
ABSTRACT
Antigen-specific immunotherapies (ASITs) address important clinical needs in treating autoimmune diseases. However, Type 1 diabetes is a heterogeneous disease wherein patient characteristics influence responsiveness to ASITs. Targeting not only disease-relevant T cell populations, but also specific groups of patients using precision medicine is a new goal toward achieving effective treatment. HLA-restricted peptides provide advantages over protein as antigens, however, methods for profiling antigen-specific T cells need to improve in sensitivity, depth, and throughput to facilitate epitope selection. Delivery approaches are highly diverse, illustrating the many ways relevant antigen-presenting cell populations and anatomical locations can be targeted for tolerance induction. The role of persistence of antigen presentation in promoting durable antigen-specific tolerance requires further investigation. Based on the outcome of ASIT trials, the field is moving toward using patient-specific variations to improve efficacy, but challenges still lie on the path to delivering more effective and safer treatment to the T1D patient population.
GRAPHICAL ABSTRACT
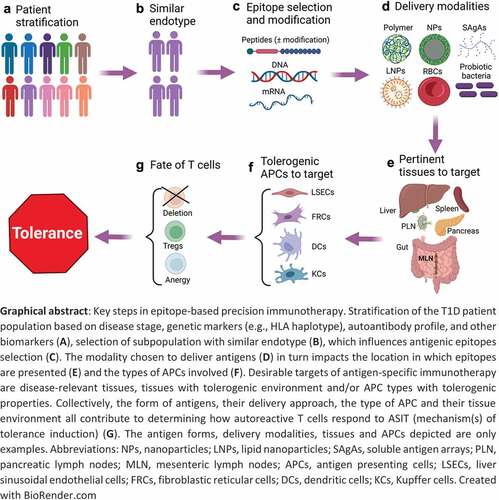
Type 1 diabetes and the need for antigen-specific therapies
Type 1 diabetes (T1D) is characterized by a progressive loss of insulin-producing β-cells, which is primarily caused by autoreactive CD4+ and CD8+ T cells, as a result of impaired immune tolerance to β-cell antigens. Several genetic factors contribute to the disease in at least two different ways. First, they may increase β-cell fragility (increased oxidative and endoplasmic reticulum stress, sensitivity to proinflammatory cytokines).Citation1 Second, they may impair general tolerance mechanisms (as suggested by the occurrence of other autoimmune conditions in both T1D patientsCitation2 and its animal model, the non-obese diabetic (NOD) mouse.Citation3 Administration of exogenous insulin for the last 100 years has saved countless lives; however, it can rarely achieve tight regulation of blood glucose levels, such that episodes of hyper- and hypoglycemia remain commonplace, and their recurrence leads to long-term complications.Citation4 Even when perfect glycemic control is achieved, there is still a residual 2- to 3-fold hazard ratio of death from any cause, particularly from cardiovascular disease, in T1D vs. non-diabetic individuals.Citation5 Transplantation of pancreatic islets is limited by the paucity of donors and vigorous allogeneic immune responses to grafted islets requiring strong immunosuppression that substantially raises the risk of opportunistic infections, malignancies, and beta-cell toxicity.Citation6–8 While the use of autologous stem cell-derived β-cells appears feasible in the future, these cells will remain vulnerable to autoreactive T cells.
Targeted immunotherapies aim to disable only relevant elements of the immune system in order to obviate global immunosuppression. Targeting specific immune cells or pathways is mostly about safety and leaving most of the immune system untouched, though this may increase efficacy in some cases. Biologics have supplanted immunosuppressive drugs in the last 10–20 years of clinical trials of T1D treatments, with the removal of specific immune cell types or blockade of specific immune pathways.Citation9 While more selective, these approaches still do not make any distinction between autoimmune cells and other immune cells. In contrast, antigen-specific immunotherapies (ASITs) offer a way to selectively engage autoreactive cells and neutralize them through various mechanisms of tolerance. For this to work, however, autoreactive T cells need to be engaged under appropriate conditions that favor tolerance. It is challenging to achieve these conditions in patients, not only because of the underlying local inflammation associated with the disease, but also because of genetic risk factors, some of which impair the induction and maintenance of tolerance (partly explaining how T1D developed in the first place). There are multiple stages of T1D disease progression (), and ASITs are useful at stages where there are enough residual β-cells left (or transplanted β-cells) to protect by blocking autoimmune responses. Otherwise, controlled exogenous insulin delivery is required.
Figure 1. Stages of human T1D disease, corresponding endotype characteristics and windows of opportunities for ASIT.
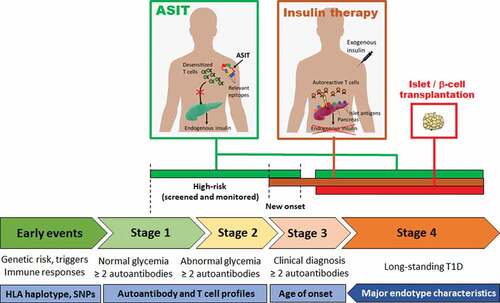
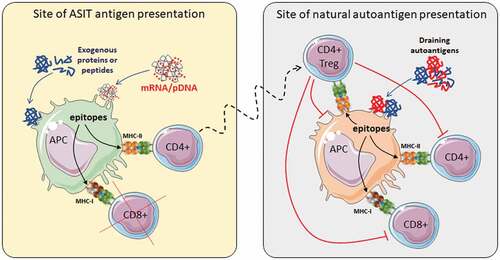
Figure 2. Bystander suppression.
Why a need for precision therapy as well?
Precision therapy is a therapeutic approach that is tailored to an individual or group of patients according to the specific biological and molecular features of the disease (e.g. targeting a specific gene mutation in cancer). It constitutes another aspect of targeted therapy that is more about efficacy and maximizing responsiveness by taking the unique characteristics of patients into account.Citation10 Some patients go through the different stages of disease progression () quite rapidly and are often diagnosed at a younger age, while other patients progress more slowly and develop diabetes well into adulthood. As T1D is both a multifactorial and polygenic autoimmune disease, it is likely that the number of genetic risk factors and the exposure to environmental influences and triggers, in combination, dictate the pace of disease progression. Due to this complexity, the factors that drive rapid versus slow progression remain poorly understood. Patients are also characterized by different autoantibody (AAb) profiles, with four major AAbs (against insulin, GAD65, IA-2 and ZnT8) being detectable in the serum prior to the onset of diabetes.Citation11 Individuals with two or more AAbs have a high risk of progressing to clinical T1D and those with one are at an intermediate risk.Citation11 Among genetic risk factors, the HLA class II locus shows the greatest association with T1D. For example, HLA-DRB1*04:01/2/4/5 (HLA-DR4, often inherited in linkage disequilibrium with HLA-DQB1*03:02 (HLA-DQ8)) and HLA-DRB1*03 (HLA-DR3, often linked with HLA-DQB1*02 (HLA-DQ2)) are most frequently found in T1D patients of European descent.Citation12,Citation13 HLA haplotypes, both class I and class II, limit the number of peptides that can be presented for each antigen, and it is highly probable that key epitopes are preferentially presented by those aforementioned HLA-DR and DQ haplotypes. Moreover, early onset was associated with an AAb profile dominated by anti-insulin (primarily in individuals with at least one allele of HLA-DR4/DQ8 haplotype), whereas later onset featured more anti-GAD65 (particularly in subjects with two alleles of HLA-DR3/DQ2 haplotypes, although the HLA genetic background seems more heterogeneous in this subgroup).Citation14,Citation15 It seems however that the faster clinical progression in early seroconverters reflects the earlier age of the first AAb appearance rather than an anti-insulin vs. anti-GAD65 pattern.Citation16 There are other gene variants, environmental exposures and biomarkers that further characterize patients, and when all these characteristics are considered together, they can delineate discrete subtypes termed endotypes,Citation13,Citation17 whereby certain characteristics often tend to correlate with one another in subsets of patients. Two patterns correlating HLA haplotype and AAb type have been noted so far in groups of patients:Citation13 one associating HLA-DR4(-DQ8) and anti-insulin as predominant and/or first and early appearing AAb, and another associating HLA-DR3(-DQ2) and anti-GAD65 as the predominant and/or first AAb (though appearing later in childhood).Citation14,Citation18 However, it should be acknowledged that these endotypes do not encompass the whole range of heterogeneity of T1D.
The NOD mouse is a rare example of a polygenic and spontaneous model of autoimmune disease, and shows many commonalities with human T1D, including a large overlap in genetic susceptibility features, the β-cell antigens targeted by T cells and other autoimmune conditions.Citation3,Citation19 However, although it has been used extensively in T1D preclinical studies, it has been criticized for limited translatability of the efficacy of immunotherapies to T1D patients. This shortfall is related to the fact that the NOD mouse, as an inbred and genetically homogeneous strain, simply cannot represent the heterogeneity of the human T1D population. Interestingly, NOD mice have a unique MHC class II haplotype (I-Ag7) that is highly similar to HLA-DQ8,Citation20 and they only develop anti-insulin AAb consistently.Citation21 Both I-Ag7 and DQ8 are known to present the same key epitope of insulin (B9:23), which was found to be essential for T1D to develop in NOD mice.Citation22 Therefore, the NOD mouse may constitute a reasonable model for the human endotype featuring HLA-DQ8 and anti-insulin, until humanized mice can be produced to model other endotypes.Citation23
In clinical trials that did not achieve their target, post-hoc data analyses were performed, whereby one objective was to determine what the best responders had in common. In the case of ASITs, it turns out that the HLA haplotype and/or the AAb profile could be indicators of responsiveness. In the Diabetes Prevention Type 1 (DPT-1) trial testing oral insulin to induce tolerance, protection was not significant when comparing all patients in treatment and placebo groups, but became significant when only patients with anti-insulin AAbs above 80 nU/mL were considered.Citation24 This suggests that patients who have a more pronounced AAb reactivity to an antigen may be more responsive to this antigen in ASIT. Mere positivity for the AAbs may not be correlated with responsiveness to therapy, as confirmed by a follow-up trial.Citation25 Across three trials involving the delivery of alum-formulated GAD65, it was generally observed that patients with the HLA-DR3-DQ2 haplotype were responsive, particularly if they also did not have the HLA-DR4-DQ8 haplotype.Citation26 These two examples support the idea that patient characteristics, which can be ascertained prior to treatment, can constitute useful predictors of responsiveness. As a result, newer ASIT trials are increasingly using these parameters as inclusion criteria (), and if this practice improves responsiveness, additional post-hoc analyses may identify other parameters that correlate with efficacy.
Table 1. Examples of clinical trials for antigen-specific immunotherapy of T1D (current and completed within the past five years).
In sum, the success of ASIT for the treatment of T1D and other autoimmune diseases will benefit from being employed in a targeted manner, not only at the cellular level (targeting only autoreactive lymphocytes), but also at the individual level (tailored to endotype groups). Furthermore, we propose that the NOD mouse should be regarded and utilized more as a disease endotype model, and that promising treatments be next evaluated in the HLA-DR4-DQ8 endotype group, which is prominent in the T1D patient population.
Why use peptides over full antigens?
As the studies described above used full proteins, the nature and number of epitopes that are ultimately presented on each HLA haplotype is not known. Because T cells selected as natural Tregs tend to have distinct T cell receptors (TCRs) from conventional T cells,Citation27 some MHC/peptide complexes may be more likely recognized by Tregs, in which case epitopes can be considered as “useful” for eliciting regulation. Peptides that engage conventional T cells may also be useful if they elicit deletion, anergy or conversion to peripheral Tregs, but that would depend more on the context of presentation rather than the nature of the peptide. Considering the full array of peptides that can be presented on different HLA haplotypes from a protein antigen, it is tempting to speculate that more useful GAD65 peptides are presented on HLA-DR3-DQ2 than on HLA-DR4-DQ8, leading to a better outcome in the GAD65-alum studies. Moreover, some peptides may not be as useful if they interfere with the display of useful peptides by competing for HLA occupancy. Thus, there may be an advantage of using a selection of peptides over the full antigen if these peptides are well-characterized and anticipated to preferentially stimulate Tregs and/or engage low-affinity T cells more effectively under tolerogenic conditions. Because of the ability of Tregs to suppress T cells across multiple specificities within and across antigens, provided that those other epitopes are displayed by the same antigen-presenting cells (APCs), it is not necessary to attempt to cover all possible epitopes in ASITs ().
The array of epitopes that have been identified from T1D-relevant proteins is constantly growing, although their level of characterization and validation is highly varied. A search in the Immune Epitope Database (IEDB) yielded 8,234 MHC class I and 3,146 MHC class II human linear epitopes, the great majority of which were eluted from MHC. A more focused search on epitopes discovered by T cell assays returned 182 MHC class I and 401 MHC class II epitopes, as of the time of writing (). These were typically identified by stimulating islet-infiltrating T cell clones with peptide libraries encompassing major β-cell antigens.Citation28–30 The recent and comprehensive review by James et al. addresses most of these epitopes, providing a detailed analysis of their antigen source and HLA-restriction.Citation31 About 75% of epitopes recognized by CD8+ T cells are derived from insulin, ZnT8 and GAD65, and about the same percentage is restricted to the common HLA-A2 allele. About 80% of epitopes recognized by CD4+ T cells are derived from GAD65, insulin and IA-2, 1/3 are restricted to HLA-DR4 and 1/4 are restricted to HLA-DQ8. Still, these numbers do not reflect the reality but rather the current knowledge, which is itself biased toward those haplotypes most commonly found in T1D patients. The IEDB also provides detailed information about TCRs recognizing some of these epitopes on specific HLA haplotypes (V region usage and CDR3 sequence) sufficient to produce TCR-transgenic T cell transductants that may be used to validate the presentation of epitopes from various ASIT delivery platforms in vitro or using in vivo models.Citation23
Table 2. Human T cell linear epitopes deposited in the Immune Epitope Database (IEDB) and validated by T cell assays, and corresponding TCRs identified. Filters applied: linear structure for epitope structure; Homo sapiens for both organism and host; type 1 diabetes mellitus for disease; include positive T cell assay; exclude B cell assays and MHC assays; further filtered on MHC class I, class II or specific HLA protein complex as indicated in the table). A more limited number of epitopes (third column) were from assays in which the TCR sequence of responding T cells was characterized (at least one chain in fourth column; or both alpha and beta chains in fifth column). One epitope can be recognized by multiple T cell clones (TCR alpha/beta pairs). Searches including both T cell and MHC assays (eluted peptides) yielded 8,234 epitopes for HLA class I and 3,146 epitopes for HLA class II. Data are accurate as of 11/02/2022.
The identification of neoepitopes targeted by diabetogenic T cells has been a major advance in the T1D field. Aside from “native” peptides resulting from the simple proteolytic cleavage of known antigenic proteins, there are other types of peptides that have been hypothesized to play a significant role in expanding the pool of pathogenic T cells and/or in targeting them in ASIT:Citation32,Citation33
Peptides resulting from the post-translational modification (PTM) of native peptides (deamidation, citrullination, etc.).Citation32
Peptides resulting from the fusion of peptides from different antigens (hybrid peptides) creating new epitopes centered on the junctions of the original two peptides.Citation34–36
Peptides encompassing new junctions produced from alternatively spliced mRNA occurring in islets but not in the thymus and other lymphoid tissues.Citation37–40
Peptides originating from defective ribosomal products (DRiP) whose expression may be increased under the influence of T1D-associated gene polymorphisms.Citation41
Artificial peptides (often termed mimotopes or altered peptide ligands) that either correspond to native peptides mutated to help fix anchoring on MHC in a particular conformation or register to enhance recognition by a specific type of T cellsCitation42 or that were identified for their ability to stimulate islet-infiltrating T cells of unknown specificity.Citation43
These neoepitopes greatly increase the antigenic display that make up the specificities of diabetogenic T cells to the point that the use of a single protein may appear preposterous, no matter how many MHC-binding epitopes that single protein may produce. It is not yet clear whether certain types of neoepitopes are associated with specific endotypes, and whether some are early drivers of the disease or appear during later stages of disease. Because some of these neoepitopes may arise under certain stress or inflammatory conditions, it is probable that they are not produced and presented under steady state conditions and that tolerance to these neoepitopes is limited. Moreover, they are not templated in the genome and may therefore be preferentially recognized as “non-self.” Thus, the inclusion of neoepitopes in ASIT to build this tolerance may prove important. Using mimotopes, as well as native epitopes that are generally ignored,Citation44 increases the chance of targeting antigen-specific T cells that are more naïve (and likely easier to manipulate phenotypically) and possibly more frequent.
There are other advantages of using peptides over proteins for ASIT:
It is easier to synthesize peptides than to produce recombinant proteins. Peptides can be synthesized with specific mutations or chemical modifications that correspond to PTMs, and the chemical synthesis process circumvents issues of biological contaminants from protein-producing cells.
The use of peptides is more amenable to customizing the treatment of T1D patients grouped by endotypes, using a “mix & match” approach.
Some delivery strategies are more compatible with the use of peptides than proteins.
Despite their shorter in-vivo half-life compared to proteins, potency and persistence of peptides can be improved by chemical modifications.Citation45
Moreover, just like proteins can be encoded by DNA vectors (as in DNA vaccines for T1DCitation46–50), it is possible to express a wide variety of epitopes from a single vector,Citation51–53 with the same previously mentioned advantages. Endogenously encoded proteins and polypeptides have the added benefit of allowing the host cells to perform PTMs on them naturally under the appropriate conditions. DNA vectors are also cheaper to produce, and while the efficiency of DNA delivery remains relatively limited, one molecule generates many mRNA molecules, which in turn produces hundreds of proteins or polypeptides.Citation54
One limitation of using peptides over proteins is that they are unable to engage autoreactive B cells that only recognize conformational epitopes. It is unclear to what extent targeting β-cell antigen-specific B cells for tolerance can help in blunting the overall autoimmune response, but one may speculate that anergized B cells serving as APCs may contribute to tolerization of T cells and/or regulatory B cells may help suppress autoreactive T cells. Several studies have suggested a beneficial involvement of regulatory B cells in treatment of T1D.Citation55–58
What immune biomarkers and epitope characteristics may be used for customization?
The concept of endotype being relatively new, associations between patient characteristics and targetable antigen specificities are yet to be made. AAbs are reliable biomarkers that are routinely measured. While the number of AAbs informs on the relative risk of progressing to overt disease, the relative titer of each AAb may provide clues on dominant immune reactivities. As AAb-producing B cells require initial help from antigen-specific CD4+ T cells, this can provide an indirect indication of dominant T cell responses. However, it remains to be seen in other trials if the DPT-1 trial post-hoc results are generalizable and if AAb titers can be used for antigen selection to achieve significant responsiveness.
A more direct measurement of T cell specificities would be to perform multiplex MHC multimer analysis to determine whether certain antigen-specific T cell populations are expanded in the circulation, possibly delineating populations that are worth targeting. In this approach, MHC multimers are used, sometimes in pairs (with two different detectable conjugates) for most accurate detection of antigen-specific T cells, using conventional or spectral flow cytometry (fluorochrome-labeled), mass cytometry (heavy metal-labeled) or DNA sequencing (DNA barcode-labeled).Citation59 In T1D, approaches with combinatorial MHC multimers validated the existence of autoreactive CD8+ T cells specific to a wider array of antigens than previously anticipated.Citation40,Citation60 The same approach can be used to analyze CD4+ T cells,Citation61 but has yet to be applied for T1D-relevant specificities. There are, however, several challenges to this approach:
Detection of autoreactive T cells is complicated by the fact that many of their TCRs are of relatively low affinity and thus signal may be weak.
This is an expensive approach. Even with high-throughput methods utilizing DNA barcodes, the high cost of MHC multimer manufacturing dramatically limits how comprehensive the analysis panel can be. However, the panels would be limited to using only MHC multimers matching the HLA haplotype of the tested individuals.
The frequency of relevant antigen-specific T cells is very low in the blood, and it remains debated whether circulating autoreactive T cells provide a useful reflection of those infiltrating the target tissue (pancreatic islets in the case of T1D).
Studies using MHC multimers also reported that autoantigen-specific T cells are found in the circulation of healthy individuals, typically with similar frequencies,Citation40,Citation60,Citation62,Citation63 which suggests that negative selection in the thymus may be limited to the highest affinity autoreactive T cells in all individuals, and that in T1D patients, it is mainly a defective regulation of the remaining T cells that allows them to be activated and become pathogenic.Citation64 Beyond detecting those T cells, it may be more important to determine their phenotype (i.e., whether these circulating T cells are more activated and antigen-experienced). Again, in MHC multimer-based analyses of circulating autoreactive CD8+ T cells comparing control and T1D donors, differences are difficult to pinpoint. Recently, a method combining DNA-barcode-labeled MHC tetramers, CITE-seq and TCR-seq (termed TetTCR-SeqHD) was employed to profile CD8+ T cells in control and T1D individuals.Citation65 While this approach provides a much deeper profiling of these rare T cells at the clonal level (thanks to TCR sequences), it is challenging to apply on a large scale (broad antigen-coverage by MHC multimers and large number of patients for screening purposes).
Another assessment of T cell specificities can be done by restimulating PBMCs with specific peptides and measuring cytokines produced in response using ELISPOT assays. In such study using insulin mimotopes, CD4+ T cells from both control and T1D donors generated IFN-γ+ spots.Citation66 However, the CD4+ T cells from control donors produced IL-10+ spots more frequently than those from T1D donors,Citation66 indicating that differences in phenotypes may be discernable to ascertain the relevance of certain antigen specificities.
Regardless of the approach used, building a comprehensive immune profile for patients and attempting to identify major/driver antigens may be a daunting task at this time, and greater automation and cost reduction will be required. While having more options (epitopes/antigens) to choose from may be helpful, using a large number of peptides for immunotherapy may not be required. The question is then: how many epitopes are enough in ASIT to achieve control of the disease? Several studies in NOD mice suggest that one or few peptides may be enough to achieve significant protection from disease onset, likely through bystander suppression and infectious tolerance (), and these peptides were generally strong binders (). In humans, clinical trials have been conducted with up to six HLA-DR4-restricted insulin- and IA2-derived peptides, which were believed to engage Tregs (). These studies have in common that all peptides used targeted CD4+ T cells, with the rationale that induction/stimulation of antigen-specific Tregs can suppress across specificities, and with the presumption that the number of functional Tregs may be more important than their diversity.
Table 3. Antigen-specific approaches pursued by biotech companies and academic laboratories.
The choice of peptides may also be based on their properties, beyond their ability to bind specific HLA haplotypes. As alluded to earlier, certain PTMs and mutations can affect the position and strength of binding, but they can also enable recognition by additional and otherwise unresponsive populations of T cells. The outcome of the T cell response can be influenced by the affinity of TCR binding to the peptide/MHC complex, and similarly, by the avidity of TCR recognition, which can be modulated by varying the dose of peptide administered. For example, when two epitopes stimulating the same T cell clone (BDC2.5) were tested in soluble antigen array (SAgA) form in NOD mice, the stronger one (p79 mimotopeCitation43 primarily induced a Tr1 (IL-10-producing) type of Tregs, with a lower dose needed to stimulate Foxp3+ Tregs, whereas the 2.5HIP peptideCitation34 (about 10-fold weaker than p79 in vitro) at the same dose only stimulated Foxp3+ Tregs.Citation71 The two peptides did not change the frequency of IFN-γ+ antigen-specific T cells, but rather induced IL-10+ T cells or stimulated Foxp3+ Tregs. In some cases, chemical modifications may change the properties of the peptide to functionally alter the phenotype of responding T cells. For example, Imcyse’s ImotopesTM are HLA class II-binding peptides fused with a small peptide motif with oxido-reductase activity, which confers cytolytic activity to the activated CD4+ T cells, resulting in the killing of autoantigen-presenting cells and autoreactive T cells around themCitation69 ().
Once pre-filtering based on known HLA restriction is accomplished, epitopes may be selected based on the source antigen (with a focus on one or few antigens) and on how well they are characterized. Epitopes that are simply predicted or eluted from MHC do not provide as much confidence as those that have been demonstrated to induce a T cell response. In particular, those found to be recognized by islet-infiltrating T cells would be prioritized as highly relevant, and the list is growing,Citation85 especially with recent efforts on identifying and validating neoepitopes using T cells from pancreatic islets and lymphoid tissues rather than blood. Once a group of disease-relevant epitopes has been defined for a specific HLA haplotype, could a mix of epitopes covering multiple antigens be used as a blanket treatment? In other words, if a patient’s autoimmune response is dominated by reactivity against insulin, would introducing other antigens by ASIT be beneficial or detrimental? This is a question that has not yet been addressed because patients have so far only been treated with one antigen at a time, except in the MultiPepT1De trial in which they received peptides from both proinsulin and IA-2 (),Citation70 and there is only one spontaneous mouse model, representing one endotype, available for testing. If epitopes from other antigens induce Tregs and all epitopes tend to be naturally presented by the same APCs, then one may speculate that covering many antigens would have a beneficial outcome. In contrast, if epitopes are presented under inappropriate conditions (leading to preferential stimulation of effector T cells), then adding other epitopes could be detrimental. Blanket coverage with diverse peptides from multiple antigens is costly, unless those peptides can all be produced from a single molecule (DNA or mRNA)Citation51 or recombinant protein.Citation86 For example, the Endotope platform enables expression of multiple MHC class I and II restricted epitopes from a DNA vaccine, whereby MHC class II epitopes are separately funneled from cytosol to endosome.Citation51,Citation52 An Endotope DNA vaccine including native epitopes and mimotopes and covering at least three different antigens was comparable in efficacy to a proinsulin-encoding DNA vaccine in preventing disease in NOD mice (insulin-dominated endotype).Citation53
Another outstanding question that remains unanswered is whether it is sufficient to target CD4+ T cells (if resulting in suppressive antigen-specific Tregs) in a disease like T1D wherein CD4+ and CD8+ T cells play a critical role. While there is evidence that some ASITs can result in deletion of autoreactive CD8+ T cells in patients, this appears to be limited to those reactive to the administered antigen.Citation47 The current favored approach for epitope-based ASIT is to achieve a sufficient level of suppression with a minimal set of key HLA class II epitopes that would be effective across all patients within the same HLA haplotype group. However, given that Treg stability may be an issue in some T1D patients, targeting and eliminating certain CD8+ T cells that are consistently expanded in T1D patients may function as a debulking therapy and increase protection from disease. The use of disease-irrelevant epitopes that activate Tregs and are co-delivered with disease-relevant epitopes may also be considered. For example, Tregitopes are IgG epitopes recognized by Tregs, which become mobilized around diabetogenic T cells when Tregitopes are co-delivered and co-presented with preproinsulin peptides, resulting in disease improvement.Citation87
How can we deliver peptides to suitable locations and APCs in vivo?
Once epitopes are selected, the next challenge is to find suitable modalities to deliver them to appropriate locations where APCs and their environment are conducive to tolerance. summarizes delivery strategies by companies and academic laboratories. The simplest form of delivery is by injection of peptides in soluble form. These peptides are most vulnerable to extracellular degradation and may diffuse widely (unless injected in a dense tissue), potentially resulting in relatively low density of epitope presentation. Multimerization of peptides or formulation in small nanoparticles (<200 nm) achieves higher molecular weights that are more conducive of draining through lymphatics to achieve more specific accumulation in T cell-rich lymph nodes, and possibly spleen. Some nanoparticles, based on their chemical composition and net charge, are preferentially taken up by specific APCs that are tolerogenic in nature (e.g., marginal zone macrophages, liver sinusoidal endothelial cells).Citation88,Citation89 Larger microparticles (as well as large macromolecules such as DNA plasmids) remain trapped in the tissue in which they are injected and rely on tissue APCs with the capacity to migrate to draining lymph nodes.Citation52,Citation90 Small and large particles have also been used to remodel the environment of the antigen uptake and/or presentation sites (with nanoparticles influencing lymph node environmentCitation91 and microparticles modulating the inoculation site,Citation92 for example). provides examples illustrating the variety of nanoparticle formulations that differ in size, properties, and content (co-delivery of drugs along with antigens). While some formulations are compatible with oral delivery,Citation93 antigens have also been delivered inside commensal bacteria for oral tolerance.Citation79,Citation80,Citation94 Alternative to using nanoparticles, peptides may be grafted onto natural polymers (hyaluronic acid, to produce SAgA),Citation71 conjugated to glycopeptides that enhance uptake in liverCitation95 or to antibodies against APC surface markersCitation72–96–Citation98 or associated with erythrocytes that are taken up by tolerogenic splenic macrophages by efferocytosis.Citation99 Peptides may be expressed from DNA or mRNA vaccines, which often require formulation into nanoparticles or liposomes to improve the delivery and control the biodistribution.Citation77,Citation100 Peptides can also be provided as already presented on MHC molecules, either on the surface of tolerogenic dendritic cellsCitation83 or on the surface of small nanoparticles,Citation84 the latter providing TCR stimulation without any costimulation to tolerize T cells. These can be used for direct presentation to T cells following administration, without requiring any expression and/or processing. Finally, instead of inducing tolerance or eliciting Tregs with specific epitopes, it is possible to use epitope-specific TCRs to engineer Tregs for cell therapy.Citation101 This approach requires identification of islet antigen-specific TCRs from islet-infiltrating T cells with the appropriate HLA restriction and any Treg, and the patient’s T cells can be engineered ex vivo to replace the preexisting TCR with a new disease-relevant TCR, then expanded prior to re-infusion. The use of chimeric antigen receptor (CAR) Tregs affords the additional advantages of whole antigen recognition rather than a specific peptide/MHC complex and independence from HLA haplotype,Citation101 although this may also be a limitation when the antigen is not a surface antigen (like many β-cell antigens) and different targets are needed. The issue of Treg stability is crucial as conversion of transferred epitope-specific Tregs to effector T cells in vivo could exacerbate the disease, and additional engineering methods or treatment to stabilize Treg function are actively sought.
Thoughts on ensuring appropriate conditions of epitope presentation
When antigenic proteins or peptides are administered to individuals, it is difficult to tightly control the type of APCs that participate in their presentation, and the tissue environment in which these antigens are taken up which influences the phenotype of these APCs. Sites like liver and gut-associated lymphoid tissues generally provide a more tolerogenic environment,Citation102,Citation103 whereas in spleen and lymph nodes, tolerance induction is more dependent on the type of APCs. When professional APCs (cells capable of maturing to elicit immunogenic responses) are involved, it is desirable to target non-inflamed environments and ensure that nothing administered with the antigens (e.g., chemical modifications, nanodelivery formulations, DNA/RNA motifs when expressing antigens) acts as immunogenic adjuvant while maximizing exposure to the extensive pool of circulating T cells. Specific dendritic cell populations (e.g., DEC205+ vs DCIR2+) have been targeted with specific antibodies that were conjugated with peptidesCitation72–Citation96–98 and reported to promote tolerance. Alternatively, nonprofessional APCs have been targeted, including hepatocytes,Citation95,Citation104 liver sinusoidal endothelial cells (e.g., Topas’ SPIONSTM, ), and lymph node stromal cells.Citation105 For example, hepatocyte-restricted delivery can be achieved using N-acetylgalactosamine-modified antigensCitation95 (approach used by Anokion, ) or using liver-specific promoter coupled with micro-RNA targets restricting expression in non-hematopoietic cells in the case of transgene-encoded antigens.Citation104 When the type of APCs cannot be restricted, codelivery of immunomodulatory drugs (e.g., rapamycinCitation76,Citation106 or cytokines (e.g., IL-10)Citation79,Citation107 with antigens helps maintain the tolerogenic function of APCs. Importantly, a single DNA vaccine encoding antigen and multiple cytokines (IL-2, IL-10 and TGF-β) has been validatedCitation49 and is being tested clinically, providing a 4-in-1 drug option that is easy and economical to manufacture.
A more broadly distributed exposure of epitopes via systemic delivery may help with engaging more autoreactive T cells in a shorter time frame, while local delivery that results in presentation at limited sites may require more prolonged exposure or more frequent administrations. Persistence of self-antigen presentation appears critical for the induction of immune tolerance (anergy/exhaustion and deletion) in CD4+ T cellsCitation108–115 and CD8+ T cells,Citation116,Citation117 regardless of whether APCs are DCsCitation112,Citation114 or B cells.Citation110,Citation111 In fact, prolonged antigen exposure can lead to tolerance even when APCs were initially in an immunogenic state.Citation111,Citation117 However, in the case of DCs, T cells can maintain effector functions after at least 3.5 days of continuous antigen presentation,Citation112 suggesting that antigen exposure should be sustained longer. These studies used continuous or regulated transgenic expression of antigens, but in ASIT, the frequency of delivery and duration of antigen presentation after each delivery differ between delivery modalities, resulting in continuous or intermittent antigen exposure. Peptides have a short half-life, whereas those delivered via nanoparticles persist longer. Multivalent delivery of peptides as SAgAs also significantly prolonged antigen presentation in vivo.Citation71 Controlled release of peptides can be achieved by osmotic pumps,Citation67 but peptides remain subject to extracellular degradation, resulting in sustained yet limited peptide delivery. DNA vaccines can sustain antigen presentation for at least two weeks in NOD mice,Citation53 but exposure is relatively limited (local) and administration once or twice a week was required to achieve sufficient T cell engagement and significantly block disease progression.Citation49,Citation53 While frequent administrations may be required at first to achieve a sufficient level of tolerance, treatments may potentially be discontinued or reduced in dose or frequency for maintenance. The route and frequency of administration represent challenges for implementation and compliance in patients, for example if frequent injections are needed. When frequent administrations are required to maintain a sufficient and sustained exposure of epitopes to autoreactive T cells, the oral route stands out as the most attractive for patients if adequate antigen protection and T cell responses can be obtained.
Outlook: what are major challenges to overcome?
The field of ASIT in T1D (and other autoimmune diseases) has witnessed significant advances with new methods that accelerate the discovery and validation of disease-relevant epitopes, new biomarkers and signatures. These strides enable the stratification of patients into endotypes and the prediction of responsiveness to therapy. In parallel, a burst of biotech activities that follow a diverse and promising array of approaches () to achieve adequate delivery and presentation of epitopes for tolerance has taken place.
There remain a series of challenges that most approaches are confronted with and will need to be further tackled in the next few years:
Biomarkers: How do we validate whether a set of common autoantigens or epitopes is relevant for a specific patient? We need additional and reliable markers that can discern autoreactive T cells in the blood (also detected in healthy individuals) that play an active part in the disease.
Models: Preclinical evaluation of ASIT is hampered by the poor representation of patient heterogeneity in animal models. While the NOD mouse model was serendipitously discovered, the current knowledge of T1D patient genetics may inform the engineering of new mouse strains that spontaneously develop T1D. Alternatively, mice hosting human immune systems from T1D patients may be used to recapitulate certain disease characteristics and assess T cell responsiveness to therapy in comparison to control immune systems.Citation23,Citation118
Combination therapy: Protection through the use of ASIT alone in patients has proved difficult to achieve and tolerance takes time to build up. Inductive treatments that dramatically, temporarily, and selectively disable effector T cell populations (e.g., with targeted biologics) may help reset the immune system before patients are subsequently “retrained” with ASIT to elicit regulation over a more controllable effector population.
Treg instability: This remains a major issue in the T1D field. Long-term maintenance of Treg numbers and function is as important as their induction. We need to better understand the signals that control Treg stability and lifespan. Continuous (and tolerogenic) antigen exposure and low-dose IL-2 may be some of the requirements, and ex vivo engineered epitope-specific Tregs might provide the ultimate weapon to potently regulate diabetogenic T cells.
Persistence of antigen presentation: For ASIT platforms in which the duration of antigen presentation is shorter than the frequency of administration, antigen exposure is intermittent. How this affects the tolerization process and T cell responses as compared to continuous exposure remains to be investigated more closely and deciphered to understand the requirements for durable tolerance.
Autoreactive T cell heterogeneity: While different flavors of Tregs may be induced, which may work synergistically, effector T cells may be more difficult to curb than previously thought. In addition to possible resistance to regulation by effector T cells,Citation119,Citation120 a reservoir of self-replenishing stem-like autoreactive CD8+ T cells was recently uncovered in NOD miceCitation121,Citation122 and are likely to exist also in humans,Citation123 which may subsist in the background and require its own targeting approach.
Author contributions
RJC wrote the first draft. RFF, CBG and RM all contributed insights and references. RJC, RFF and CBG produced the tables and figures.
Acknowledgments
We thank Michael E. James for critical reading of our manuscript.
Disclosure statement
RJC is the inventor of the Endotope platform. The authors do not have other conflicts of interest to report in this area.
Additional information
Funding
References
- Mallone R, Eizirik DL. Presumption of innocence for beta cells: why are they vulnerable autoimmune targets in Type 1 diabetes? Diabetologia. 2020;63(10):1999–15. doi:10.1007/s00125-020-05176-7.
- Hughes JW, Riddlesworth TD, DiMeglio LA, Miller KM, Rickels MR, McGill JB. Autoimmune diseases in children and adults with Type 1 diabetes from the T1D exchange clinic registry. J Clin Endocrinol Metab. 2016;101:4931–37. doi:10.1210/jc.2016-2478.
- Aubin AM, Lombard-Vadnais F, Collin R, Aliesky HA, McLachlan SM, Lesage S. The NOD mouse beyond autoimmune diabetes. Front Immunol. 2022;13:874769. doi:10.3389/fimmu.2022.874769.
- Khadilkar A, Oza C. Glycaemic control in youth and young adults: challenges and solutions. Diabetes Metab Syndr Obes. 2022;15:121–29. doi:10.2147/DMSO.S304347.
- Lind M, Svensson AM, Kosiborod M, Gudbjornsdottir S, Pivodic A, Wedel H, Dahlqvist S, Clements M, Rosengren A. Glycemic control and excess mortality in Type 1 diabetes. N Engl J Med. 2014;371:1972–82. doi:10.1056/NEJMoa1408214.
- Nellore A, Fishman JA. The microbiome, systemic immune function, and allotransplantation. Clin Microbiol Rev. 2016;29:191–99. doi:10.1128/CMR.00063-15.
- Fishman JA. Infection in organ transplantation. Am J Transplant. 2017;17:856–79. doi:10.1111/ajt.14208.
- Shen Y, Lian D, Shi K, Gao Y, Hu X, Yu K, Zhao Q, Feng C. Cancer risk and mutational patterns following organ transplantation. Front Cell Dev Biol. 2022;10:956334. doi:10.3389/fcell.2022.956334.
- Atkinson MA, Roep BO, Posgai A, Wheeler DCS, Peakman M. The challenge of modulating beta-cell autoimmunity in Type 1 diabetes. Lancet Diabetes Endocrinol. 2019;7:52–64. doi:10.1016/S2213-8587(18)30112-8.
- Deligne C, You S, Mallone R. Personalized immunotherapies for Type 1 diabetes: who, what, when, and how? J Pers Med. 2022;12:542. doi:10.3390/jpm12040542.
- Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, Greenbaum CJ, Herold KC, Krischer JP, Lernmark Å, et al. Staging presymptomatic Type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. 2015;38:1964–74. doi:10.2337/dc15-1419.
- Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, et al. HLA DR-DQ haplotypes and genotypes and Type 1 diabetes risk: analysis of the Type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084–92. doi:10.2337/db07-1331.
- Ilonen J, Lempainen J, Veijola R. The heterogeneous pathogenesis of Type 1 diabetes mellitus. Nat Rev Endocrinol. 2019;15:635–50. doi:10.1038/s41574-019-0254-y.
- Krischer JP, Lynch KF, Lernmark A, Hagopian WA, Rewers MJ, She JX, Toppari J, Ziegler A-G, Akolkar B. Genetic and environmental interactions modify the risk of diabetes-related autoimmunity by 6 years of age: the TEDDY study. Diabetes Care. 2017;40:1194–202. doi:10.2337/dc17-0238.
- Krischer JP, Lynch KF, Schatz DA, Ilonen J, Lernmark A, Hagopian WA, Rewers MJ, She J-X, Simell OG, Toppari J, et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: the TEDDY study. Diabetologia. 2015;58:980–87. doi:10.1007/s00125-015-3514-y.
- Vehik K, Bonifacio E, Lernmark A, Yu L, Williams A, Schatz D, Rewers M, She J-X, Toppari J, Hagopian W, et al. Hierarchical order of distinct autoantibody spreading and progression to Type 1 diabetes in the TEDDY study. Diabetes Care. 2020;43:2066–73. doi:10.2337/dc19-2547.
- Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, Bosi E, Brusko TM, DiMeglio LA, Evans-Molina C, et al. Introducing the endotype concept to address the challenge of disease heterogeneity in Type 1 diabetes. Diabetes Care. 2020;43:5–12. doi:10.2337/dc19-0880.
- Giannopoulou EZ, Winkler C, Chmiel R, Matzke C, Scholz M, Beyerlein A, Achenbach P, Bonifacio E, Ziegler A-G. Islet autoantibody phenotypes and incidence in children at increased risk for Type 1 diabetes. Diabetologia. 2015;58:2317–23. doi:10.1007/s00125-015-3672-y.
- Anderson M, Bluestone J. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85. doi:10.1146/annurev.immunol.23.021704.115643.
- Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to Type 1 diabetes. Nat Immunol. 2001;2:501–07. doi:10.1038/88694.
- Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. 2008;20:111–18. doi:10.1016/j.coi.2007.11.005.
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, et al. Prime role for an insulin epitope in the development of Type 1 diabetes in NOD mice. Nature. 2005;435:220–23. doi:10.1038/nature03523.
- Khosravi-Maharlooei M, Madley R, Borsotti C, Ferreira LMR, Sharp RC, Brehm MA, Greiner DL, Parent AV, Anderson MS, Sykes M, et al. Modeling human T1D-associated autoimmune processes. Mol Metab. 2022;56:101417. doi:10.1016/j.molmet.2021.101417.
- Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, Cuthbertson, D, Rafkin-Mervis, LE, Chase, HP, Leschek, E. Effects of oral insulin in relatives of patients with Type 1 diabetes: the diabetes prevention trial–Type 1. Diabetes Care. 2005;28:1068–76. doi:10.2337/diacare.28.5.1068.
- Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study G, Krischer JP, Schatz DA, Bundy B, Skyler JS, Greenbaum CJ. Effect of oral insulin on prevention of diabetes in relatives of patients with Type 1 diabetes: a randomized clinical trial. JAMA. 2017;318:1891–902. doi:10.1001/jama.2017.17070.
- Hannelius U, Beam CA, Ludvigsson J. Efficacy of GAD-alum immunotherapy associated with HLA-DR3-DQ2 in recently diagnosed Type 1 diabetes. Diabetologia. 2020;63:2177–81. doi:10.1007/s00125-020-05227-z.
- Lagattuta KA, Kang JB, Nathan A, Pauken KE, Jonsson AH, Rao DA, Sharpe AH, Ishigaki K, Raychaudhuri S. Repertoire analyses reveal T cell antigen receptor sequence features that influence T cell fate. Nat Immunol. 2022;23:446–57. doi:10.1038/s41590-022-01129-x.
- Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PT, Brodnicki TC, O’Connell PJ, Kedzierska K, Rodda C, et al. Proinsulin-specific, HLA-DQ8, and HLA-DQ8-transdimer–restricted CD4+ T cells infiltrate islets in Type 1 diabetes. Diabetes. 2015;64:172–82. doi:10.2337/db14-0858.
- Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, Bottino R, Naji A, Kaddis J, Elyaman W, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with Type 1 diabetes. Nat Med. 2016;22:1482–87. doi:10.1038/nm.4203.
- Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, Jones KL, Gottlieb PA, Kappler JW, Tang Q, et al. Islet-derived CD4 T-cells targeting proinsulin in human autoimmune diabetes. Diabetes. 2016;66:722–34. doi:10.2337/db16-1025.
- James EA, Mallone R, Kent SC, DiLorenzo TP. T-cell epitopes and neo-epitopes in Type 1 diabetes: a comprehensive update and reappraisal. Diabetes. 2020;69:1311–35. doi:10.2337/dbi19-0022.
- James EA, Pietropaolo M, Mamula MJ. Immune recognition of beta-cells: neoepitopes as key players in the loss of tolerance. Diabetes. 2018;67:1035–42. doi:10.2337/dbi17-0030.
- Piganelli JD, Mamula MJ, James EA. The role of beta cell stress and neo-epitopes in the immunopathology of Type 1 diabetes. Front Endocrinol (Lausanne). 2020;11:624590. doi:10.3389/fendo.2020.624590.
- Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, Armstrong M, Powell RL, Reisdorph N, Kumar N, et al. Pathogenic CD4 T cells in Type 1 diabetes recognize epitopes formed by peptide fusion. Science. 2016;351:711–14. doi:10.1126/science.aad2791.
- Jin N, Wang Y, Crawford F, White J, Marrack P, Dai S, Kappler JW. N-terminal additions to the WE14 peptide of chromogranin a create strong autoantigen agonists in Type 1 diabetes. Proc Natl Acad Sci USA. 2015;112:13318–23. doi:10.1073/pnas.1517862112.
- Baker RL, Jamison BL, Haskins K. Hybrid insulin peptides are neo-epitopes for CD4 T cells in autoimmune diabetes. Curr Opin Endocrinol Diabetes Obes. 2019;26:195–200. doi:10.1097/MED.0000000000000490.
- Diez J, Park Y, Zeller M, Brown D, Garza D, Ricordi C, Hutton J, Eisenbarth GS, Pugliese A. Differential splicing of the IA-2 mRNA in pancreas and lymphoid organs as a permissive genetic mechanism for autoimmunity against the IA-2 Type 1 diabetes autoantigen. Diabetes. 2001;50:895–900. doi:10.2337/diabetes.50.4.895.
- Dogra RS, Vaidyanathan P, Prabakar KR, Marshall KE, Hutton JC, Pugliese A. Alternative splicing of G6PC2, the gene coding for the islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), results in differential expression in human thymus and spleen compared with pancreas. Diabetologia. 2006;49:953–57. doi:10.1007/s00125-006-0185-8.
- de Jong VM, Abreu JR, Verrijn Stuart AA, van der Slik AR, Verhaeghen K, Engelse MA, Blom B, Staal FJT, Gorus FK, Roep BO. Alternative splicing and differential expression of the islet autoantigen IGRP between pancreas and thymus contributes to immunogenicity of pancreatic islets but not diabetogenicity in humans. Diabetologia. 2013;56:2651–58. doi:10.1007/s00125-013-3034-6.
- Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze JV, Nigi L, Lalanne AI, Sebastiani G, Carré A, Pinto S, et al. Conventional and neo-antigenic peptides presented by β cells are targeted by circulating naïve CD8+ T cells in Type 1 diabetic and healthy donors. Cell Metab. 2018;28:946–60 e6. doi:10.1016/j.cmet.2018.07.007.
- Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, van Veelen PA, Carlotti F, de Koning EJP, Hoeben RC, et al. Autoimmunity against a defective ribosomal insulin gene product in Type 1 diabetes. Nat Med. 2017;23:501–07. doi:10.1038/nm.4289.
- Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, Marrack P, Eisenbarth G, Kappler JW. Specificity and detection of insulin-reactive CD4+ T cells in Type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci USA. 2011;108:16729–34. doi:10.1073/pnas.1113954108.
- Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. 2001;166:908–17. doi:10.4049/jimmunol.166.2.908.
- Tian J, Song M, Kaufman DL. Designing personalized antigen-specific immunotherapies for autoimmune diseases—the case for using ignored target cell antigen determinants. Cells. 2022;11:1081. doi:10.3390/cells11071081.
- Skwarczynski M, Toth I. Peptide-based synthetic vaccines. Chem Sci. 2016;7:842–54. doi:10.1039/C5SC03892H.
- Solvason N, Lou YP, Peters W, Evans E, Martinez J, Ramirez U, Ocampo A, Yun R, Ahmad S, Liu E, et al. Improved efficacy of a tolerizing DNA vaccine for reversal of hyperglycemia through enhancement of gene expression and localization to intracellular sites. J Immunol. 2008;181:8298–307. doi:10.4049/jimmunol.181.12.8298.
- Roep BO, Solvason N, Gottlieb PA, Abreu JR, Harrison LC, Eisenbarth GS, Yu L, Leviten M, Hagopian WA, Buse JB, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8 + T cells in Type 1 diabetes. Sci Transl Med. 2013;5:191ra82. doi:10.1126/scitranslmed.3006103.
- Bot A, Smith D, Bot S, Hughes A, Wolfe T, Wang L, Woods C, von Herrath M. Plasmid vaccination with insulin B chain prevents autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:2950–55. doi:10.4049/jimmunol.167.5.2950.
- Pagni PP, Chaplin J, Wijaranakula M, Wesley JD, Granger J, Cracraft J, O‘Brien, C, Perdue, N, Kumar, V, Li, S, et al. Multicomponent plasmid protects mice from spontaneous autoimmune diabetes. Diabetes. 2021;71:157-169. doi:10.2337/db21-0327.
- Tisch R, Wang B, Weaver DJ, Liu B, Bui T, Arthos J, Serreze DV. Antigen-specific mediated suppression of β cell autoimmunity by plasmid DNA vaccination. J Immunol. 2001;166:2122–32. doi:10.4049/jimmunol.166.3.2122.
- Dastagir SR, Postigo-Fernandez J, Xu C, Stoeckle JH, Firdessa-Fite R, Creusot RJ. Efficient presentation of multiple endogenous epitopes to both CD4+ and CD8+ diabetogenic T cells for tolerance. Mol Ther Methods Clin Dev. 2017;4:27–38. doi:10.1016/j.omtm.2016.12.002.
- Postigo-Fernandez J, Creusot RJ. A multi-epitope DNA vaccine enables a broad engagement of diabetogenic T cells for tolerance in Type 1 diabetes. J Autoimmun. 2019;98:13–23. doi:10.1016/j.jaut.2018.11.003.
- Postigo-Fernandez J, Firdessa-Fite R, Creusot RJ. Preclinical evaluation of a precision medicine approach to DNA vaccination in Type 1 diabetes. Proc Natl Acad Sci USA. 2022;119:e2110987119. doi:10.1073/pnas.2110987119.
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–42. doi:10.1038/nature10098.
- Di Caro V, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N, Fiorina P. Involvement of suppressive B-lymphocytes in the mechanism of tolerogenic dendritic cell reversal of Type 1 diabetes in NOD mice. PLoS One. 2014;9:e83575. doi:10.1371/journal.pone.0083575.
- Boldison J, Da Rosa LC, Davies J, Wen L, Wong FS. Dendritic cells license regulatory B cells to produce IL-10 and mediate suppression of antigen-specific CD8 T cells. Cell Mol Immunol. 2020;17:843–55. doi:10.1038/s41423-019-0324-z.
- Luo Z, Lundin S, Mejia-Cordova M, Hassani I, Blixt M, Hjelmqvist D, Lau J, Espes D, Carlsson P-O, Sandler S, et al. Interleukin-35 prevents development of autoimmune diabetes possibly by maintaining the phenotype of regulatory B cells. Int J Mol Sci. 2021;22:22. doi:10.3390/ijms222312988.
- Hamad AR, Ahmed R, Donner T, Fousteri G. B cell-targeted immunotherapy for Type 1 diabetes: what can make it work? Discov Med. 2016;21:213–19.
- Bentzen AK, Hadrup SR. Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol Immunother. 2017;66:657–66. doi:10.1007/s00262-017-1971-5.
- Azoury ME, Tarayrah M, Afonso G, Pais A, Colli ML, Maillard C, Lavaud C, Alexandre-Heymann L, Gonzalez-Duque S, Verdier Y, et al. Peptides derived from insulin granule proteins are targeted by CD8(+) T cells across MHC class I restrictions in humans and NOD mice. Diabetes. 2020;69:2678–90. doi:10.2337/db20-0013.
- Uchtenhagen H, Rims C, Blahnik G, Chow IT, Kwok WW, Buckner JH, James EA. Efficient ex vivo analysis of CD4+ T-cell responses using combinatorial HLA class II tetramer staining. Nat Commun. 2016;7:12614. doi:10.1038/ncomms12614.
- Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, Kuranda K, Nigi L, Eugster A, Østerbye T, et al. Islet-reactive CD8 + T cell frequencies in the pancreas, but not in blood, distinguish Type 1 diabetic patients from healthy donors. Sci Immunol. 2018;3. doi:10.1126/sciimmunol.aao4013.
- Wiedeman AE, Muir VS, Rosasco MG, DeBerg HA, Presnell S, Haas B, Dufort MJ, Speake C, Greenbaum CJ, Serti E, et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow Type 1 diabetes progression. J Clin Invest. 2020;130:480–90. doi:10.1172/JCI126595.
- Samassa F, Mallone R. Self-antigens, benign autoimmunity and Type 1 diabetes: a beta-cell and T-cell perspective. Curr Opin Endocrinol Diabetes Obes. 2022;29:370–78. doi:10.1097/MED.0000000000000735.
- Ma KY, Schonnesen AA, He C, Xia AY, Sun E, Chen E, Sebastian KR, Guo Y-W, Balderas R, Kulkarni-Date M, et al. High-throughput and high-dimensional single-cell analysis of antigen-specific CD8(+) T cells. Nat Immunol. 2021;22:1590–98. doi:10.1038/s41590-021-01073-2.
- Nakayama M, McDaniel K, Fitzgerald-Miller L, Kiekhaefer C, Snell-Bergeon JK, Davidson HW, Rewers M, Yu L, Gottlieb P, Kappler JW, et al. Regulatory vs. inflammatory cytokine T-cell responses to mutated insulin peptides in healthy and Type 1 diabetic subjects. Proc Natl Acad Sci USA. 2015;112:4429–34. doi:10.1073/pnas.1502967112.
- Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of Type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med. 2011;208:1501–10. doi:10.1084/jem.20110574.
- Serr I, Furst RW, Achenbach P, Scherm MG, Gokmen F, Haupt F, Sedlmeier E-M, Knopff A, Shultz L, Willis RA, et al. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nat Commun. 2016;7:10991. doi:10.1038/ncomms10991.
- Malek Abrahimians E, Vander Elst L, Carlier VA, Saint-Remy JM. Thioreductase-containing epitopes inhibit the development of Type 1 diabetes in the NOD mouse model. Front Immunol. 2016;7:67. doi:10.3389/fimmu.2016.00067.
- Liu YF, Powrie J, Arif S, Yang JHM, Williams E, Khatri L, Joshi M, Lhuillier L, Fountoulakis N, Smith E, et al. Immune and metabolic effects of antigen-specific immunotherapy using multiple β-cell peptides in Type 1 diabetes. Diabetes. 2022;71:722–32. doi:10.2337/db21-0728.
- Firdessa-Fite R, Johnson SN, Leon MA, Khosravi-Maharlooei M, Baker RL, Sestak JO, Berkland C, Creusot RJ. Soluble antigen arrays efficiently deliver peptides and arrest spontaneous autoimmune diabetes. Diabetes. 2021;70:1334–46. doi:10.2337/db20-0845.
- Price JD, Hotta-Iwamura C, Zhao Y, Beauchamp NM, Tarbell KV. DCIR2+ cDC2 DCs and Zbtb32 restore CD4+ T-cell tolerance and inhibit diabetes. Diabetes. 2015;64:3521–31. doi:10.2337/db14-1880.
- Kenison JE, Jhaveri A, Li Z, Khadse N, Tjon E, Tezza S, Nowakowska D, Plasencia A, Stanton VP, Sherr DH, et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc Natl Acad Sci USA. 2020;117:32017–28. doi:10.1073/pnas.2016451117.
- Villalba A, Rodriguez-Fernandez S, Ampudia RM, Cano-Sarabia M, Perna-Barrull D, Bertran-Cobo C, Ehrenberg C, Maspoch D, Vives-Pi M. Preclinical evaluation of antigen-specific nanotherapy based on phosphatidylserine-liposomes for Type 1 diabetes. Artif Cells Nanomed Biotechnol. 2020;48:77–83. doi:10.1080/21691401.2019.1699812.
- Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, Haskins K. Nanoparticles containing an insulin–ChgA hybrid peptide protect from transfer of autoimmune diabetes by shifting the balance between effector T cells and regulatory T cells. J Immunol. 2019;203:48–57. doi:10.4049/jimmunol.1900127.
- Lee JS, Han P, Chaudhury R, Khan S, Bickerton S, McHugh MD, Park HB, Siefert AL, Rea G, Carballido JM, et al. Metabolic and immunomodulatory control of Type 1 diabetes via orally delivered bile-acid-polymer nanocarriers of insulin or rapamycin. Nat Biomed Eng. 2021;5:983–97. doi:10.1038/s41551-021-00791-0.
- Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, Akilli-Öztürk Ö, Kranz LM, Berger H, Petschenka J, et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science. 2021;371:145–53. doi:10.1126/science.aay3638.
- Rivas EI, Driver JP, Garabatos N, Presa M, Mora C, Rodriguez F, Serreze DV, Stratmann T. Targeting of a T cell agonist peptide to lysosomes by DNA vaccination induces tolerance in the nonobese diabetic mouse. J Immunol. 2011;186:4078–87. doi:10.4049/jimmunol.0902395.
- Takiishi T, Korf H, Van Belle TL, Robert S, Grieco FA, Caluwaerts S, Galleri L, Spagnuolo I, Steidler L, Van Huynegem K, et al. Reversal of autoimmune diabetes by restoration of antigen-specific tolerance using genetically modified Lactococcus lactis in mice. J Clin Invest. 2012;122:1717–25. doi:10.1172/JCI60530.
- Robert S, Gysemans C, Takiishi T, Korf H, Spagnuolo I, Sebastiani G, Van Huynegem K, Steidler L, Caluwaerts S, Demetter P, et al. Oral delivery of glutamic acid decarboxylase (GAD)-65 and IL10 by Lactococcus lactis reverses diabetes in recent-onset NOD mice. Diabetes. 2014;63:2876–87. doi:10.2337/db13-1236.
- Mao R, Yang M, Yang R, Chen Y, Diao E, Zhang T, Li D, Chang X, Chi Z, Wang Y. Oral delivery of the intracellular domain of the insulinoma-associated protein 2 (IA-2ic) by bacterium-like particles (BLPs) prevents Type 1 diabetes mellitus in NOD mice. Drug Deliv. 2022;29:925–36. doi:10.1080/10717544.2022.2053760.
- Lin M, Stoica-Nazarov C, Surls J, Kehl M, Bona C, Olsen C, Brumeanu TD, Casares S. Reversal of Type 1 diabetes by a new MHC II-peptide chimera: “Single-epitope-mediated suppression” to stabilize a polyclonal autoimmune T-cell process. Eur J Immunol. 2010;40:2277–88. doi:10.1002/eji.200940094.
- Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot HJ, Roep, BO. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide-for Type 1 diabetes. Lancet Diabetes Endocrinol. 2020;8:470–72. doi:10.1016/S2213-8587(20)30104-2.
- Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016;530:434–40. doi:10.1038/nature16962.
- Anderson AM, Landry LG, Alkanani AA, Pyle L, Powers AC, Atkinson MA, Mathews CE, Roep BO, Michels AW, Nakayama M. Human islet T cells are highly reactive to preproinsulin in Type 1 diabetes. Proc Natl Acad Sci USA. 2021;118:118. doi:10.1073/pnas.2107208118.
- Kaushansky N, Kerlero de Rosbo N, Zilkha-Falb R, Yosef-Hemo R, Cohen L, Ben-Nun A, Klein R. ‘Multi-epitope-targeted’ immune-specific therapy for a multiple sclerosis-like disease via engineered multi-epitope protein is superior to peptides. PLoS One. 2011;6(11):e27860. doi:10.1371/journal.pone.0027860.
- De Groot AS, Skowron G, White JR, Boyle C, Richard G, Serreze D, Martin WD. Therapeutic administration of tregitope-human albumin fusion with insulin peptides to promote antigen-specific adaptive tolerance induction. Sci Rep. 2019;9:16103. doi:10.1038/s41598-019-52331-1.
- Nagy NA, de Haas AM, Geijtenbeek TBH, van Ree R, Tas SW, van Kooyk Y, de Jong EC. Therapeutic liposomal vaccines for dendritic cell activation or tolerance. Front Immunol. 2021;12:674048. doi:10.3389/fimmu.2021.674048.
- Benne N, Ter Braake D, Stoppelenburg AJ, Broere F. Nanoparticles for inducing antigen-specific T cell tolerance in autoimmune diseases. Front Immunol. 2022;13:864403. doi:10.3389/fimmu.2022.864403.
- Kwiatkowski AJ, Stewart JM, Cho JJ, Avram D, Keselowsky BG. Nano and microparticle emerging strategies for treatment of autoimmune diseases: multiple sclerosis and Type 1 diabetes. Adv Healthc Mater. 2020;9:e2000164. doi:10.1002/adhm.202000164.
- Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell, CM. Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific. Cell Rep. 2016;16:2940–52. doi:10.1016/j.celrep.2016.08.033.
- Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, Xia C, Brusko TM, Wasserfall CH, Clare-Salzler MJ, et al. Dual-sized microparticle system for generating suppressive dendritic cells prevents and reverses Type 1 diabetes in the nonobese diabetic mouse model. ACS Biomater Sci Eng. 2019;5:2631–46. doi:10.1021/acsbiomaterials.9b00332.
- Lee Y, Kamada N, Moon JJ. Oral nanomedicine for modulating immunity, intestinal barrier functions, and gut microbiome. Adv Drug Deliv Rev. 2021;179:114021. doi:10.1016/j.addr.2021.114021.
- Mallone R, Culina S. Of bugs and men: antigen-fortified Lactococcus lactis for Type 1 diabetes immunotherapy. Diabetes. 2014;63:2603–05. doi:10.2337/db14-0587.
- Damo M, Wilson DS, Watkins EA, Hubbell JA. Soluble N-acetylgalactosamine-modified antigens enhance hepatocyte-dependent antigen cross-presentation and result in antigen-specific CD8(+) T cell tolerance development. Front Immunol. 2021;12:555095. doi:10.3389/fimmu.2021.555095.
- Mukhopadhaya A, Hanafusa T, Jarchum I, Chen YG, Iwai Y, Serreze DV, Steinman RM, Tarbell KV, DiLorenzo TP. Selective delivery of β cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8 + T cells in NOD mice. Proc Natl Acad Sci USA. 2008;105:6374–79. doi:10.1073/pnas.0802644105.
- Petzold C, Riewaldt J, Koenig T, Schallenberg S, Kretschmer K. Dendritic cell-targeted pancreatic beta-cell antigen leads to conversion of self-reactive CD4(+) T cells into regulatory T cells and promotes immunotolerance in NOD mice. Rev Diabet Stud. 2010;7:47–61. doi:10.1900/RDS.2010.7.47.
- Tabansky I, Keskin DB, Watts D, Petzold C, Funaro M, Sands W, Wright P, Yunis EJ, Najjar S, Diamond B, et al. Targeting DEC-205−DCIR2+ dendritic cells promotes immunological tolerance in proteolipid protein-induced experimental autoimmune encephalomyelitis. Mol Med. 2018;24:17. doi:10.1186/s10020-018-0017-6.
- Watkins EA, Antane JT, Roberts JL, Lorentz KM, Zuerndorfer S, Dunaif AC, Bailey LJ, Tremain AC, Nguyen M, De Loera RC, et al. Persistent antigen exposure via the eryptotic pathway drives terminal T cell dysfunction. Sci Immunol. 2021;6:6. doi:10.1126/sciimmunol.abe1801.
- Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for Immunotherapy. Mol Ther Methods Clin Dev. 2020;16:50–62. doi:10.1016/j.omtm.2019.10.015.
- Adair PR, Kim YC, Zhang AH, Yoon J, Scott DW. Human tregs made antigen specific by gene modification: the power to treat autoimmunity and antidrug antibodies with precision. Front Immunol. 2017;8:1117. doi:10.3389/fimmu.2017.01117.
- Gottwick C, Carambia A, Herkel J. Harnessing the liver to induce antigen-specific immune tolerance. Semin Immunopathol. 2022;44:475–84. doi:10.1007/s00281-022-00942-8.
- Rezende RM, Weiner HL. Oral tolerance: an updated review. Immunol Lett. 2022;245:29–37. doi:10.1016/j.imlet.2022.03.007.
- Akbarpour M, Goudy KS, Cantore A, Russo F, Sanvito F, Naldini L, Annoni A, Roncarolo MG. Insulin B chain 9–23 gene transfer to hepatocytes protects from Type 1 diabetes by inducing Ag-specific FoxP3 + T regs. Sci Transl Med. 2015;7:289ra81. doi:10.1126/scitranslmed.aaa3032.
- Cire S, Da Rocha S, Ferrand M, Collins MK, Galy A. In vivo gene delivery to lymph node stromal cells leads to transgene-specific CD8+ T cell anergy in mice. Mol Ther. 2016;24:1965–73. doi:10.1038/mt.2016.168.
- Carey ST, Gammon JM, Jewell CM. Biomaterial-enabled induction of pancreatic-specific regulatory T cells through distinct signal transduction pathways. Drug Deliv Transl Res. 2021;11:2468–81. doi:10.1007/s13346-021-01075-5.
- Liu X, Zhang S, Li X, Zheng P, Hu F, Zhou Z. Vaccination with a co-expression DNA plasmid containing GAD65 fragment gene and IL-10 gene induces regulatory CD4(+) T cells that prevent experimental autoimmune diabetes. Diabetes Metab Res Rev. 2016;32:522–33. doi:10.1002/dmrr.2780.
- Tanchot C, Barber DL, Chiodetti L, Schwartz RH. Adaptive tolerance of CD4+ T cells in vivo: multiple thresholds in response to a constant level of antigen presentation. J Immunol. 2001;167:2030–39. doi:10.4049/jimmunol.167.4.2030.
- Singh NJ, Schwartz RH. The strength of persistent antigenic stimulation modulates adaptive tolerance in peripheral CD4+ T cells. J Exp Med. 2003;198:1107–17. doi:10.1084/jem.20030913.
- Raimondi G, Zanoni I, Citterio S, Ricciardi-Castagnoli P, Granucci F. Induction of peripheral T cell tolerance by antigen-presenting B cells. I. Relevance of antigen presentation persistence. J Immunol. 2006;176:4012–20. doi:10.4049/jimmunol.176.7.4012.
- Raimondi G, Zanoni I, Citterio S, Ricciardi-Castagnoli P, Granucci F. Induction of peripheral T cell tolerance by antigen-presenting B cells. II. Chronic antigen presentation overrules antigen-presenting B cell activation. J Immunol. 2006;176:4021–28. doi:10.4049/jimmunol.176.7.4021.
- Obst R, van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D, van Santen H-M. Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation. Proc Natl Acad Sci USA. 2007;104:15460–65. doi:10.1073/pnas.0707331104.
- Noval Rivas M, Weatherly K, Hazzan M, Vokaer B, Dremier S, Gaudray F, Goldman M, Salmon I, Braun MY. Reviving function in CD4+ T cells adapted to persistent systemic antigen. J Immunol. 2009;183:4284–91. doi:10.4049/jimmunol.0901408.
- Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci USA. 2010;107:20453–58. doi:10.1073/pnas.1008437107.
- Trefzer A, Kadam P, Wang SH, Pennavaria S, Lober B, Akcabozan B, Kranich J, Brocker T, Nakano N, Irmler M, et al. Dynamic adoption of anergy by antigen-exhausted CD4+ T cells. Cell Rep. 2021;34:108748. doi:10.1016/j.celrep.2021.108748.
- den Boer AT, Diehl L, van Mierlo GJ, van der Voort EI, Fransen MF, Krimpenfort P, den Boer A, van Mierlo GJD, van der Voort EIH, Melief CJM, et al. Longevity of antigen presentation and activation status of APC are decisive factors in the balance between CTL immunity versus tolerance. J Immunol. 2001;167:2522–28. doi:10.4049/jimmunol.167.5.2522.
- Redmond WL, Hernandez J, Sherman LA. Deletion of naive CD8 T cells requires persistent antigen and is not programmed by an initial signal from the tolerogenic APC. J Immunol. 2003;171:6349–54. doi:10.4049/jimmunol.171.12.6349.
- Borsotti C, Danzl NM, Nauman G, Holzl MA, French C, Chavez E, Khosravi-Maharlooei M, Glauzy S, Delmotte FR, Meffre E, et al. HSC extrinsic sex-related and intrinsic autoimmune disease–related human B-cell variation is recapitulated in humanized mice. Blood Adv. 2017;1:2007–18. doi:10.1182/bloodadvances.2017006932.
- Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol. 2008;181:7350–55. doi:10.4049/jimmunol.181.10.7350.
- Buckner JH, Nepom GT. Obstacles and opportunities for targeting the effector T cell response in Type 1 diabetes. J Autoimmun. 2016;71:44–50. doi:10.1016/j.jaut.2016.02.009.
- Gearty SV, Dundar F, Zumbo P, Espinosa-Carrasco G, Shakiba M, Sanchez-Rivera FJ, Socci ND, Trivedi P, Lowe SW, Lauer P, et al. An autoimmune stem-like CD8 T cell population drives Type 1 diabetes. Nature. 2022;602:156–61. doi:10.1038/s41586-021-04248-x.
- Grebinoski S, Zhang Q, Cillo AR, Manne S, Xiao H, Brunazzi EA, Tabib T, Cardello C, Lian CG, Murphy GF, et al. Autoreactive CD8(+) T cells are restrained by an exhaustion-like program that is maintained by LAG3. Nat Immunol. 2022;23:868–77. doi:10.1038/s41590-022-01210-5.
- Abdelsamed HA, Zebley CC, Nguyen H, Rutishauser RL, Fan Y, Ghoneim HE, Crawford JC, Alfei F, Alli S, Ribeiro SP, et al. Beta cell-specific CD8(+) T cells maintain stem cell memory-associated epigenetic programs during Type 1 diabetes. Nat Immunol. 2020;21:578–87. doi:10.1038/s41590-020-0633-5.