the E7/pIRES2-EGFP plasmid, respectively. Thus, p3× E7/RIES2-EGFP, pHSP110/RIES2-EGFP and p3× E7-HSP110/RIES2-EGFP plasmids were prepared.
2.2. Peptides
The cationic peptide RGD-GGG-K18 (hereafter referred to as AK), GGG-K18 (hereafter referred to as GK) and RGD-GGG were synthesized, and the latter two served as controls formulation according to the published study.Citation27 The purity of the peptides determined by HPLC (reverse-phase high performance liquid chromatography) was >95%. The E749-57 peptide was selected as a mouse H-2 Db CTL epitope ().
Table 1. Sequence of peptides used in the present study
2.3. Mice and cell lines
TC-1 cells, which were derived from primary epithelial cells of C57BL/6 mice transformed with HPV16 E6, E7 and c-Ha-ras oncogenes,Citation28 were purchased from the Biospes Biotechnology company in Chongqing, China, and cultured in DMEM (HyClone, Logan, UT, USA) with 10% fetal bovine serum (FBS; SERENA), containing 1% antibiotics in 5% CO2 at 37°C. HAVSMC (human aortic smooth muscle cell) cells were obtained from Institute of Immunology of Third Military Medical University, and were grown in DMEM supplemented with 10% FBS, 1% antibiotics. C57BL/6 mice (female, six- to eight-week old) were purchased from the laboratory animal center of Third Military Medical University in Chongqing, China. All animals were maintained in pathogen-free conditions. All of the animal studies were performed under the Guidelines for Animal Care and Use Committee of the Third Military Medical University.
2.4. Nanoparticle preparation
The peptide/DNA charge ratio was calculated according to the published method.Citation29 One microgram of pIRES2-3× E7-EGFP (hereafter referred to as E7), pIRES2-HSP110-EGFP (hereafter referred to as HSP110) or pIRES2-3× E7-HSP110-EGFP (hereafter referred to as E7-HSP110) plasmid with a series of different amounts of AK or GK polypeptide were dissolved in 60 μl Hank’s balanced salt solution (HBS; 150mMNaCl, 10 mM HEPES, pH 7.2). After vortexing for 30 min and subsequent incubation for 30 min at room temperature, the plasmids and cationic peptides self-assembled into nanoparticles. The particle properties were identified by DNase I protection assay, gel retardation assay, TEM (transmission electron microscopy) and particle size analysis as our previous report.Citation29
2.5 Targeting ability by immunofluorescence microscope detection
Cell vitality was evaluated before targeting ability detection. TC-1 cells were seeded in 96-well plates (1 × 106cells/well) and incubated in DMEM containing 10% FBS (DMEM/FBS, 100 μl) for 24 h. The cells were exposed to AK/(E7, HSP110, E7-HSP110), GK/(E7, HSP110, E7-HSP110), peptide and plasmids for 48 h, respectively, and the annexin-V-FLUOS Staining was performed to assess cell vitality. Subsequently, to test whether the nanoparticle vaccine types were able to target tumor cells, TC-1 and HAVSMC cells were seeded in 12-well plates (5 × 106 cells/well) and incubated in 1 ml of DMEM/FBS for 24 h. The culture medium of TC-1 cells was then exchanged with fresh DMEM/FBS containing FITC-labeled AK and FITC-labeled GK, and culture medium of HAVSMC cells was then exchanged with fresh DMEM/FBS containing FITC-labeled AK. The cells were incubated for 48 h. Thereafter, cells were washed three times with PBS to remove unbound peptide, and observed by positive fluorescence microscope (OLYMPUS, Japan).
2.6. Competitive inhibition experiment
TC-1 cells were seeded in 12-well plates (5 × 106 cells/well) (microscope cover glasses put in before) and incubated in 1 ml of DMEM/FBS for 24 h. The culture medium was then exchanged with fresh DMEM/FBS, 2 μg/μl AK-labeled with Rhodamine and a different concentration of free RGD-GGG cyclic peptides were incubated with TC-1 cells for 48 h. After that, cells were washed three times with PBS to remove unbound peptide, and the fluorescence on cells was then determined by fluorescence microscope (OLYMPUS, Japan).
2.7. Cell transfection and Western blotting
TC-1 cells were inoculated with nanoparticles of AK/(E7, HSP110, E7-HSP110) and GK/(E7, HSP110, E7-HSP110) at charge ratios of 2.0 in a serum-free medium for 4 h, followed by replacement with complete DMEM medium. Forty-eight hours after inoculation, the cells were harvested for Western blotting, to verify the expression of HSP110 by using an anti-mouse HSPH110 antibody (R&D Systems, Minneapolis, USA) as the primary antibody.
2.8. Vaccine formulations and immunizations
Nanoparticles were prepared at charge ratio of 2.0, containing 74 μg of plasmid E7-HSP110 or 73 μg of HSP110 (equal moles to 50 μg of E7 plasmid) combined with proportional amounts of AK/(E7, HSP110, E7-HSP110) and GK/(E7, HSP110, E7-HSP110), control groups included immunizations with 74 μg E7-HSP110 or 73 μg HSP110 or 50 μg E7 plasmid in HBS alone (). The final volume of each vaccination formulation was adjusted to 100 μl with HBS, and then was used for mouse immunization via intraperitoneal injection at the right flank, followed by the same regimen two weeks later. At ten days after the second immunization, intracellular cytokine staining and LDH (the lactate dehydrogenase) release assay were applied.
Table 2. Immunization protocols of nanoparticle vaccine in C57BL/6 mice
2.9. In vitro splenocyte proliferation assay
As our previous study,Citation8 splenocytes from immunized mice of indicated group were seeded in triplicate in 96-well culture plates and incubated with E749–57 peptides (10 μg/ml) at 37°C in a humidified 5% CO2 atmosphere for 5 days. The assays were performed according to the MTT (methyl thiazolyl tetrazolium) test kit protocol (Sigma-Aldrich). Briefly, after 5 days incubation, the cells in each well were pulsed with MTT (50 µg/well) for the 4 h, followed by DMSO addition and the record of OD570 value in each well. Proliferation index (ratio) was reported as the number of cells when stimulated with E749–57 over that without E749–57 stimulation.
2.10. In vitro specific cytolysis test by LDH release assay
Splenocytes were collected from the vaccinated mice, and restimulated with E749-57 (10 μg/ml) and mIL-2 (10 μg/ml) for five days. The viable splenocytes were used as effector cells. The TC-1 cells were used as the target cells. According to the manufacturer’s protocol, the Non-Radioactive Cytotoxicity assay (Promega, USA) was used to detect the cytolytic effect of effector cells, which target on TC-1 cells at indicated E/T ratios (effector/target cell ratios). Specific lysis was calculated according to the formula: % specific lysis = [(Experimental release-spontaneous release/(Maximal release-spontaneous release)] x100%. The experiments were repeated three times.
2.11. Detection of E749-57-specific T cell response by tetramers tracing and flow cytometry analysis
At ten days after the second vaccination, splenocytes were collected from the immunized mice (five per group). 25 μl pooled splenocytes (2 × 107/ml) from each vaccination group were seeded in 48-well culture plates and stained with 25 μl 2× Staining Cocktail with PE-conjugated E749-57 peptide-MHC class I tetramers (HelixGen bio-company, China) and FITC-conjugated CD4 antibody (Biolegend, USA) for 1 h ice-cryopreservation. And the staining of PE-conjugated E749-57 peptide-MHC class I tetramers and FITC-conjugated CD8a antibody (Biolegend, USA) was performed independently. The cells were washed twice with 150 μl FACS buffer. Then, cells were re-suspended in fixed solution. Analysis was performed on a BD Biosciences FACS can with CellQuest software (BD Immunocytometry Systems).
To explore the IFN-γ secretion of T cells, similarly, at ten days after the second vaccination, splenocytes were collected from the immunized mice (five per group). First, 1 ml pooled splenocytes (5 × 106/ml) from each vaccination group with E749-57 peptide (10 μg/ml) and mIL-2 (10 μg/ml) were incubated for 16 h, then incubated with Golgistop (Biolegend, USA) for 6 h. Cells were stained with FITC-conjugated-CD8a antibody for 30 min. Subsequently, the cells were washed twice with PBS, and permeabilized by Permeability buffer for 40 min. Last, the cells were stained with PE-conjugated-IFN-γ (Biolegend, USA) antibody for 40 min. Cells were subjected to flow cytometry.
2.13. Mouse tumor model
In tumor challenge experiments, C57BL/6 mice (n = 10 per group) were immunized by the dosage regimens described above and a two-week interval. Ten days after the second immunization, the mice were injected s.c. (subcutaneous) in the right flank with 5 × 105 TC-1 cells in PBS. The mice were observed twice a week for survivals. In tumor therapeutic experiments, C57BL/6 mice (n = 10 per group) were initially injected s.c. in the right flank with 5 × 105 TC-1 cells. The first vaccination was performed when the tumor tissue could be palpated (around 7 days post tumor inoculation). On day 7 and 14 after tumor inoculation, the mice were vaccinated s.c. in the right flank with nanoparticles or a series of indicated formulations by the doses described above and then monitored twice a week for tumor growth. The tumor size was observed for 30 days post tumor inoculation. Tumor volume was calculated based on the following equation: V (tumor volume) = L(length) ×W (width) ×D (depth). Starting at day 21 after challenging the established tumor cells, the tumors were measured every other day, and mice with tumor sizes >2700 mm3 were considered moribund and sacrificed.
2.14. Statistical analysis
The experimental data were evaluated by ANOVA. P-values <0.05 were considered as statistically significant.
3. Results
3.1. Assembly and characterization of the nanoparticles
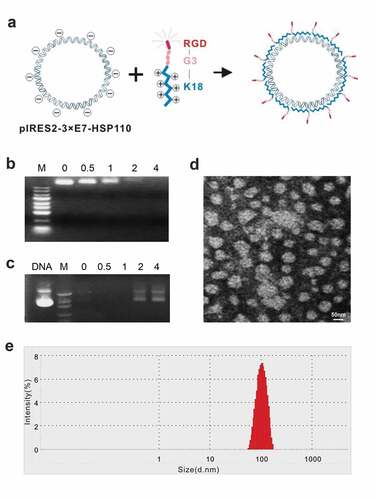
Nanoparticles were self-assembled from positively charged polypeptides and negatively charged plasmids () at the indicated peptide (+)/plasmid (−) charge ratios. The DNA retardation assay showed that the negative charge of plasmid DNA was not completely neutralized if the charge ratios of peptide/plasmid were <2 because a certain amount of DNA was detected in agarose. But when the charge ratio was ≥2, the DNA band disappeared completely, and the plasmid DNA was neutralized by the peptide and was completely blocked (). The DNase I protection assay indicated that DNase I easily digested the plasmid DNA just ratios were <2; however, the peptide effectively protected the plasmid DNA when the ratio ≥2 (). Therefore, the ratio 2 was taken as the suitable ratio of peptide/plasmid and was used for the nanoparticle preparation. The TEM showed that most of the nanoparticles were nearly circular, with a diameter of about 100 nm (). The particle size analyzer exhibited the diameter of most particles ranging from 70 to 130 nm, with a peak at 100 nm ().
Figure 1. Identification of biological features of nanoparticles
3.2. Nanoparticles AK/E7-HSP110 effectively entered TC-1 cells and induced exogenous HSP110 expression in TC-1 cell line
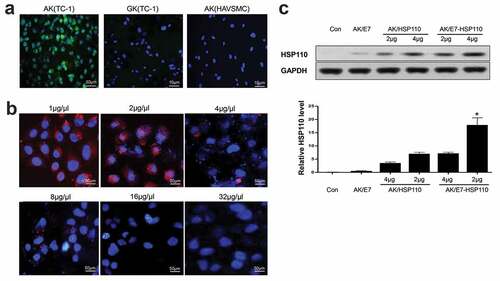
The targeting ability of nanoparticles to cancer cells overexpressing αvβ3 integrin as a receptor of RGD peptide,Citation30,Citation31 was particularly emphasized. Prior to evaluate the delivery efficacy, the viability of TC-1 and HAVSMC cells was measured after 48 h treatment with the nanoparticles. No significant changes in TC-1 and HAVSMC cell proliferation and apoptosis between the experimental group and the controls (data no show), indicating the negligible cytotoxicity of nanoparticles to the experimental cell lines. Then, the TC-1 or HAVSMC cells were incubated with nanoparticles for 48 h, and were analyzed by fluorescence microscope. Results demonstrated that AK containing nanoparticles entered TC-1 cells more effectively than GK; however, AK particles hardly entered HAVSMC cells that express undetectable αvβ3 integrin (). The specific recognition of AK to αvβ3 integrin was further evaluated by a competition assay. After AK-labeled Rhodamine and a series of concentrations of free RGD-GGG cyclic peptides were incubated with TC-1 cells, the mean fluorescence intensity of AK in TC-1 cells gradually but significantly decreased to the background level (), suggesting that the targeting feature of nanoparticles was attributed to the specific binding of RGD with the αvβ3 integrin. Importantly, the internalized nanoparticles could efficiently express the exogenous gene because nanoparticles AK/E7-HSP110 displayed higher HSP110 (116KD) protein expression than the controls ().
Figure 2. AK-nanoparticles targeted TC-1 cells and expressed exogenous protein
3.3. Nanoparticle AK/E7-HSP110 elicited E749-57-specific lymphocyte immune responses in vitro
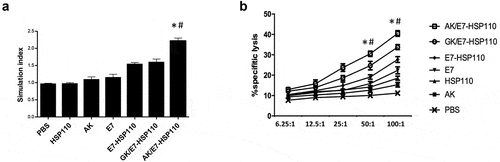
To investigate whether E749-57 specific immune responses can be induced in C57BL/6 mice, ten days after the last immunization with indicated immunogens, splenocytes were collected from the immunized mice, followed by in vitro re-stimulation with E749-57 peptide for five days. The proliferation assay demonstrated that the splenocytes from mice immunized with AK/E7-HSP110 proliferated more efficiently than various control groups (). Lymphocyte cytolytic capability was further assessed by LDH release assay using TC-1 cells as a target. Accordingly, the AK/E7-HSP110 nanoparticles showed significantly stronger cytolytic ability than controls when the target ratio (E/T) was 50:1 and 100:1 (). Of note, AK/E7-HSP110 nanoparticles showed markedly higher proliferation and cytolytic capability than GK/E7-HSP110 nanoparticles, indicating the significance of tumor targeting in anti-tumor immunity.
Figure 3. Nanoparticle AK/E7-HSP110 elicited E749-57-specific lymphocyte proliferation and cytolytic capacity
3.4. MHC class I tetramers recognized functional E749-57-specific epitope of CD8+ T cells
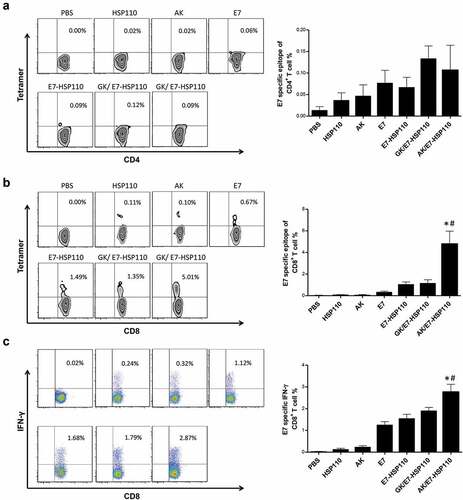
MHC I tetramers tracing and FCM assays were used to evaluate whether the nanoparticles AK/E7-HSP110 vaccine can trigger the expansion of E749-57-specific CD8+T or CD4+ T cells in mice. The splenocytes were collected from immunized C57BL/6 mice at ten days after the last immunization. FCM assays indicated that there was no detectable E749-57-specific CD4+ T cells of mice among mice immunized with nanoparticles AK/E7-HSP110 or other control nanoparticles (). However, the results showed the frequency of E749-57-specific CD8+ T cells in AK/E7-HSP110 group (5.01 ± 1.22%) markedly increased compared with other groups (). To address the functional activity of the E749-57-specific CD8+ T cells in each group, the splenocytes were collected from the immunized mice and re-stimulated with the E749-57 peptide in vitro, and FCM assay indicated that the frequency of CD8+IFN-γ+ T cells in mice immunized with nanoparticles AK/E7-HSP110 was much higher than GK/E7-HSP110 group (P< .05) and all other control groups (P< .01) ().
Figure 4. Specific CD8+T-cell immune response promoted by nanoparticle AK/E7-HSP110 in mice
3.5. Vaccination with AK/E7-HSP110 nanoparticles inhibited tumor growth in wild type but failed in CD8-knockout C57BL/6 mice
To determine whether the AK/E7-HSP110 nanoparticles exert efficient anti-tumor effects in vivo, we observed the survival rate of C57BL/6 mice inoculated with TC-1 cells in a tumor prophylactic experiment. We found that 80% of mice immunized with AK/E7-HSP110 nanoparticles and 50% of mice immunized with E7-HSP110 nanoparticles successfully survived for 60 days, the in vivo experimental end of this study. In contrast, all mice immunized with other control immunogens died before 50 days post immunization (). We further performed the tumor treatment experiments to evaluate whether the AK/E7-HSP110 nanoparticles display the therapeutic anti-tumor effects in the same animal tumor model. Results showed that the tumors volume of the AK/E7-HSP110 immunization group were significantly smaller than all other groups at day 20, 23, 27 ().
Figure 5. Preventative and therapeutic effects of vaccines on mice inoculated with TC-1 tumor cells
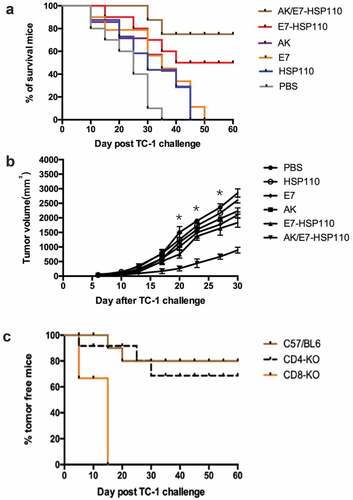
To investigate whether CD4+ or CD8 + T cells are responsible for the anti-tumor effects of the nanoparticles AK/E7-HSP110, the tumor challenge experiment was conducted in CD8- and CD4-knockout mice (n = 10 each group). Mice were vaccinated with the AK/E7-HSP110 nanoparticles twice with 2-week intervals, and followed by inoculated with TC-1 cells. The tumor growth was observed for 60 days post tumor inoculation. The bar graph demonstrated that nanoparticles AK/E7-HSP110 immunization protect 80% of C57BL/6 mice and 70% of CD4-knockout mice from the tumor plantation, but had particularly lowest anti-tumor ability against TC-1 tumor in CD8-knockout mice (), indicating CD8+ T cells are essential for immune response against tumor challenge elicited by immunization of nanoparticles AK/E7-HSP110.
4. Discussion
Vaccination is an attractive strategy to direct the immune system against specific cancer-associated antigens in tumors. CD8+ T cells play a fundamental role in tumor rejection, and thus induction of a potent CD8+ T-cell response is a major goal of vaccines. HPV positive cervical cancer is attractive prototype for immunotherapy in vaccines, given that the viral oncogenes encode neoantigenic oncoproteins that drive the disease. In patients with HPV+ tumors, generating therapeutically efficacious CTL responses has proven difficult to achieve with classical vaccination strategies involving peptide vaccines.Citation32 To evoke more potent immunity against tumor, in this study we design a new vaccine format by combination of the fusion gene fragment encoding both the immuno-dominant CTL epitope E749-57 and the potent chaperone molecule HSP110, and the cationic peptide RGD-GGG-K18, which will self-assemble into nanoparticles with tumor cell targeting ability. Our results shown that such nanoparticle vaccine AK/E7-HSP110 significantly improved the epitope-specific immunity both ex vivo and in vivo via CD8-dependent manner in mice. The vaccination of nanoparticle vaccine also prevented TC-1 tumors growth and prolonged the survival time of the immunized mice.
The over expression of integrin is the unique characteristic structure and biochemistry of tumor cells. The integrin ligands can thereby selectively direct molecules or drugs to tumors.Citation33 RGD has been widely used as the ligand of integrin ανβ3 which is highly expressed in tumor cells.Citation21 Therefore, the tumor targeting strategy, RGD binding drugs or DNA plasmid could facilitate them to enter tumor cells.Citation21–23 It has been well recognized that nanoparticles have the capability of condensing plasmid DNA into small particles to augment the expression of objective DNA. RGD conjugated nanoparticle as the specific delivery system, will facilitate the delivery efficacy and the targeting accuracy. Sun Y and Long Q have verified that nanoparticle systems are effective both in delivering antigens linked to cationic peptide and in transferring genes into target cells.Citation20,Citation23 However, the use of the cationic peptide RGD in self-assembly nanoparticles for HPV16 associated cervical cancer has not been addressed to date yet, which however has been addressed in this study. We find that the prepared nanoparticle AK/E7-HSP110 has no impact to TC-1 cells vitality. Furthermore, as a classic tumor targeting ligand,Citation23,Citation34 the targeting accuracy of RGD has been also verified in this study by the analysis of competitive test in which free RGD-GGG cyclic peptides can effectively inhibit the entry of nanoparticles AK/E7-HSP110 into TC-1 cells.
Heat shock protein has been identified to play a significant role in tumor antigen presentation and in triggering antigen cross expression in dendritic cells (DC).Citation35,Citation36 In this process known as cross presentation, the immature DCs capture antigens carried by HSP and develop into a fully mature DC (immunogenic DC) with potential T cell stimulation. Therefore, the molecular chaperone seems to be able to involve in the intracellular antigen processing pathway in DC and trigger the cross-presentation of CD8+ associated antigens and trigger the CTL response. In addition to enhancing the transportation and processing of antigens, it is also found that APC-HSP interaction can promote the phenotype and functional maturity of APC. Although the intrinsic stimulation effect of stress protein is controversial, there is plenty of evidence that they can be used as a “danger signal” in the extracellular environment after injury or stress cells are released. It has been reported that HSP110 DNA vaccine inhibits CT26 colorectal cancer and B16 melanoma, and activates HSP110 specific CD4+ and CD8+ T cells.Citation37 When used for a vaccine adjuvant, HSP110 can significantly induce the body’s immune response to its associated antigen and induce or promote the secretion of TNF-α andIL-12. These pro-inflammatory factors can also be used as “dangerous signals” to further induce immune cells to aggregate and stimulate the immune response.Citation38 Strengthened by the direction of RGD and the chaperone property of HSP110, the nanoparticles AK/E7-HSP110 has been shown the capacity of eliciting strong lymphocyte proliferation and triggering potent cytotoxic lysis to TC-1 cells when restimulated with E749-57 peptide in vitro.
In addition, we have also identified that the elevated immune response mainly attributed to specific CD8+ T lymphocytes, evidenced by MHC tetramer tracing test. Specific IFN-γ secretion by CD8+ T lymphocytes can be induced by E749-57 peptide restimulation as determined by MHC tetramer analysis. In addition, AK/E7-HSP110 nanoparticles have been shown to completely protect CD4-KO mice from subsequent tumor challenge; in contrast, such nanoparticle vaccination of the CD8-KO mice does not provide any protection from further tumor challenge. These findings indicated that the CD8+ T cells other than the CD4+ T cells were absolutely required for the induction of a protective immune response by vaccination with AK/E7-HSP110 in this tumor model.
In conclusion, in this study we combine the application of the RGD-GGG-K18 and fusion double expression plasmid IRES2-3× E7-HSP110-EGFP to form a novel nanoparticle vaccine. Data in this study indicate that the nanoparticle AK/E7-HSP110 significantly boosts the CD8+ T cell dependent anti-tumor immune response, possibly by enhancing MHC-I antigen presentation by efficient delivery of the antigenic payload to cross-presenting pathway in DCs. Though the vaccine format in this study can efficiently decrease tumor growth and extend the survival time in mice model, further modified vaccine format with more T cell epitopes and B cell epitopes included to develop multiple peptides-based vaccine strategy might evoke stronger anti-tumor immune response, which could be tested in the future.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. Lancet. 2013;382(9895):889–99. doi:10.1016/S0140-6736(13)60022-7.
- Luria L, Cardoza-Favarato G. Human papillomavirus. In: StatPearls. Treasure Island (FL); 2020. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK448132/#article-23034.s2
- Iljazovic E, Mena M, Tous S, Alemany L, Omeragić F, Sadiković A, Clavero O, Vergara M, Bosch FX, De Sanjosé S, et al. Human papillomavirus genotype distribution in invasive cervical cancer in Bosnia and Herzegovina. Cancer Epidemiol. 2014;38(5):504–10. doi:10.1016/j.canep.2014.06.004.
- Munoz N, Franceschi S, Bosetti C, Moreno V, Herrero R, Smith JS, Shah KV, Meijer CJ, Bosch FX. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet. 2002;359(9312):1093–101. doi:10.1016/S0140-6736(02)08151-5.
- Smalley Rumfield C, Roller N, Pellom ST, Schlom J, Jochems C. Therapeutic vaccines for HPV-associated malignancies. Immunotargets Ther. 2020;9:167–200. doi:10.2147/ITT.S273327.
- Feltkamp MC, Smits HL, Vierboom MP, Minnaar RP, De Jongh BM, Drijfhout JW, Schegget JT, Melief CJM, Kast WM. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur J Immunol. 1993;23(9):2242–49. doi:10.1002/eji.1830230929.
- Khammanivong V, Liu XS, Liu WJ, Rodda SJ, Leggatt GR, Tindle RW, Frazer IH, Fernando GJP. Paucity of functional CTL epitopes in the E7 oncoprotein of cervical cancer associated human papillomavirus type 16. Immunol Cell Biol. 2003;81(1):1–7. doi:10.1046/j.1440-1711.2003.01130.x.
- Ren F, Xu Y, Mao L, Ou R, Ding Z, Zhang X, Tang J, Li B, Jia Z, Tian Z, et al. Heat shock protein 110 improves the antitumor effects of the cytotoxic T lymphocyte epitope E7(49-57) in mice. Cancer Biol Ther. 2010;9(2):134–41. doi:10.4161/cbt.9.2.10391.
- Chen S, Ou R, Tang J, Deng X, Wu Y, Van Velkinburgh JC, Ni B, Xu Y. Enhanced anti-tumor effects of HPV16E7(49-57)-based vaccine by combined immunization with poly(I:C) and oxygen-regulated protein 150. Cancer Epidemiol. 2013;37(2):172–78. doi:10.1016/j.canep.2012.10.005.
- Barrios K, Celis E. TriVax-HPV: an improved peptide-based therapeutic vaccination strategy against human papillomavirus-induced cancers. Cancer Immunol Immunother. 2012;61(8):1307–17. doi:10.1007/s00262-012-1259-8.
- Goeckeler JL, Petruso AP, Aguirre J, Clement CC, Chiosis G, Brodsky JL. The yeast Hsp110, Sse1p, exhibits high-affinity peptide binding. FEBS Lett. 2008;582(16):2393–96. doi:10.1016/j.febslet.2008.05.047.
- Murshid A, Gong J, Calderwood SK. The role of heat shock proteins in antigen cross presentation. Front Immunol. 2012;3:63. doi:10.3389/fimmu.2012.00063.
- Segal BH, Wang XY, Dennis CG, Youn R, Repasky EA, Manjili MH, Subjeck JR. Heat shock proteins as vaccine adjuvants in infections and cancer. Drug Discov Today. 2006;11(11–12):534–40. doi:10.1016/j.drudis.2006.04.016.
- Guo QY, Yuan M, Peng J, Cui X-M, Song G, Sui X, Lu S-B. Antitumor activity of mixed heat shock protein/peptide vaccine and cyclophosphamide plus interleukin-12 in mice sarcoma. J Exp Clin Cancer Res. 2011;30:24. doi:10.1186/1756-9966-30-24.
- Wang XY, Kazim L, Repasky EA, Subjeck JR. Characterization of heat shock protein 110 and glucose-regulated protein 170 as cancer vaccines and the effect of fever-range hyperthermia on vaccine activity. J Immunol. 2001;166(1):490–97. doi:10.4049/jimmunol.166.1.490.
- Wang XY, Chen X, Manjili MH, Repasky E, Henderson R, Subjeck JR. Targeted immunotherapy using reconstituted chaperone complexes of heat shock protein 110 and melanoma-associated antigen gp100. Cancer Res. 2003;63:2553–60.
- Kim HL, Sun X, Subjeck JR, Wang XY. Evaluation of renal cell carcinoma vaccines targeting carbonic anhydrase IX using heat shock protein 110. Cancer Immunol Immunother. 2007;56(7):1097–105. doi:10.1007/s00262-006-0258-z.
- Ding Z, Ou R, Ni B, Tang J, Xu Y. Cytolytic activity of the human papillomavirus type 16 E711-20 epitope-specific cytotoxic T lymphocyte is enhanced by heat shock protein 110 in HLA-A*0201 transgenic mice. Clin Vaccine Immunol. 2013;20(7):1027–33. doi:10.1128/CVI.00721-12.
- Danhier F, Le Breton A, Preat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9(11):2961–73. doi:10.1021/mp3002733.
- Sun Y, Kang C, Liu F, Zhou Y, Luo L, Qiao H. RGD peptide-based target drug delivery of doxorubicin nanomedicine. Drug Dev Res. 2017;78(6):283–91. doi:10.1002/ddr.21399.
- Katsamakas S, Chatzisideri T, Thysiadis S, Sarli V. RGD-mediated delivery of small-molecule drugs. Future Med Chem. 2017;9(6):579–604. doi:10.4155/fmc-2017-0008.
- Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13(17):5043–54. doi:10.1016/j.bmc.2005.04.084.
- Long Q, Zhu W, Guo L, Pu L. RGD-conjugated resveratrol HSA nanoparticles as a novel delivery system in ovarian cancer therapy. Drug Des Devel Ther. 2020;14:5747–56. doi:10.2147/DDDT.S248950.
- Luo D, Saltzman WM. Synthetic DNA delivery systems. Nat Biotechnol. 2000;18(1):33–37. doi:10.1038/71889.
- Wu YZ, Zhao JP, Wan Y, Jia Z-C, Zhou W, Bian J, Ni B, Zou L-Y, Tang Y. Mimovirus: a novel form of vaccine that induces hepatitis B virus-specific cytotoxic T-lymphocyte responses in vivo. J Virol. 2002;76(20):10264–69. doi:10.1128/JVI.76.20.10264-10269.2002.
- Awasthi S, Cox RA. Transfection of murine dendritic cell line (JAWS II) by a nonviral transfection reagent. Biotechniques. 2003;35(3):600–602, 604. doi:10.2144/03353dd03.
- Tian Z, Wang H, Jia Z, Shi J, Tang J, Mao L, Liu H, Deng Y, He Y, Ruan Z, et al. Tumor-targeted inhibition by a novel strategy - mimoretrovirus expressing siRNA targeting the Pokemon gene. Curr Cancer Drug Targets. 2010;10(8):932–41. doi:10.2174/156800910793357907.
- Domingos-Pereira S, Roh V, Hiou-Feige A, Galliverti G, Simon C, Tolstonog GV, Nardelli-Haefliger D. Vaccination with a nanoparticle E7 vaccine can prevent tumor recurrence following surgery in a human papillomavirus head and neck cancer model. Oncoimmunology. 2021;10(1):1912473. doi:10.1080/2162402X.2021.1912473.
- Tang J, Yin R, Tian Y, Huang Z, Shi J, Fu X, Wang L, Wu Y, Hao F, Ni B, et al. A novel self-assembled nanoparticle vaccine with HIV-1 Tat(4)(9)(-)(5)(7)/HPV16 E7(4)(9)(-)(5)(7) fusion peptide and GM-CSF DNA elicits potent and prolonged CD8(+) T cell-dependent anti-tumor immunity in mice. Vaccine. 2012;30(6):1071–82. doi:10.1016/j.vaccine.2011.12.029.
- Oba M, Fukushima S, Kanayama N, Aoyagi K, Nishiyama N, Koyama H, Kataoka K. Cyclic RGD peptide-conjugated polyplex micelles as a targetable gene delivery system directed to cells possessing α v β3 and αvβ 5integrins. Bioconjug Chem. 2007;18(5):1415–23. doi:10.1021/bc0700133.
- Shayakhmetov DM, Eberly AM, Li ZY, Lieber A. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J Virol. 2005;79(2):1053–61. doi:10.1128/JVI.79.2.1053-1061.2005.
- Khong H, Overwijk WW. Adjuvants for peptide-based cancer vaccines. J Immunother Cancer. 2016;4:56. doi:10.1186/s40425-016-0160-y.
- Marelli UK, Rechenmacher F, Sobahi TR, Mas-Moruno C, Kessler H. Tumor targeting via integrin ligands. Front Oncol. 2013;3:222. doi:10.3389/fonc.2013.00222.
- Zhou X, Liu HY, Zhao H, Wang T. RGD-modified nanoliposomes containing quercetin for lung cancer targeted treatment. Onco Targets Ther. 2018;11:5397–405. doi:10.2147/OTT.S169555.
- Calderwood SK, Stevenson MA, Murshid A. Heat shock proteins, autoimmunity, and cancer treatment. Autoimmune Dis. 2012;2012:486069. doi:10.1155/2012/486069.
- Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14(1):105–11. doi:10.1007/s12192-008-0068-7.
- Miyazaki M, Nakatsura T, Yokomine K, Senju S, Monji M, Hosaka S, Komori H, Yoshitake Y, Motomura Y, Minohara M, et al. DNA vaccination of HSP105 leads to tumor rejection of colorectal cancer and melanoma in mice through activation of both CD4 T cells and CD8 T cells. Cancer Sci. 2005;96(10):695–705. doi:10.1111/j.1349-7006.2005.00093.x.
- Manjili MH, Park J, Facciponte JG, Subjeck JR. HSP110 induces “danger signals” upon interaction with antigen presenting cells and mouse mammary carcinoma. Immunobiology. 2005;210(5):295–303. doi:10.1016/j.imbio.2005.04.002.