
ABSTRACT
A promising personal immunotherapy is autologous dendritic cells (DC) loaded ex vivo with autologous tumor antigens (ATA) derived from self-renewing autologous cancer cells. DC-ATA are suspended in granulocyte-macrophage colony stimulating factor at the time of each subcutaneous injection. Previously, irradiated autologous tumor cell vaccines have produced encouraging results in 150 cancer patients, but the DC-ATA vaccine demonstrated superiority in single-arm and randomized trials in metastatic melanoma. DC-ATA have been injected into more than 200 patients with melanoma, glioblastoma, and ovarian, hepatocellular, and renal cell cancers. Key observations include: [1] greater than 95% success rates for tumor cell cultures and monocyte collection for dendritic cell production; [2] injections are well-tolerated; [3] the immune response is rapid and includes primarily TH1/TH17 cellular responses; [4] efficacy has been suggested by delayed but durable complete tumor regressions in patients with measurable disease, by progression-free survival in glioblastoma, and by overall survival in melanoma.
Introduction
Immunotherapy is now firmly established as a therapeutic modality for the treatment of widespread malignancy.Citation1–3 Because of inherent interpatient heterogeneity of cancer and individual immune systems, there is increasing interest in personalized and personal immunotherapies.Citation4,Citation5 Although these terms are often used synonymously, in this review, we distinguish between “personalized” and “personal.” Personalized immunotherapies are those that use a menu for selection of a combination of agents that are applicable to that specific patient. These may be chosen based on specific genomic or phenotypic markers, or by the availability of specific agents that are potentially additive or synergistic without increasing toxicity. One example of a personalized approach is the selection of a combination of monoclonal antibodies based on the phenotypic expression of that patient’s cancer cells. A second example of a personalized approach is the use of autologous antigen-presenting cells to present one or more tumor-associated antigens that are selected based on their expression on each patient’s cancer cells. A third example is the combining of immune modulating agents selected as per the characteristics of a specific patient’s tumor microenvironment (TME). In contrast, personal immunotherapies consist entirely of autologous biological material. The first such product that was granted commercial marketing approval was sipuleucel-T (Provenge®), autologous mononuclear cells incubated with a fusion protein that expresses granulocyte macrophage colony stimulating factor (GM-CSF) to differentiate monocytes (MC) into dendritic cells (DC) in vitro, and prostatic acid phosphatase (PAP) as an antigenic target.Citation6 The final product consists of autologous DC, lymphocytes, and undifferentiated MC, hence the generic name sipuleucel-T rather than sipuldencel-T. Other examples are adoptive cell therapies that utilize autologous immune cells including natural killer cell-based therapies such as lymphokine activated killer (LAK) cells and cytokine-induced killer (CIK) cells,Citation7,Citation8 tumor infiltrating lymphocytes (TIL),Citation9,Citation10 and autologous immune cells modified by chimeric antigen receptor (CAR),Citation11,Citation12 or T cell receptor (TCR) engineering.Citation13,Citation14 Other examples include purely autologous vaccines, such as those derived from autologous tumor, including tumor lysates,Citation15–18 heat shock proteins,Citation19 and mRNA.Citation20 For vaccine purposes, these autologous products can be injected directly as immunogens or delivered via autologous DC.
This article focuses on a singular personal immunotherapy that utilizes autologous tumor cells (TC) and autologous DC as personal therapeutic cancer vaccines.Citation21 The product is unique such that the antigen source is autologous cancer cells that are self-renewing in short-term tissue culture and have characteristics of “tumor initiating cells (TIC),” a term that encompasses cells with features of cancer stem cells and early progenitor cells.Citation22,Citation23 A generic term for the product is DC-ATA based on the autologous DC and autologous tumor antigens (ATA). Other autologous DC-ATA approaches have used lysates of whole tumor,Citation17,Citation18 mRNA from whole tumor,Citation20 or manufactured antigens selected after genomic analysis of whole tumor and computer selection of antigens predicted to illicit a good immune response in that patient.Citation24–28
Theoretical advantages of tumor initiating cells as an antigen source
There are several potential advantages to the autologous TIC approach, but one of the most important one may be the elimination of normal cells from the population of cells from which tumor antigen is derived. Tumor masses contain not only malignant cells, but also hematopoietic, immune, and stromal cells. Specific tumor masses may consist almost entirely of malignant cells at one extreme, or a predominance of stroma and nonmalignant cells at the other extreme. Cell counts on histologic sections of cancer specimens have estimated that anywhere from 5% to 95% of a sample are cancer cells, and conversely, 95% to 5% normal cells.Citation29,Citation30 A higher stroma to TC ratio is associated with a worse prognosis.Citation31 Because of the interplay between malignant and nonmalignant cells in the TME, there is interest in targeting non-cancerous TME components.Citation32 However, nonselective immunization against these normal cells has not been considered a desirable strategy because of the theoretical potential to destroy anti-cancer cells such as natural killer and cytotoxic T cells, and/or induce autoimmune responses against normal tissue.Citation33
The other unique aspect of this approach is the focus on TICs, which typically make up only a small percentage of malignant cells in both hematologic malignancies and solid tumors, compared to the percentage of terminally differentiated cells that constitute the vast majority of the malignant cells.Citation34–36 The identification and isolation of TICs for therapeutic targeting has proved challenging,Citation37,Citation38 but short-term (30-d) cell cultures under conditions that favor survival of TC result in the elimination of all but self-renewing cancer cells, unless specific cytokines such as interleukin-2 (IL-2) are added to favor survival and propagation of lymphocytes.Citation9,Citation39,Citation40 TICs are believed to be responsible for resistance to standard anti-cancer therapies and to account for the propagation of metastatic lesions, and therefore are desirable targets for anti-cancer therapy including immunotherapy.Citation41–47 The DC-ATA approach is designed to induce or enhance endogenous immune responses against antigens on TICs that result from translation and transcription of mRNA resulting from non-synonymous mutations that are unique to each cancer patient and therefore are tumor specific antigens.Citation48,Citation49 Furthermore, if there are tumor-associated antigens associated with driver mutations or other tumor-associated antigens that the immune system can recognize, these may also be safely targeted (e.g. melanin, gp100, and thyroid antigens associated with melanoma). Antigens from TICs have induced immune responses and anti-tumor effects in animal models.Citation50–53 Various phenotypic markers have been associated with TICs, including CD133, CD24, CD44, CXCR4, EpCAM, ALDH-1, and CD166,Citation54 but expression varies among different tissue types, and there is no unique TIC phenotype.Citation55–57 We continue to test cell lines for expression of various markers, but we do not use them as a quality or release criteria. The non-synonymous mutations in TICs are preserved in subclones of more differentiated malignant cells, although the latter will contain greater numbers of acquired mutations, including driver mutations that are not present on the TICs. These mutations may or may not be associated with some differences in phenotype.
From a vaccine perspective, TIC antigens could be introduced directly or indirectly (e.g. mRNA) as injections that would result in endogenous DC processing and antigen presentation to stimulate antigen-specific immune responses. However, there are numerous theoretical advantages to loading autologous DC ex vivo with relevant antigen. This includes loading of a much larger number of DC than might be attracted to an injection site, and loading of antigens ex vivo, away from any inhibitory factors that might interfere with antigen loading in vivo, such as happens in the milieu of the immunosuppressive TME. After injection, only a small percentage of DC will traffic to regional lymph nodes where they activate T lymphocytes that have T-cell receptors that are cognate for tumor antigens.Citation58–64 Furthermore, non-trafficking DC that undergo apoptosis after injection, but contain antigenic peptides in their lysosomes, may induce a desired antigen-specific anti-tumor immune response after phagocytosis and protein processing by endogenous DC.Citation65–68
Components of the personal DC-ATA vaccines
Autologous tumor antigens (ATA)
The most important component of any vaccine is antigen.Citation69–71 The evolution of short-term cell cultures as the source of ATA was reviewed previously.Citation72 Prior to 2000, our vaccine efforts focused on irradiated autologous TC, and specifically self-renewing cells from autologous short-term cell lines,Citation73–78 rather than whole autologous tumor as was being pursued most notably in colon cancer and melanoma.Citation15,Citation16,Citation79,Citation80 Additional perceived advantages of this approach were: (1) the use of short-term autologous cancer cell cultures as the source of cancer antigens, rather than long-term allogeneic cell lines that focused on antigens that might be commonly shared; (2) the cell culture conditions and time in culture eliminated nonmalignant cells that were inevitably present in the initial tumor sample that had been surgically resected, and therefore reduced the quantity of irrelevant antigens; (3) the cell culture conditions promoted the survival of cells that could self-renew, which later were found to be consistent with cancer stem cells, and more rapidly proliferating early progenitor cells; (4) culturing allowed selection of ATA from cells with the capacity to self-renew, rather than more differentiated malignant cells that eventually undergo apoptosis and do not contribute to propagation of the cancer; (5) the use of intact TC rather than secreted antigens might facilitate antigen recognition in the cancer cell membrane; (6) the number of tumor cells used for ATA might serve as a surrogate for the quantity of antigen being presented; (7) the cell lines provided an unlimited supply of pure TC that could be used for genetic analysis and for in vitro anti-tumor assays; and (8) because the cells were proliferating, there was the potential to transfect the cells with DNA for specific cytokines that might enhance immune recognition of the TC.Citation73,Citation81 In recent years, we used lysates of irradiated TC (ITC) rather than intact ITC as the source of antigen.Citation82 This results in a final product that consists only of DC rather than DC plus residual ITC that were not phagocytosed during coincubation.Citation83 Human studies indicate that DC loaded with autologous antigen by incubating with either ITC or ITC lysate results in an immunogenic vaccine.Citation84
The rationale for this personal approach was further enhanced when it was shown that patient-specific neoantigens were generally more important immunologic targets on cancer cells than shared tumor-associated antigens.Citation48,Citation49,Citation85–87 It is now known that all cancers are naturally immunogenic because each contain tens to thousands of non-synonymous mutations from which proteins potentially can be transcribed and translated.Citation48,Citation49,Citation88 As such mutations have taken place during the life span of the patient, rather than existing at birth, the resulting proteins are likely to appear foreign to the patient’s immune system, and therefore antigenic.Citation89–91 The existence of these natural immune responses to neoantigens is the basis for the success of immune checkpoint inhibitors of programmed death molecule 1 (PD-1) and its ligand (PDL-1) and human cytotoxic T cell antigen-4 (anti-CTLA4).Citation92–99 The unique antigens encoded by such singular mutations have been loaded onto autologous DC, and when injected, induce and/or enhance immune recognition of such antigens.Citation100 We have continued to use self-renewing autologous TC as the source of unique patient-specific neoantigens, rather than the more complex and expensive process of performing genomic analysis and using computer programs to predict what may be the optimal neoantigen targets in each patient’s tumor, and then manufacturing these antigens.Citation26,Citation28,Citation101
DC-ATA: antigen-loaded autologous dendritic cells
The second component of the DC-ATA vaccine is the autologous DC that will be loaded with antigen ex vivo by incubating with ITC, or with a lysate of ITC. As reviewed elsewhere, DC are the most important of the antigen-presenting cells that are crucial for initiation of anti-tumor immune responses.Citation102–105 We and others have been testing DC vaccines in the clinic for more than two decades.Citation106–108 By 2000, there was great interest in autologous DC loaded with antigen as an alternative to injecting antigen directly. The growing enthusiasm for DC was based on increasing awareness of the extensive preclinical studies of DC that culminated in a Nobel Prize for Ralph Steinman in 2011Citation109,Citation110 By 2000 it was established that DC could be reliably generated from peripheral blood mononuclear cells (PBMC) by incubating in the presence of interleukin-4 (IL-4) and GM-CSF.Citation111 Important for potential commercial application, it was demonstrated that both MC and DC could be cryopreserved and subsequently thawed with retention of viability and biological activity.Citation112 Studies in murine tumor models suggested that DC vaccines were superior to TC vaccines as antigen delivery strategies.Citation113 Recent animal studies suggest that immunizing with personal neoantigens via DC is also more effective than direct injection of personal neoantigens.Citation114 Early clinical reports demonstrated safety and some efficacy for DC loaded with anti-idiotype antigens in patients with B cell lymphoma,Citation115 and for DC loaded with ATA or tumor associated peptide antigens in patients with metastatic melanoma.Citation116 When we started treating patients with DC-ATA vaccines, it was quickly apparent that the clinical outcomes were superior to what we had observed with TC vaccines.Citation117,Citation118 This was subsequently confirmed in a single arm trial in melanoma in which overall survival was compared to that of historical controls,Citation119 and in a randomized trial in melanoma that directly compared personal DC vaccines to personal TC vaccines with self-renewing ITC serving as the antigen source for both products.Citation120
DC can be loaded with ATA by various means including creation of a DC-TC fusion hybridoma cell, incubation of DC with TC, incubation of DC with TC lysate, incubation with free antigen, and electroporation with mRNA.Citation84,Citation121 We have utilized both incubation of DC with ITC and incubation of DC with ITC lysates. Between 2001 and 2015, DC were incubated with intact ITC,Citation119,Citation120,Citation122–124 during 2017–2021 DC were incubated with ITC lysates.Citation125,Citation126
Manufacturing of personal DC-ATA vaccines
The methodology for manufacturing personal DC-ATA was published previously.Citation117–120,Citation122–124 illustrates the manufacturing procedure for the two intermediate products (autologous self-renewing TC and autologous DC), and the final product (antigen-loaded DC, i.e., DC-ATA). A surgical tumor specimen is processed into a cell suspension and incubated for two to four weeks in media that sustains TICs and supports their propagation. Peripheral blood mononuclear cells (PBMC) are collected by leukapheresis.Citation127 The PBMC are enriched for MC from which DC are differentiated by incubating MC with IL-4 and GM-CSF. ATA are loaded by incubating DC for 18 to 24 hours with either whole ITC, or with ITC lysate. In recent years, DC were incubated with ITC lysate for trials in glioblastoma (GBM),Citation125 and ovarian cancer.Citation126 The final DC-ATA product is divided into aliquots and cryopreserved in liquid nitrogen for eventual administration. At the time of each treatment, a vial of thawed DC-ATA cell product is admixed with 500-µg GM-CSF shortly before each subcutaneous injection.
Figure 1. Manufacturing of dendritic cell-autologous tumor antigen (DC-ATA) vaccine.
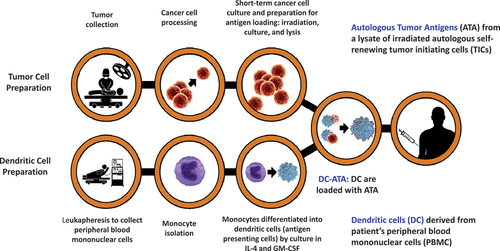
Over the years, there have been improvements in the media used to grow TC including adding growth factors and utilizing serum-free media specifically selected because of its ability to enhance stem cell growth in vitro, which also eliminates or minimizes the use of fetal bovine serum (FBS).Citation72 Intact ITC or ITC lysate have served as the ATA source.Citation72 The use of lysate rather than intact ITC yields a more uniform final product and eliminates the remote possibility that tumorigenic TC might survive and propagate. Using an ITC lysate rather than whole ITC results in a product that is 97–100% DC-ATA.Citation72,Citation125,Citation126 Methodology to enrich for MC from PBMC collections has evolved from plastic adherence and scraping,Citation116,Citation118,Citation122 to using mechanical flow procedures based on cell size.Citation120,Citation122,Citation124,Citation125,Citation128 Vaccine doses for individual patients have ranged from 1 to 30 million cells per subcutaneous injection given weekly for 3 weeks and then at weeks 8, 12, 16, 20, and 24 which results in eight doses over about 6 months.
Adjuvant granulocyte-macrophage colony stimulating factor
The third component of the DC-ATA vaccine is adjuvant GM-CSF. When we started clinical investigation into personal ATA vaccines, it was generally accepted that an immune-stimulating adjuvant was necessary for a vaccine to be effective.Citation129,Citation130 Alum and incomplete Freund’s adjuvant (mineral oil) and complete Freund’s adjuvant (mineral oil with dried mycobacteria) were available,Citation131 but more popular at the time in cancer vaccine investigation was Bacille-Calmette-Guérin (BCG) which was being used with allogeneic cell lines in melanoma,Citation132 and with lysates of whole autologous tumor in colorectal cancer and melanoma.Citation15,Citation16 IL-2 had not yet been approved, but the cytokines interferon-alpha (INF-a), interferon gamma (IFN-γ), and GM-CSF had recently become commercially available and were perceived as potentially superior to BCG as adjuvants.Citation133–135 Since then additional adjuvants have been developed, and adjuvants are still considered to be an important component for optimizing cancer vaccines.Citation136,Citation137
IFN-γ and GM-CSF were tested as adjuvants in a 98-patient randomized phase 2 trial in which patients were stratified by tumor type and by whether they had measurable metastatic disease prior to randomization.Citation138 All patients received a personal vaccine consisting of self-renewing autologous ITC. Because of existing regulatory restrictions, these adjuvants were not admixed with the ITC, but instead they were injected subcutaneously at an adjacent site at the time of each TC vaccine injection. As an alternative to participating in this randomized trial, physicians who wanted to treat their patients with the patient-specific vaccine, but did not want to enroll their patients in the randomized trial, had the option of using the adjuvant of their choice, typically BCG, IFN-α, or the hapten dinitrophenol, or no adjuvant at all.Citation138 Most of the patients in the randomized trial had melanoma, renal cell cancer (RCC), or sarcoma; about half had measurable disease. Injections of ITC vaccine and adjuvant were well-tolerated and similar in both arms. The most common adverse events (AEs) were mild-to-moderate local injection site reactions or systemic symptoms such as fatigue, fever, chills, rash, and/or pruritus, myalgias, arthralgias, and nausea and/or vomiting. About 25% of patients in each arm had a positive delayed-type hypersensitivity reaction to an intradermal injection of one million autologous ITC. There were no differences in objective response rates (ORRs), progression-free survival (PFS), or overall survival (OS).Citation138 In a retrospective analysis of 74 patients with metastatic melanoma who were treated with autologous TC vaccine, the 53 patients who received GM-CSF or IFN-γ as adjuvant had longer PFS (p < .0001) compared to 21 who received a different adjuvant (n = 14) or no adjuvant (n = 7).Citation78 It was concluded that either of these cytokines would be suitable as an adjuvant in subsequent vaccine trials.
In 2000, when we switched our research focus to DC loaded with ATA, it was decided to proceed with GM-CSF as the adjuvant, largely based on work by others.Citation109,Citation139–141 It had been shown that GM-CSF had important effects on the maturation and function of DC, and even though the immunostimulatory function of DC in the skin was enhanced by various cytokines, GM-CSF had the most profound effects.Citation109 In a seminal study performed in murine tumor models, cancer cells were transfected with genetic messages for 10 potential adjuvants, including IL-2, IL-4, IL-5, IL-6, GM-CSF, IFNγ, ICAM-1, CD2, IL-1 receptor antagonist, and human TNF-α, and then the cells were irradiated.Citation140 GM-CSF was the most powerful adjuvant of the 10 tested. Other studies also suggested that GM-CSF was an effective vaccine adjuvant.Citation142,Citation143 Subsequently, additional studies in mice,Citation144,Citation145 and in humans,Citation146,Citation147 have provided more evidence for beneficial adjuvant effects of GM-CSF. In all the personal DC-ATA trials, the cells were admixed with 500 µg of GM-CSF just prior to each injection.
Two phase 2 trials conducted in melanoma patients suggested that repeated subcutaneous injections of GM-CSF monotherapy daily in alternating 2-week cycles might be an effective immunotherapy with acceptable toxicity.Citation148,Citation149 However, efficacy as defined by PFS and OS was not confirmed in two large randomized trials.Citation150–152 Although it is generally well-tolerated, GM-CSF does have predictable mild-to-moderate, self-limited, local and systemic side effects.Citation153,Citation154 These are more troublesome in association with consecutive daily injections compared to single injections. Individuals with known hypersensitivity to GM-CSF were ineligible to participate in DC-ATA vaccine trials, but during screening no patient has ever been excluded because of a history of such hypersensitivity. We have observed two acute hypersensitivity reactions during our trials. In 2003, aggregates in a single lot of a GM-CSF formulation that was being produced by a new manufacturer, were determined to be the cause of anaphylactoid reactions in one patient with RCC and one with melanoma.Citation119,Citation123 There have been no acute anaphylactoid reactions following DC-ATA vaccinations since that time. In a randomized trial in ovarian cancer, one patient in the control arm, who was receiving injections of autologous MC suspended in GM-CSF, had recurrent and refractory urticaria after her eighth and final injection, which led to a brief hospitalization.Citation155
Issues for DC-ATA vaccines
There are many questions that arise for a product such as personal DC-ATA. The first three are feasibility questions, the fourth addresses safety, and the last two are related to efficacy. First, what is the feasibility of collecting tumors and successfully establishing cell lines? The second question is what is the feasibility and success rate for collecting sufficient numbers of MC and differentiating them into DCs? Third, what is the success rate for converting the intermediate TC and DC products into satisfactory final DC-ATA products? The fourth question is: are subcutaneous injections of DC-ATA admixed in GM-CSF well-tolerated? Fifth, are injections of DC-ATA associated with desirable immune responses? Sixth, are subcutaneous injections of personal DC-ATA vaccines associated with clinical benefit, such as ORR, PFS, and OS?
The remainder of this review addresses these six questions using data generated during the past 22 years in a series of clinical trials conducted in patients with a variety of advanced cancers. Those trials are summarized in . The clinical data emanated from a 9-patient phase 1–2 trial in distant metastatic or locally advanced RCC,Citation123 an 8-patient phase 1 trial in locally advanced hepatocellular cancer (HCC) in Chinese patients who had hepatitis B,Citation122 a 54-patient single arm adaptive phase 2 trial in metastatic melanoma,Citation119 a 42-patient randomized phase 2 trial in metastatic melanoma in which 18 were randomized to DC-ATA and 24 to the ITC vaccine control arm,Citation124 a 45-patient randomized phase 2 trial in newly diagnosed stages 3 and 4 ovarian cancer in which 29 were randomized to DC-ATA (28 treated) and 16 to the MC control arm,Citation155 and a 60-patient single-arm phase 2 trial in patients with newly diagnosed GBM.Citation125
Table 1. Summary of clinical trials in which patients were treated with personal dendritic cell-autologous tumor antigen (DC-ATA) vaccines.
*Yes, cell lines can be reliably established from surgically resected fresh tumor specimens.
For this personal vaccine approach, there can be no treatment product unless a short-term cell line is successfully established to serve as the ATA source. ATA is the first intermediate product needed in order to manufacture this personal vaccine. If ATA cannot be produced, then there can be no DC-ATA therapeutic vaccine product. The logistics associated with tissue acquisition and shipping specimens can be challenging. Logistical coordination is required to obtain approvals and permissions at various levels in order to collect sufficient quantities of fresh, sterile, tumor tissue and transport it to the manufacturing facility in a timely manner. The clinical team must be sure that each clinical site has been appropriately “activated,” which includes approved consent forms, local regulatory approvals, and education of local personnel who will be involved in the study. Clinical sites are provided with kits specifically designed for collection and transport of fresh tumor tissue. These kits are validated with shelf-lives of about 90 d from the time the transport media is made. Sections of tumor are placed into conical tubes that contain transport media, then shipped by overnight courier to the manufacturing laboratory. Quality of tumor, fresh (not frozen or formalin fixed) and non-necrotic, is more important than quantity. About a cubic centimeter of tissue is recommended, but a melanoma cell line was established from a needle aspirate of ocular melanoma.Citation156
In addition to the successful implementation of the logistics needed for tissue acquisition, this metric also takes into account the appropriateness of standard operating procedures for growing short-term cancer cell lines and the training and competency of laboratory staff. Historically cell lines have been successfully established from samples that took up to 3 d to arrive from foreign countries.Citation156 Tumor samples are processed and either placed directly into cell culture and/or cryopreserved. If appropriate freezing procedures are used, the success rate is comparable for fresh and cryopreserved samples.Citation72,Citation156,Citation157 Overgrowth of fibroblasts can be problematic for certain tissues, especially if FBS is needed for cell propagation; therefore, special procedures are sometimes needed to limit the growth of fibroblasts.Citation158 Between 2000 and 2011, in trials with ITC and DC vaccines, enough TC were grown to insure that 10 million ITC per dose would be available for at least eight doses; so the goal was to grow 100 million TC. The success rate using this criterion was about 45%, with success rates of more than 50% in RCC,Citation75,Citation76,Citation123 melanoma,Citation119,Citation120,Citation124 sarcoma,Citation77 and primary GBM,Citation156 with lower success rates for recurrent GBM and for other cancers.Citation156 Using media supplemented with factors to encourage propagation of TICs, the success rate in the China HCC study was 17/17 (100%) including 15/15 for hepatoma, 1/1 for cholangiocarcinoma, and 1/1 for metastatic colon adenocarcinoma.Citation122
Based on lack of clinical correlation with the numbers of TC injected in TC vaccines, or incubated with DC for DC vaccines, in recent years, we have attempted to provide ATA from one million ITC per dose and have grown cell lines for either four weeks or to 10 million cells, whichever occurs first, with the caveat that accurate cell counting is difficult when spheroids are present. The best data regarding cell line success using current methods are from clinical trials recently conducted during 2018–2021 in patients with newly diagnosed GBM or ovarian cancer.Citation125,Citation126 In the GBM trial, 106 patients signed a consent for tumor acquisition, but many of these proved to be other than GBM. Ultimately, 80 GBM tumors were received at the manufacturing site, but 7 patients withdrew consent for participation; so, their cell cultures were discontinued. Efforts to establish cell lines were attempted for 73 patients with success in 71 (97%).Citation125 The two unsuccessful samples were due to contamination. Samples for treatment were prepared for 60 patients and 58 (97%) had lysates from more than one million ITC per dose. Similarly, in patients with newly diagnosed stage 3 or 4 ovarian cancer, efforts were successful for 70/72 (97%) of ovarian samples that resulted from 92 pre-surgical consents for tissue acquisition.Citation126 Twelve of those samples were not malignant ovarian cancer, one patient was ineligible because of low-stage disease, no tumor was submitted from three patients, and four patients withdrew consent for participation. Of the two unsuccessful samples, one was contaminated, and one could not be grown. All 56 (100%) of the successful cell cultures provided more than one million lysed ITC per dose.
Despite the numerous obstacles and challenges, data indicate that it is feasible to reliably collect tumor tissue and successfully establish short-term cell lines for use in generating ATA for the manufacturing of the autologous DC-ATA vaccines. However, there is an inherent inefficiency due to uncontrollable clinical issues, including lack of a specific tissue and/or stage diagnosis until after the surgical procedure, and the subsequent withdrawal of consent by patients for various reasons. Thus collectively, in the ovary and GBM trials, out of a total of 198 consents to collect tissue, an effort to establish a cell line was only attempted on behalf of 145 patients (73%), but cell lines were successfully established for 141/145 (97.2%).
*Yes, it is feasible to collect sufficient quantities of monocytes and differentiate them into dendritic cells
DCs are the second critical intermediate component needed for manufacturing DC-ATA. Sufficient numbers of autologous MC must be collected and differentiated into DC in order to manufacture personal DC-ATA vaccines. Similar to tissue acquisition, obtaining autologous MC also requires permissions, agreements, standard operating procedures, coordination of PBMC collection and shipping of PBMC to the manufacturing site, and competency of laboratory personnel who isolate the MC and differentiate them into DC in cell culture. If there are insufficient MC, or they cannot be differentiated into DC, then there can be no DC-ATA for that patient.
In order to collect PBMC, all patients underwent a 2–4-h leukapheresis procedure on a commercially available cell collecting machine.Citation127 Leukapheresis procedures were performed without prior injection of colony stimulating factors for mobilization, in order to minimize the number of granulocytes collected, which is a problem for both granulocyte colony stimulating factor and GM-CSF, and to avoid premature differentiation of MC to DC prior to in vitro antigen exposure, which occurs with GM-CSF. In our RCC and melanoma DC-ATA trials conducted between 2000 and 2011, PBMC were collected at one location using leukapheresis procedures and a specialized team that was originally established for the collection of PBMC from patients who were to be treated with high-dose chemotherapy and autologous stem cell rescue, and for clinical trials of adoptive cell therapies utilizing cells derived from PBMC, and later for generation of DC for the DC-ATA vaccine trials. The success rate was 100%, including 11/11 for RCC patientsCitation123 and 87/87 for melanoma patients.Citation83,Citation119,Citation124
The recent trials in ovary and GBM involved multiple clinical institutions (six ovary, seven GBM) with PBMC products collected by local leukapheresis teams and shipped by overnight courier from as far away as Colorado, New Jersey, and Kentucky. Samples always arrived for further processing within 48 h of collection. A target goal was set at 450,000 CD14+ cells following an elutriation procedure to enrich for MC with removal of erythrocytes, platelets, and neutrophils. This number was chosen based on the number of DC differentiated from MC in previous studies. It was intended to assure final products containing at least one million DC per dose. The success rates for PBMC collection were 63/65 (97%) for GBM patientsCitation125 and 47/50 (94%) for ovarian cancer patients.Citation128 Ten GBM patients required a second leukapheresis in order to achieve an adequate MC number, but one patient declined to repeat the pheresis procedure, and one patient had three unsuccessful collection attempts. Among the ovarian cancer patients, two required a second leukapheresis for success, and three declined to undergo a second leukapheresis. DC were successfully generated from the MC for all 89 patients (60 GBM and 29 ovarian) for whom manufacturing of DC-ATA was attempted.
*Yes, once intermediate tumor cell and monocyte products are available, a final DC-ATA product can be reliably manufactured
For a commercial product and reliable treatment planning, it is important that when the intermediate ITC lysate and DC are available, the final DC-ATA product can be reliably manufactured. However, missteps can occur during manufacturing, and the final DC-ATA product must meet pre-specified quality assurance criteria before the personal product is released for clinical use. Final DC-ATA products using intact ITC were generated for 11/11 RCC patients with nine administered,Citation123 56/56 melanoma patients in the single arm trial with 54 administered,Citation119 18/18 in the melanoma randomized trial with all 18 DC-ATA products administered,Citation120,Citation124 and 17/17 in the HCC trial with eight administered.Citation122 In the multicenter trials using the ITC-lysate DC-ATA products, success rates were 60/60 in GBM with 57 administeredCitation125 and 28/29 in ovarian cancer with all 28 administered with the one failure due to contamination.Citation128 The combined results were 181/182 (99.4%).
*Yes, subcutaneous injections of DC-ATA admixed in GM-CSF are well-tolerated
Safety is a critical issue for all therapeutic products. Although it is presumed that autologous products should be well-tolerated, this has had to be confirmed in clinical subjects. GM-CSF is commercially available in the United States and has a well-known side-effect profile,Citation153,Citation154 but the safety of DC-ATA admixed with GM-CSF had to be established. In all of these trials, study agents were injected subcutaneously at weeks 1, 2, 3, 8, 12, 16, 20, and 24, i.e., up to eight doses over 6 months.
summarizes AE in the larger trials and shows that patients received an average of more than seven of the eight planned injections in each study. In addition to the 180 patients who received DC-ATA, there were 24 patients injected with ITC admixed with GM-CSF in the randomized melanoma trial,Citation120,Citation124 and 16 patients injected with autologous MC admixed in GM-CSF in the randomized ovarian trial.Citation155 Data in confirm that the personal vaccines administered in GM-CSF are well-tolerated. No patient discontinued treatment because of an AE. There were no life-threatening (grade 4) or lethal toxicities, and severe (grade 3) AEs were rare. As shown in , the most common AEs were local injection site reactions (ISR), including erythema, discomfort, and swelling, with frequencies of 54%–80%, except in GBM in which the frequency of ISR was only 16%, perhaps because so many GBM patients were taking corticosteroids.Citation125 AE tended to be mild to moderate in intensity, brief in duration, and self-limited. Occasional patients took acetaminophen or non-steroidal anti-inflammatory agents briefly, and/or diphenhydramine for rashes that were not readily controlled with topical creams.
Table 2. Summary of adverse events reported in DC-ATA clinical trials in which up to 8 doses were injected.
In all of these trials, the cellular vaccines were admixed in GM-CSF; therefore, it is hard to determine whether AE are all due to GM-CSF or are some AE unique for the DC-ATA. The vast majority of AE were grade 1 or 2, and many of the grade 3 were attributed to other than the study agents. For instance, the grade 3 AE in the DC-ATA and MC arms of the ovarian trial were cytopenias attributed to concurrent chemotherapy.Citation155 Interestingly, in the randomized melanoma trial, although DC-ATA and ITC were both well-tolerated, there was a higher frequency of grade 2 or 3 AE in the ITC arm perhaps because of residual FBS in the ITC product.Citation124 In the two randomized trials, the frequency and severity of AE were similar between treatment arms,Citation124,Citation155 which suggest that vaccine-related AEs are probably due to GM-CSF. There were only two hospitalizations because of serious adverse events (SAEs) attributed to vaccine injections. One occurred in a melanoma patient treated with DC-ATA in 2003, who experienced an anaphylactoid reaction that led to her being hospitalized overnight.Citation119 This proved to be due to aggregates in the GM-CSF product. The other was an ovarian cancer patient in the MC control arm who was hospitalized after her final injection because of refractory urticaria that were treated with corticosteroids.Citation155
Disease progression was always the reason for discontinuing treatment except for two patients who died from other medical causes. There was a similar lack of toxicity among the nine RCC patients in that trial.Citation123 There was no exacerbation of hepatitis or viremia among eight hepatitis-B-virus positive HCC patients.Citation122 There has been no evidence of cumulative toxicity except for rare patients who have developed more frequent and bothersome skin rashes after receiving several injections. In two patients in whom this has occurred, there were no such symptoms when the DC product was injected by itself rather than admixed in GM-CSF.Citation119
*Yes, DC-ATA injections are associated with desired immune responses
The mechanism of action (MOA) of all vaccines including DC-ATA is the induction and/or enhancement of anti-cancer immune responses that result in targeting and killing of cancer cells.Citation69–71 Most important ones are direct cytotoxic T cell effects, but there may also be a role for antibody-dependent cell-mediated cytotoxicity based on anti-tumor antibodies in conjunction with various immune cells with appropriate Fc receptors.Citation69–71 Detection of desirable immune responses is supportive evidence for the in vivo bioactivity of DC-ATA and evidence that the MOA is taking place, and ideally might be a surrogate marker for clinical efficacy.
In vaccine trials in which a small number of specific antigens are being used for immunization, it is relatively easy to measure immune responses to the specific antigens using enzyme linked immunosorbent spot (ELISpot) assays.Citation159–161 This is more complicated when one is using a vaccine in which the various antigens are not defined. We have not looked for humoral and/or cellular responses to specific tumor-associated antigens in patients treated with our personal DC-ATA vaccines, because to date we have not attempted to identify all of the possible neoantigens or shared antigens expressed on each patient’s self-renewing TC, although this theoretically can be done. The initiation of such responses starts shortly after injection, but efforts to document B cell and/or T cell immune responses typically take place 1–4 weeks after the first injection. There was demonstration of both humoral B-cell and cellular T-cell antigen-specific immune responses induced by personal DC-based COVID-19 vaccines.Citation162 In a 31-subject phase 1 trial, there was an increase in IgG antibodies specific for the receptor binding domain of the SARS-CoV-2 spike protein in 70% of patients on day 28.Citation162 In a 145-subject phase 2 trial, T cell responses to the spike protein were present by day 14 in 94% of patients.Citation162 Such antigen-specific tests are believed to better reflect the immune response than serum or plasma proteomics since the nexus for immune responses reside in lymph nodes rather than in the blood stream, and blood levels may be only transiently elevated, if at all. For these reasons, measurement of serum or plasma cytokines days to weeks following vaccinations may not reliably capture the immune response.
The most convincing serologic data for desirable immune responses in our cancer trials came from the melanoma randomized trial in which there were obvious changes in clusters of cytokines between a week-0 baseline, 1 week prior to initiating the vaccines, and week-4, 1 week after the third of three weekly vaccinations. There were decreases in clusters of certain immune inhibiting cytokines and angiogenesis factors.Citation124 A more sophisticated principal component analysis confirmed statistically significant clustering of cytotoxic CD4 and CD17 cytokines in the DC-ATA arm, but not in the ITC arm.Citation163 In this analysis, there was also some correlation between an enhanced immune response and survival.Citation163 In the randomized ovarian trial and single-arm GBM trial, blood samples were obtained before and 1 week and 2 weeks after initiating vaccination (1 and 2 weeks after the first injection), and the second time point was 1 week after the second injection). In the ovarian trial, compared to baseline levels there was a statistically significant doubling in the number of interferon-γ ELISpots within 2 weeks of starting vaccination in the DC-ATA arm, but there was no change in the number of interferon-γ ELISpots within 2 weeks of starting vaccination in the MC control arm.Citation164 It is planned to analyze changes in cytokine levels and T cell immune responses in GBM patients treated with DC-ATA in the single arm trial.
Because DC-ATA has been admixed with GM-CSF in all of these cancer trials, it is difficult to distinguish the immune effects induced by DC-ATA as opposed to those induced by GM-CSF. However, the antigen-specific T cell responses documented following the anti-covid DC vaccine are noteworthy because GM-CSF was not given with that product.Citation162 Both mouse experiments and samples from the phase I trial of the anti-covid vaccine suggested that there might be some benefit for the addition of GM-CSF in terms of immunoglobulin responses to the receptor binding domain of the spike protein.Citation162 As noted above, in the melanoma randomized trial, based on cytokine clustering, there were substantial differences in immune responses between the two arms, even though GM-CSF was included with both products, which suggests the differences were related to the vaccine cell products.Citation124,Citation162 In the single-arm melanoma trial, an analysis of changes in levels of serum thymus-and activation-regulated chemokine (TARC/CCL17) suggested a correlation between PFS and increases in TARC levels.Citation165 TARC is secreted by DC,Citation166 but also is induced by GM-CSF.Citation167 In the randomized phase 2 melanoma trial, there were similar large increases in serum levels of TARC in both study arms,Citation124 which suggests that this was an effect of GM-CSF rather than due to secretion of TARC by the large number of DC that were injected in the DC-ATA treatment arm.
*Yes, subcutaneous injections of personal DC-ATA vaccines are associated with clinical benefit in some patients
In cancer patients, a suggestion of clinical efficacy may be inferred from ORR, based on regression of tumors that were present and measurable prior to starting treatment, and by prolonged PFS or OS compared to historical or contemporary controls.Citation168 Ultimately, safety and clinical efficacy must be established by large randomized well-controlled phase 3 clinical trials, typically with OS as the primary endpoint. However, it is hard to justify the costs of such trials and the participation of large numbers of human subjects in the absence of some suggestion of efficacy in earlier phase trials. Of the three metrics, ORR, PFS, and OS, OS is considered the gold standard of clinical endpoints because the date of death is definitive and easily determined.Citation168,Citation169 However, inference regarding causality and OS can be confounded by subsequent treatment; therefore, other therapies must be tracked until the time of death or censoring. ORR and PFS are not always surrogate markers for improved OS. Documentation of tumor regression is useful, but is only applicable to patients with measurable disease, and anything other than a complete response is somewhat arbitrary, which is why standard definitions of response and tumor progression continue to evolve.Citation170,Citation171 Because of changes in definitions, it is often inappropriate to compare response rates observed in recent trials to those reported in earlier trials. In modern trials, by convention PFS, also referred to as event-free survival or time to progression (TTP), is defined by whichever occurs first: the date of disease progression or the date of death, regardless of cause. In phase 2 trials, PFS has an advantage over ORR because it includes all patients regardless of disease measurability, and an advantage over OS because it is not confounded by subsequent therapies.Citation168,Citation172 However, PFS is the least definitive of these endpoints because it includes arbitrary definitions of disease progression, confirming that cancer progression actually has occurred, is confounded by deaths from non-cancer causes, and is affected by the frequency of observations during which progression might be recognized.Citation173
For several reasons, our DC-ATA trials have emphasized OS more than PFS or ORR as a clinical trial endpoint. First, most patients have not had measurable disease at the time of treatment because of previous surgical resection of tumor in order to make DC-ATA, and the challenges in conducting trials in patients with readily detectable progressive metastatic disease that has been refractory to other therapies. Second, PFS may be misleading because immune therapies may cause tumor inflammation that can be confused with disease progression. For instance, in the large, randomized trial that resulted in regulatory approval of ipilimumab, there were no significant differences in response rates or PFS compared to the control arm, but the OS was superior in the immunotherapy arms.Citation92 Third, it is generally believed that therapeutic cancer vaccines are most likely to provide benefit if administered in the setting of no measurable tumor or minimal tumor burden.Citation174,Citation175
There was no tumor response rate data obtained in the recent GBM and ovary trials because DC-ATA was given as an adjunctive therapy after primary therapy was completed; therefore, nearly all patients lacked measurable disease when vaccine therapy was initiated. Response rate was not an objective of the HCC phase I trial because the focus was on short-term follow up for safety.Citation122 Only eight patients in the DC-ATA arm of the randomized melanoma trial had measurable disease.Citation120,Citation124 In earlier trials, estimations of ORR were only possible in the subsets of patients who had measurable disease at the time vaccine treatment was initiated. Therefore, the only formal response data per RECIST criteria are 0/7 in metastatic RCCCitation123 and 0/15 in metastatic melanoma.Citation119 However, there have been delayed durable complete responses in patients who had measurable disease at baseline, including two delayed complete regressions in RCC and one in melanoma. A 49-year-old male with recurrent RCC had measurable pulmonary lymphadenopathy that was stable during 6 months of vaccination, but gradually regressed completely.Citation123 He subsequently received gamma knife treatment to a solitary small brain lesion that was presumed to be a metastasis, but was cancer free 5 years from the start of vaccine therapy. A 74-year-old male had a 1-cm lung metastasis from RCC when he started treatment.Citation123 The lesion was regressing but technically stable during the 6 months of vaccine therapy, but totally resolved 1 month later. This patient also was subsequently treated for a single small brain lesion that was presumed to be a metastasis, and he also was cancer-free 5 years after starting the vaccine. In the randomized melanoma trial, at diagnosis, a 59-year-old woman had metastases to the cervical spine and, subsequently, had repeated recurrences of widespread metastatic disease including axilla, bowel, brain, gallbladder, and soft tissue metastases, despite multiple surgeries, radiation, chemotherapy, tyrosine kinase therapy, and IL-2 based immunotherapy.Citation176 She had five new sites of progressing soft-tissue metastases at the time she started vaccine therapy. These five lesions stabilized during the 6 months of DC-ATA injections; 6 months later, she was declared to have a partial response per radiologic scans, and 3 months later, all lesions had totally resolved. She received no other anti-cancer therapy and was still disease-free 5 years after starting the vaccine.Citation124,Citation177 Based on these three patients, the delayed durable complete response rate is a respectable 3/30 (10%) for patients treated with DC-ATA who had measurable disease at the time vaccine therapy was initiated.
In terms of PFS, in the randomized melanoma trial median PFS was somewhat longer in the DC-ATA arm (5.4 vs 3.7 months), but the difference was not statistically significant, while the difference in OS was signficant.Citation124 The converse was seen in the GBM trial in which the median PFS of 10.4 months was 50% greater than the medians reported from each of six other GBM trials, all of which had median PFS of less than eight months.Citation178 However, this prolonged PFS, which persisted only while DC-ATA was being administered, did not translate into a longer OS compared to the historical data.Citation124 It was hypothesized that OS may have been longer had DC-ATA injections been continued for a longer duration.
OS is generally considered the most reliable endpoint for establishing efficacy in cancer clinical trials, but OS is difficult to interpret unless there is a control arm, and potentially can be affected by effective salvage therapies. In the 54-patient single-arm melanoma trial, DC-ATA treatment was associated with a median OS of 5 y compared to about 20 months in a historical control group of 74 patients who were treated with ITC vaccine.Citation78,Citation119 A large and statistically robust difference persisted even when the historical control group was limited to patients who received at least the first three weekly vaccine injections.Citation119 The only randomized trial data available at this time is from the 42-patient melanoma trial.Citation120,Citation124 Even though early closure of this trial meant that it was severely underpowered to detect a difference in OS, survival results were much better in the DC-ATA arm with median OS of 43.4 vs. 20.5 months, 3-y OS of 61% vs 25% (p = .018), and a hazard ratio of 0.304, or a 70% reduction in the risk of death (p = .0053) with the DC-ATA vaccine.Citation124 In addition to this data, there have been encouraging survival outcomes in several patients with liver metastases who were rendered disease free by surgery with or without radiofrequency ablation, and then treated with a personal DC vaccine derived from the resected tumor.Citation179 One such patient, who had also previously had lung and small bowel metastases prior to the liver metastases, has now been disease free for 20 y from DC-ATA initiation.
Definitive proof of therapeutic efficacy requires a large, randomized, and preferably double-blinded, phase 3 trial. Such a trial was approved for patients with metastatic melanoma with a DC-ATA treatment arm and an MC control arm.Citation180,Citation181 However, because of a financial shortfall, the sponsoring company discontinued this trial after only six patients had started treatment. A randomized, double-blind phase 3 trial has been granted regulatory approval for newly diagnosed GBM patients with OS as the primary endpoint (ClinicalTrials.gov NCT05100641).
Other issues
There is a strong rationale for combining monoclonal antibody immune checkpoint inhibitors with therapeutic anti-cancer vaccines.Citation182–185 Agents such as nivolumab, pembrolizumab, and ipilimumab are most in effective in patients who have a high tumor mutation load, and therefore have large numbers of neoantigens and existing anti-tumor immune responses that have been suppressed via PD-1/PDL-1 and/or CTLA-4 receptors.Citation186,Citation187 Even in an immune-responsive cancer such as melanoma, a limitation of immune checkpoint inhibitors is that more than one-third of patients have tumors that are not associated with tumor infiltrating lymphocytes.Citation183 The proportion of non-infiltrated tumors is typically even higher in adenocarcinomas. Histologic assessment has defined three general immune signatures that are both prognostic for survival and predictive of a response to PD-1/PDL-1. These include (1) T cell infiltration with elevated PDL-1 expression, (2) T cells limited to the tumor periphery, but not infiltrating the tumor, and (3) neither T cell infiltration nor elevated PDL-1 expression.Citation188–191 The last signature is considered an immunologically “cold tumor,” with no potential for response to anti-PD-1/PDL-1 therapy.Citation192 For this reason, there is great interest in the potential for vaccines that might convert an immunologically “cold” tumor into a “hot” tumor,Citation193,Citation194 which might then benefit from checkpoint immunotherapy,Citation183,Citation184 or other immune stimulating therapies, such as IL-2.Citation195,Citation196 Furthermore, studies in B16 melanoma mouse tumor models showed that concomitant vaccine therapy enhances the clinical effects of both anti-CTLA-4 and anti-PD-1 therapies.Citation144,Citation145 In the randomized phase 2 melanoma trial, there was an interesting relationship between baseline serum PD-1 levels and changes in those levels after therapy that were associated with improved survival, but only in the DC vaccine arm.Citation197 For this reason, an exploratory safety trial has been initiated for the combination of personal DC-vaccine therapy and anti-PD-1 therapy in patients with metastatic melanoma [ClinicalTrials.gov NCT03743298]
Another issue is the dose of DC-ATA administered. shows the average number of cells injected during each of the first three weekly injections in each trial. In all trials to date, the dose of DC-ATA has been dictated by the number of DC generated from MC obtained by leukapheresis rather than a predetermined dose. Typically, the total product was aliquoted into 10 separate vials with up to eight being injected during the course of vaccine therapy. In the first four trials, cells were counted at the time of thawing just prior to injection;Citation119,Citation122–124 in the two recent trials a cell count was performed on a single representative thawed sample of each personal DC-ATA batch lot.Citation125,Citation128 In the DC-ATA trials in which DC were incubated with intact ITC,Citation119,Citation122–124 the final treatment product included unphagocytosed ITC, and as a consequence, up to 20% of the cells were residual ITC.Citation83 In contrast, in the recent randomized ovary trial and single-arm GBM trial, products were 97%–100% DC and 0%–3% lymphocytes.Citation125,Citation128
Table 3. Numbers of DC-ATA products injected by clinical trial. Numbers reported are the average numbers of cells injected during each of the first three injections.
There was no correlation between dose and clinical outcome in any of these trials, including delayed tumor response in the first three trials,Citation119,Citation123,Citation124 and PFS and OS in the five trials with such data.Citation119,Citation123–125,Citation128 It is unclear what a minimum effective dose may be, but in the GBM trial, the five patients who received less than 1 million cells per dose have survived 10.4, 13.3, 22.9, 36.3+, and 37.5+ months,Citation198 which suggests that even these seemingly low doses are still above a minimum threshold that might define a clinically potent dose. In the covid vaccine phase 2 trial, the average number of DC injected was only 2.1 million with a range of 0.2 to 16.5 million, and 43 received a single injection that contained fewer than 1 million DC, but this was sufficient to rapidly induce antigen-specific T-cell responses.Citation163 One study by others suggested that injection of no more than five million DC is associated with better trafficking to regional lymph nodes.Citation63 Based on these observations, for the GBM phase 3 randomized trial, it is planned to treat with fixed dose of two million cells per injection based on the number of cells at the time of freezing (estimated one million cells at the time of thawing), rather than dividing up the batch of DC-ATA cells into a fixed number of aliquots [ClinicalTrials.gov NCT05100641].
Another unsettled issue is duration of vaccine therapy. In all trials to date except for the phase 1 HCC trial,Citation122 the treatment schedule has been eight vaccines over 6 months, with injections at weeks 1, 2, 3, 8, 12, 16, 20, and 24. In a small number of patients, this has resulted in long-term disease control,Citation119,Citation123–125,Citation178 but for most, it does not. It is possible that survival would be increased if patients continued to receive DC-ATA injections until disease progression was established. It is not surprising that there is a strong correlation between OS and whether patients received all eight injections, since the only reason patients discontinued therapy was because of progressive disease.Citation125 But for all patients in the single-arm GBM trial, 68% of patients completed all eight doses and OS and PFS were increased relative to historical controls until about 2 months after vaccines were discontinued.Citation178 Since the vaccines have been so well-tolerated, and since most patients have not had progressive disease after 6 months of injections, it should be feasible to treat a high proportion of patients for a longer period of time. Because of the availability of the immortal TC lines and the ability to do repeated leukaphereses, treatment products can be generated repeatedly. This concept has been incorporated into the randomized phase 3 trial in GBM in which in the absence of confirmed progressive disease, patients may receive vaccine treatment every 4 weeks for up to 17–18 months [ClinicalTrials.gov NCT05100641]. It is anticipated that for patients who have not progressed after eight injections, in order to provide the additional vaccine doses, many likely will require one or more additional leukapheresis procedures, in addition to growing additional TC from the original TC culture.
Conclusions
Despite complex logistics and the need for technical cell culture expertise, personal patient-specific DC-ATA vaccines can be reproducibly and reliably manufactured and made available for therapeutic use in less than 8 weeks from the collection of tumor tissue. Not surprisingly, injections of personal DC-ATA vaccines are associated with only mild-to-moderate self-limited AEs that likely are due to the GM-CSF adjuvant. These AEs are less disruptive than the AE that cancer patients experience because of their disease and the effects of other therapies. Although difficult to measure because of interpatient heterogeneity and variation in personal treatment products, there is evidence that DC-ATA induces a desirable immune response that is detectable within 1–4 weeks of starting treatment. Clinical efficacy remains to be established in a randomized phase 3 trial with a primary endpoint of OS, but delayed durable complete objective responses, and promising PFS and OS have been observed in the trials that have already been completed.
Authors’ contributions
Dr. Dillman provided medical oversight for all of the DC-ATA trials referenced in this review, composed the initial draft of the manuscript, and revised the manuscript based on input from coauthors and reviewers. Dr. Nistor and Dr. Keirstead reviewed the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We would like to thank the patients who have provided tumor tissue and participated in these trials, and to acknowledge the contributions of Shankar Nayak, Linda Beutel, Andrew Cornforth, Patric Schiltz, Senthamil Selvan, Denysha Carbonell and Kelly Austin in vaccine manufacturing during 1990-2011, Andrew Cornforth, Krystal Godding, Rockelle Robles, Chris Langford, Joseph Requejo, and Remington Wong in vaccine manufacturing during 2017-2022, and the logistical and clinical research assistance of Carole Depriest, Robin Ellis, and Cristina de Leon during 1990-2011, and Jim Langford, Katrina Lopez, Candace Hsieh, and Adrienne Wang during 2017-2022.
Disclosure statement
The authors are all employees of AIVITA Biomedical and have stock equity in the company. R.O. Dillman, M.D., is Chief Medical Officer, G.I. Nistor, M.D., is Chief Scientific Officer, and H.S. Keirstead is Chairman and Chief Executive Officer.
Additional information
Funding
References
- Dillman RO. Cancer immunotherapy. Cancer Biother Radiopharm. 2011;26(1):1–17. doi:10.1089/cbr.2010.0902.
- Lohmueller J, Finn OJ. Current modalities in cancer immunotherapy: immunomodulatory antibodies, CARs and vaccines. Pharmacol Ther. 2017;178:31–47. doi:10.1016/j.pharmthera.2017.03.008.
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. doi:10.1038/s41577-020-0306-5.
- Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. doi:10.1038/nrclinonc.2017.166.
- El-Sayes N, Vito A, Mossman K. Tumor heterogeneity: a great barrier in the age of cancer immunotherapy. Cancers (Basel). 2021;13(4):806. doi:10.3390/cancers13040806.
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22. doi:10.1056/NEJMoa1001294.
- Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, Matory YL, Skibber M, Shiloni E, Vetto JT, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313(23):1485–92. doi:10.1056/NEJM198512053132327.
- Mesiano G, Todorovic M, Gammaitoni L, Leuci V, Diego LG, Carnevale-Schianca F, Fagioli F, Piacibello W, Aglietta M, Sangiolo D. Cytokine-induced killer (CIK) cells as feasible and effective adoptive immunotherapy for the treatment of solid tumors. Expert Opin Biol Ther. 2012;12(6):673–84. doi:10.1517/14712598.2012.675323.
- Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Toalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. 1988;319(25):1676–80. doi:10.1056/NEJM198812223192527.
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PR, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2009;17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116.
- June C, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. doi:10.1056/NEJMra1706169.
- Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. doi:10.1038/s41408-021-00459-7.
- Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, Gavarret J, Bianchi FC, Pumphrey NJ, Ladell K, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med. 2012;18(6):980–7. doi:10.1038/nm.2764.
- Sun Y, Li F, Sonnemann H, Jackson KR, Talukder AH, Katailiha AS, Lizee G. Evolution of CD8+ T cell receptor (TCR) engineered therapies for the treatment of cancer. Cells. 2021;10(9):2379. doi:10.3390/cells10092379.
- Hoover HC, Jr, Surdyke M, Dangel RB, Peters LC, Hanna MG. Delayed cutaneous hypersensitivity to autologous tumor cells in colorectal cancer patients immunized with an autologous tumor cell: Bacillus Calmette-Guérin vaccine. Cancer Res. 1984;44:1671–6.
- Berd D, McGuire HC, McCue P, Mastrangelo MJ. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: clinical and immunologic results in 64 patients. J Clin Oncol. 1990;8(11):1858–67. doi:10.1200/JCO.1990.8.11.1858.
- Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, Heth JA, Salacz M, Taylor S, D’Andre SD, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16(1):142. doi:10.1186/s12967-018-1507-6.
- Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, Nowosielski M, Iglseder S, Freyschlag CF, Oberndorfer S, et al. Audencel immunotherapy based on dendritic cells has no effect on overall and progression-free survival in newly diagnosed glioblastoma: a phase II randomized trial. Cancers. 2018;10(10):372. doi:10.3390/cancers10100372.
- Tesatori A, Richards J, Whitman E, Mann B, Lutzky J, Camacho L, Pariani G, Tosti G, Kirkwood JM, Hoos A, et al. Phase III comparison of Vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: the C-100-21 study group. J Clin Oncol. 2008;26(6):955–62. doi:10.1200/JCO.2007.11.9941.
- Figlin RA, Tannir NM, Uzzo RG, Tykodi SS, Chen DYT, Master V, Kappor A, Vaena D, Lowrance W, Bratslavsky J, et al. Results of the ADAPT phase 3 study of rocalpuldencel-T in combination with sunitinib as first-line therapy in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2020;26(10):2327–36. doi:10.1158/1078-0432.CCR-19-2427.
- Antonarelli G, Corti C, Tarantino P, Ascione L, Cortes J, Romero P, Mittendorf EA, Disis ML, Curigliano G. Therapeutic cancer vaccines revamping: technology advancements and pitfalls. Ann Oncol. 2021;32:1537–51.
- Bansal N, Banerjee D. Tumor initiating cells. Curr Pharm Biotechnol. 2009;10(2):192–6. doi:10.2174/138920109787315015.
- Qureshi-Baig K, Ullmann P, Haan S, Letellier E. Tumor-initiating cells: a mmunel review of isolation approaches and new challenges in targeting strategies. Mol Cancer. 2017;16(1):40. doi:10.1186/s12943-017-0602-2.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–6. doi:10.1038/nature14426.
- Türeci Ö, Vormehr M, Diken M, Kreiter S, Huber C, Sahin U. Targeting the heterogeneity of cancer with individualized neoepitope vaccines. Clin Cancer Res. 2016;22(8):1885–96. doi:10.1158/1078-0432.CCR-15-1509.
- Balan S, Finnigan J, Bhardwaj N. Dendritic cell strategies for eliciting mutation-derived tumor antigen responses in patients. Cancer J. 2017;23(2):131–7. doi:10.1097/PPO.0000000000000251.
- Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359(6382):1355–60. doi:10.1126/science.aar7112.
- Palmer CD, Rappaport AR, Davis MJ, Hart MG, Scallan CD, Hong SJ, Gitlin L, Kraemer LD, Kounlavouth S, Yang A, et al. Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Nat Med. 2022;28(8):1619–29. doi:10.1038/s41591-022-01937-6.
- West NP, Dattani M, McShane P, Hutchins G, Grabsch J, Mueller W, Treanor D, Quirke P, Grabsch H. The proportion of tumour cells is an independent predictor for survival in colorectal cancer patients. Br J Cancer. 2010;102(10):1519–23. doi:10.1038/sj.bjc.6605674.
- Smits AJJ, Kummer JA, de Bruin PC, Bol M, van den Tweel JG, Seldenrijk KA, Willems SM, Offerhaus GJA, de Weger RA, van Diest PJ, et al. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod Pathol. 2014;27(2):168–74. doi:10.1038/modpathol.2013.134.
- Wu J, Liang C, Chen M, Su W. Association between tumor-stroma ratio and prognosis in solid tumor patients: a systematic review and meta-analysis. Oncotarget. 2016;7(42):68954–65. doi:10.18632/oncotarget.12135.
- Xu M, Zhang T, Xia R, Wei Y, Wei X. Targeting the tumor stroma for cancer therapy. Mol Cancer. 2022;21(1):208. doi:10.1186/s12943-022-01670-1.
- Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20(4):840. doi:10.3390/ijms20040840.
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. doi:10.1038/35102167.
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355(12):1253–61. doi:10.1056/NEJMra061808.
- Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12(2):133–43. doi:10.1038/nrc3184.
- Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, Laino L, De Francesco F, Papaccio G. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. Faseb J. 2013;27(1):13–24. doi:10.1096/fj.12-218222.
- Abbaszadegan MR, Bagheri V, Razavi MS, Momtazi AA, Sahebkar A, Gholamin M. Isolation, identification, and characterization of cancer stem cells: a review. J Cell Physiol. 2017;232(8):2008–18. doi:10.1002/jcp.25759.
- Oldham RK, Dillman RO, Yannelli JR, Barth NM, Maleckar JR, Sferruzza A, Cohen RJ, Minor DR, Spitler L, Birch R, et al. Continuous infusion interleukin-2 and tumor-derived activated cells as treatment of advanced solid tumors: a national biotherapy study group trial. Mol Biother. 1991;68(1):68–73. doi:10.1002/1097-0142(19910701)68:1<1:AID-CNCR2820680102>3.0.CO;2-K.
- Dillman RO, Oldham RK, Barth NM, Cohen RJ, Minor DR, Birch R, Maleckar JR, Yannelli JR, Sferruzza A, Arnold J, et al. Continuous interleukin-2 and tumor-infiltrating lymphocytes as treatment of advanced melanoma. A national biotherapy study group trial. Cancer. 1991;68(1):1–8. doi:10.1002/1097-0142(19910701)68:1<1:AID-CNCR2820680102>3.0.CO;2-K.
- Dawood S, Austin L, Cristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology. 2014;28:1101–7.
- Zhou L, Lu L, Wicha MS, Chang AE, Xia JC, Ren X, Li Q. Promise of cancer stem cell vaccine. Hum Vaccines Immunother. 2015;11(12):2796–9. doi:10.1080/21645515.2015.1083661.
- Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. 2016;11(1):47–76. doi:10.1146/annurev-pathol-012615-044438.
- Codd AS, Kanaseki T, Torigo T, Zsuzsanna T. Cancer stem cells as targets for immunotherapy. Immunology. 2017;153(3):304–14. doi:10.1111/imm.12866.
- Atashzar MR, Baharlou R, Karami J, Abdollahi H, Rezaei R, Pourramezan F, Zoljalali Moghaddam SH. Cancer stem cells: a review from origin to therapeutic implications. J Cell Physiol. 2020;235(2):790–803. doi:10.1002/jcp.29044.
- Maccalli C, Rasul KI, Elawad M, Ferrone S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol. 2018;53:189–200. doi:10.1016/j.semcancer.2018.09.006.
- Chen P, Hsu WH, Han J, Xia Y, DePinho RA. Cancer stemness meets immunity: from mechanism to therapy. Cell Rep. 2021;34(1):108597. doi:10.1016/j.celrep.2020.108597.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi:10.1126/science.aaa4971.
- Ward JP, Gubin MM, Schreiber RD. The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer. Adv Immunol. 2016;130:25–74.
- Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, Li M, Ginestier C, Wicha MS, Moyer JS, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72(7):1853–64. doi:10.1158/0008-5472.CAN-11-1400.
- Lu L, Tao H, Chang AE, Hu Y, Shu G, Chen Q, Egenti M, Owen J, Moyer JS, Prince ME. Cancer stem cell vaccine inhibits metastases of primary tumors and induces humoral immune responses against cancer stem cells. Oncoimmunology. 2015;4(3):e990767. doi:10.4161/2162402X.2014.990767.
- Dashti A, Ebrahimi M, Hadjati J, Memarnejadian A, Moazzeni SM. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 2016;374(1):175–85. doi:10.1016/j.canlet.2016.01.021.
- Hirohashi Y, Torigoe T, Tsukahara T, Kanaseki T, Kochin V, Sato N. Immune responses to human cancer stem-like cells/cancer-initiating cells. Cancer Sci. 2016;107(1):12–17. doi:10.1111/cas.12830.
- So JY, Suh N. Targeting cancer stem cells in solid tumors by vitamin D. J Steroid Biochem Mol Biol. 2015;148:79–85. doi:10.1016/j.jsbmb.2014.10.007.
- Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T. Cancer stem cell hierarchy in glioblastoma multiforme. Front Surg. 2016;3:21. doi:10.3389/fsurg.2016.00021.
- Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–20. doi:10.1158/0008-5472.CAN-08-0364.
- Parmiani G. Melanoma cancer stem cells: markers and functions. Cancers (Basel). 2016;8(3):34. doi:10.3390/cancers8030034.
- Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59:56–8.
- De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, Strijk SP, Ruers TJ, Boerman OC, Oyen WJ, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63(1):12–17.
- Nair S, McLaughlin C, Weizer A, Su Z, Boczkowski D, Dannull J, Vieweg J, Gilboa E. Injection of immature dendritic cells into adjuvant-treated skin obviates the need for ex vivo maturation. J Immunol. 2003;171(11):6275–82. doi:10.4049/jimmunol.171.11.6275.
- Ridolfi R, Riccobon A, Galassi R, Giorgetti G, Petrini M, Fiammenghi L, Stefanelli M, Ridolfi L, Moretti A, Migliori G, et al. Evaluation of in vivo labelled dendritic cell migration in cancer patients. J Transl Med. 2004;2(1):27. doi:10.1186/1479-5876-2-27.
- Martin-Fontecha A, Lanzavecchia A, Sallusto F. Dendritic cell migration to peripheral lymph nodes. Handb Exp Pharmacol. 2009;188:31–49.
- Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JF, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009;15(7):2531–40. doi:10.1158/1078-0432.CCR-08-2729.
- Aarntzen EH, Srinivas M, Bonetto F, Cruz LJ, Verdijk P, Schreibelt G, van de Rakt M, Lesterhuis WJ, van Riel M, Punt CJ, et al. Targeting of 111In-labeled dendritic cell human vaccines improved by reducing number of cells. Clin Cancer Res. 2013;19(6): 1525-1523. doi:10.1158/1078-0432.CCR-12-1879.
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12(8):557–69. doi:10.1038/nri3254. Campana S, De Pasquale C, Carrega P, Ferlazzo G, Bonaccorsi I. Cross-dressing: an alternative mechanism for antigen presentation. Immunol Letters 2015;168:349-354.
- Van Endert P. Intracellular recycling and cross-presentation by MHC class I molecules. Immuno Rev. 2016;272(1):80–96. doi:10.1111/imr.12424.
- Colbert JD, Cruz F, Rock KL. Cross-presentation of exogenous antigens on MHCI molecules. Curr Opin Immunol. 2020;64:1–8. doi:10.1016/j.coi.2019.12.005.
- MacNabb BW, Chen X, Tumuluru S, Godfrey J, Kasal DN, Yu J, Jongsma ML, Spaapen R, Kline DE, Kline J. Dendritic cells can prime anti-tumor CD8+ T cell responses through major histocompatibility complex cross-dressing. Immunity. 2022;55:982–97. doi:10.1016/j.immuni.2022.04.016.
- Schlom J, Hodge JW, Palena C, Tsang KY, Jochems C, Greiner JW, Farsaci B, Madan RA, Heery CR, Gulley JL. Therapeutic cancer vaccines. Adv Cancer Res. 2014;121:67–124.
- Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015;125(9):3401–12. doi:10.1172/JCI80009.
- Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immune-therapeutics: platforms and current progress. J Hematol Oncol. 2022;15(1):28. doi:10.1186/s13045-022-01247-x.
- Dillman RO, Hsieh C, Nistor GI. A clinical odyssey: cancer stem cells as the antigen source for autologous cancer vaccines. Adv Stem Cell Res. 2019;1:1–13.
- Dillman RO, Nayak SK, Beutel L. Establishing in vitro cultures of autologous tumor cells for use in active specific immunotherapy. J Immunother Emphasis Tumor Immunol. 1993;14(1):65–9. doi:10.1097/00002371-199307000-00009.
- Dillman RO, Nayak SK, Brown JV, Mahdavi K, Beutel LD. The feasibility of using short-term cultures of ovarian cancer cells for use as autologous tumor cell vaccines as adjuvant treatment of advanced ovarian cancer. Cancer Biother Radiopharm. 1999;14(6):443–9. doi:10.1089/cbr.1999.14.443.
- Dillman RO, Beutel LD, Cornforth AN, Nayak SK. Short-term tumor cell lines from renal cell carcinoma for use as autologous tumor cell vaccines in the treatment of kidney cancer. Cancer Biother Radiopharm. 2000;15(2):161–8. doi:10.1089/cbr.2000.15.161.
- Dillman RO, Barth NM, VanderMolen LA, Mahdavi K, Beutel LD, de Leon C, DePriest C, Nayak S. Autologous tumor cell line–derived vaccine for patient-specific treatment of advanced renal cell carcinoma. Cancer Biother Radiopharm. 2004;19(5):570–80. doi:10.1089/1084978042484786.
- Dillman RO, Barth NM, Selvan SR, Beutel LD, Depriest C, de Leon C, Peterson C, Nayak S. Phase I/II trial of autologous tumor cell line–derived vaccines for recurrent or metastatic sarcomas. Cancer Biother Radiopharm. 2004;19(5):581–8. doi:10.1089/1084978042484812.
- Dillman RO, DePriest C, DeLeon C, Barth NM, Schwartzberg LS, Beutel LD, Schiltz PM, Nayak SK. Patient-specific vaccines derived from autologous tumor cell lines as active specific immunotherapy: results of exploratory phase I/II trials in patients with metastatic melanoma. Cancer Biother Radiopharm. 2007;22(3):309–21. doi:10.1089/cbr.2007.345.
- Haspel MV, McCabe RP, Pomato N, Janesch NJ, Knowlton JV, Peters LC, Hoover HC, Jr, Hanna MC, Jr. Generation of tumor cell-reactive human monoclonal antibodies using peripheral blood lymphocytes from actively immunized colorectal carcinoma patients. Cancer Res. 1985;45:1236–43.
- Hoover HC, Jr, Surdyke MG, Dangel RB, Peters LC, Hanna MG, Jr. Prospectively randomized trial of adjuvant active-specific immunotherapy for human colorectal cancer. Cancer. 1985;55:1236–43.
- Nayak SK, Schiltz PM, Dillman RO. Modulation of renal carcinoma cells in vitro: comparison after transduction with retroviral vector containing a human γ-interferon gene versus incubation with soluble γ-interferon. J Interferon Cytokine Res. 1999;19(1):49–58. doi:10.1089/107999099314414.
- González FE, Gleisner A, Falcón-Beas F, Osorio F, López MN, Salazar-Onfray F. Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum Vaccines Immunother. 2014;10(11):3261–9. doi:10.4161/21645515.2014.982996.
- Dillman RO, Cornforth AN, McClay EF, Depriest C. Patient-specific dendritic cell vaccines with autologous tumor antigens in 72 patients with metastatic melanoma. Melanoma Manag. 2019 31;6(2):MMT20. doi:10.2217/mmt-2018-0010.
- Geskin LJ, Damiano JJ, Patrone CC, Butterfield LH, Kirkwood JM, Falo LD. Three antigen-loading methods in dendritic cell vaccines for metastatic melanoma. Melanoma Res. 2018;28(3):211–21. doi:10.1097/CMR.0000000000000441.
- Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wölfel C, Huber C, Wölfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A. 2005;102(44):16013–18. doi:10.1073/pnas.0500090102.
- Desrichard A, Snyder A, Chan TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res. 2016;22(4):807–12. doi:10.1158/1078-0432.CCR-14-3175.
- Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. 2017;18(3):255–62. doi:10.1038/ni.3682.
- Bobisse S, Foukas PG, Coukos G, Harari A. Neoantigen-based cancer immunotherapy. Ann Transl Med. 2016;4(14):262. doi:10.21037/atm.2016.06.17.
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nature Med. 2013;19(6):747–52. doi:10.1038/nm.3161.
- Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014;20(13):3401–10. doi:10.1158/1078-0432.CCR-14-0433.
- Ma W, Pham B, Li T. Cancer neoantigens as potential targets for immunotherapy. Clin Exp Metastasis. 2022;39(1):51–60. doi:10.1007/s10585-021-10091-1.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi:10.1056/NEJMoa1003466.
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–61. doi:10.1016/j.ccell.2015.03.001.
- Schadendorf D, Hodi S, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–94. doi:10.1200/JCO.2014.56.2736.
- Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–30. doi:10.1200/JCO.2013.53.0105.
- Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Lao CD, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised controlled, open-label phase 3 trial. Lancet Oncol. 2015;16(4):375–84. doi:10.1016/S1470-2045(15)70076-8.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–32. doi:10.1056/NEJMoa1503093.
- Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-0002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16(8):908–18. doi:10.1016/S1470-2045(15)00083-2.
- Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16(1):223–49. doi:10.1146/annurev-pathol-042020-042741.
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–8. doi:10.1126/science.aaa3828.
- Vormehr M, Diken M, Boegel S, Kreiter S, Türeci Ö, Sahin U. Mutanome directed cancer immunotherapy. Curr Opin Immunol. 2016;39:14–22. doi:10.1016/j.coi.2015.12.001.
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77. doi:10.1038/nrc3258.
- Mellman I. Dendritic cells: master regulators of the immune response. Cancer Immunol Res. 2013;1(3):145–9. doi:10.1158/2326-6066.CIR-13-0102.
- Bonaccorsi I, Campana S, Morandi B, Ferlazzo G. Acquisition and presentation of tumor antigens by dendritic cells. Crit Rev Immunol. 2015;35(5):349–64. doi:10.1615/CritRevImmunol.v35.i5.10.
- Dillman RO, Cornforth AN, Nistor GI. Dendritic cell vaccines for melanoma: past, present, and future. Melanoma Manag. 2016;3(4):273–89. doi:10.2217/mmt-2016-0014.
- Constantino J, Gomes C, Falcão A, Neves BM, Cruz MT. Dendritic cell-based immunotherapy: a basic review and recent advances. Immunol Res. 2017;65(4):798–810. doi:10.1007/s12026-017-8931-1.
- de Rosa F, Ridolfi L, Fiammenghi L, Petrini M, Granato AM, Ancarani V, Pancisi E, Soldati V, Cassan S, Bulgarelli J, et al. Dendritic cell vaccination for metastatic melanoma: a 14-year monoinstitutional experience. Melanoma Res. 2017;27(4):351–7. doi:10.1097/CMR.0000000000000356.
- Linnette GP, Carreno BM. On the twentieth anniversary of dendritic cell vaccines – riding the next wave. Cancer Res. 2022;82(6):966–8. doi:10.1158/0008-5472.CAN-21-4440.
- Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9(1):271–96. doi:10.1146/annurev.iy.09.040191.001415.
- Steinman RM. Decisions about dendritic cells: past, present, and future. Ann Rev Immunol. 2012;30(1):1–22. doi:10.1146/annurev-immunol-100311-102839.
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179(4):1109–18. doi:10.1084/jem.179.4.1109.
- Feuerstein B, Berger TG, Maczek C, Röder C, Schreiner D, Hirsch U, Haendle I, Leisgang W, Glaser A, Kuss O, et al. A method for the production of cryopreserved aliquots of antigen-preloaded, mature dendritic cells ready for clinical use. J Immunol Methods. 2000;245(1–2):15–29. doi:10.1016/S0022-1759(00)00269-6.
- Klein C, Bueler H, Mulligan RC. Comparative analysis of genetically modified dendritic cells and tumor cells as therapeutic cancer vaccines. J Exp Med. 2000;191(10):1699–708. doi:10.1084/jem.191.10.1699.
- Zhang R, Yuan F, Shu Y, Tian Y, Zhou B, Yi L, Zhang X, Ding Z, Xu H, Yang L. Personalized neoantigen-pulsed dendritic cell vaccines show superior immunogenicity to neoantigen-adjuvant vaccines in mouse tumor models. Cancer Immunol Immunother. 2020;69(1):135–45. doi:10.1007/s00262-019-02448-z.
- Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B–cell lymphoma using autologous antigen–pulsed dendritic cells. Nat Med. 1996;2(1):52–8. doi:10.1038/nm0196-52.
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4(3):328–32. doi:10.1038/nm0398-328.
- Dillman RO, Selvan SR, Schiltz PM, Peterson C, Allen K, DePriest C, McClay E, Barth N, Sheehy P, de Leon C, et al. Phase I/II trial of melanoma patient–specific vaccine of proliferating autologous tumor cells, dendritic cells, and GM-CSF: planned interim analysis. Cancer Biother Radiopharm. 2004;19(5):658–65. doi:10.1089/1084978042484759.
- Dillman RO, Selvan SR, Schiltz PM. Patient-specific dendritic-cell vaccines for metastatic melanoma. N Engl J Med. 2006;355(11):1179–81. doi:10.1056/NEJMc061667.
- Dillman RO, Selvan SR, Schiltz PM, McClay EF, Barth NM, DePriest C, de Leon C, Mayorga C, Cornforth AN, Allen K. Phase II trial of dendritic cells loaded with antigens from self-renewing, proliferating autologous tumor cells as patient-specific anti-tumor vaccines in patients with metastatic melanoma. Final Rep Cancer Biother Radiopharm. 2009;24(3):311–9. doi:10.1089/cbr.2008.0599.
- Dillman RO, Cornforth AN, Depriest C, McClay EF, Amatruda TT, de Leon C, Ellis RE, Mayorga C, Carbonell D, Cubellis JM. Tumor stem cell antigens as consolidative active specific immunotherapy: a randomized phase II trial of dendritic cells versus tumor cells in patients with metastatic melanoma. J Immunother (1991). 2012;35(8):641–9. doi:10.1097/CJI.0b013e31826f79c8.
- Lee J, Boczkowski D, Nair S. Programming human dendritic cells with mRNA. Methods Mol Biol. 2013;969:111–25.
- Wang X, Bayer ME, Chen X, Fredrickson C, Cornforth AN, Liang G, Cannon J, He J, Fu Q, Liu J, et al. Phase I trial of active specific immunotherapy with autologous dendritic cells pulsed with autologous irradiated tumor stem cells in hepatitis B-positive patients with hepatocellular carcinoma. J Surg Oncol. 2015;111(7):862–7. doi:10.1002/jso.23897.
- Dillman RO, Depriest C. Dendritic cell vaccines presenting autologous tumor antigens from self-renewing cancer cells in metastatic renal cell cancer. J Exploratory Res Pharmacol. 2018;3(4):93–101. doi:10.14218/JERP.2018.00012.
- Dillman RO, Cornforth AN, Nistor GI, McClay EF, Amatruda TT, Depriest C. Randomized phase II trial of autologous dendritic cell vaccines versus autologous tumor cell vaccines in metastatic melanoma: 5-year follow up and additional analyses. J ImmunoTher Cancer. 2018;6(1):19. doi:10.1186/s40425-018-0330-1.
- Bota DA, Taylor TH, Piccioni DE, Duma CM, LaRocca RV, Kesari S, Carrillo JA, Abedi M, Aiken RD, Hsu FPK, et al. Phase 2 study of AV-GBM-1 (a tumor-initiating cell targeted dendritic cell vaccine) in newly diagnosed glioblastoma patients: safety and efficacy assessment. J Exp Clin Cancer Res. 2022;41(1):344. doi:10.1186/s13046-022-02552-6.
- Eskander RN, Abaid LN, Corr BR, Mason JR, Cappuccini F, Friedman RL, Robles RM, Lopez KL, Nistor GI, Dillman RO Tumor collection and establishment of tumor-initiating cell cultures to serve as the antigen source for AV-OVA-1 dendritic cell vaccines for patients with newly diagnosed advanced ovarian cancer. Poster presented at: the 37th Annual Scientific Meeting of the Society for Immunotherapy of Cancer (SITC); 2022 Nov 8-12; Boston, MA.
- Flanigan RC, Polcari AJ, Shore ND, Price TH, Sims RB, Maher JC, Whitmore JB, Corman JM. An analysis of leukapheresis and central venous catheter use in the randomized, placebo controlled, phase 3 IMPACT trial of sipuleucel-T for metastatic castrate resistant prostate cancer. J Urol. 2013;189(2):521–6. doi:10.1016/j.juro.2012.09.029.
- Mason JR, Abaid LN, Corr BR, Eskander RN, Cappuccini F, Friedman RL, Robles RM, Lopez KL, Nistor GI, Dillman RO Leukapheresis to obtain monocytes to produce dendritic cells for manufacturing AV-OVA-1 personal vaccines in a randomized phase II trial in patients with newly diagnosed advanced ovarian cancer. Poster presented at: the 37th Annual Scientific Meeting of the Society for Immunotherapy of Cancer (SITC); 2022 Nov 8-12; Boston, MA.
- Mitchell MS, Harel W, Kempf RA, Hu E, Kan-Mitchell J, Boswell WD, Dean G, Stevenson L. Active-specific immunotherapy for melanoma. J Clin Oncol. 1990;8(5):856–69. doi:10.1200/JCO.1990.8.5.856.
- Cole IJ, Gruber J. Progress and prospects for human cancer vaccines. J Natl Cancer Inst. 1992;84(1):18–23. doi:10.1093/jnci/84.1.18.
- Bystryn JC. Clinical activity of a polyvalent melanoma antigen vaccine. Recent Results Cancer Res. 1995;139:337–48.
- Morton DL, Foshag LJ, Hoon DS, Nizze JA, Famatiga E, Wanek LA, Chang C, Davtyan DG, Gupta RK, Elashoff R, et al. Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma vaccine. Ann Surg. 1992;216(4):463–82. doi:10.1097/00000658-199210000-00010.
- Weiner LM. Applications of gamma-interferon in cancer therapy. Mol Biother. 1991;3:186–91.
- Fagerberg J. Granulocyte-macrophage colony-stimulating factor as an adjuvant in tumor immunotherapy. Med Oncol. 1996;13:155–60.
- Mitchell MS, Darrah D, Stevenson L. Therapy of melanoma with allogeneic melanoma lysates alone or with interferon-alfa. Cancer Invest. 2002;20(5–6):759–68. doi:10.1081/CNV-120002493.
- Obeid J, Hu Y, Slingluff CL. Vaccines, adjuvants, and dendritic cell activators—current status and future challenges. Semin Oncol. 2015;42(4):549–61. doi:10.1053/j.seminoncol.2015.05.006.
- Temizoz B, Kuroda E, Ishii KJ. Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol. 2016;28(7):329–38. doi:10.1093/intimm/dxw015.
- Dillman RO, Wiemann M, Nayak SK, DeLeon C, Hood K, DePriest C. Interferon-gamma or granulocyte macrophage colony stimulating factor administered as adjuvants with a vaccine of irradiated autologous tumor cells from short-term cell line cultures: a randomized phase II trial of the cancer biotherapy research group. J Immunother (1991). 2003;26(4):367–73. doi:10.1097/00002371-200307000-00009.
- Schrader JW. Peptide regulatory factors and optimization of vaccines. Mol Immunol. 1991;28(3):295–9. doi:10.1016/0161-5890(91)90077-W.
- Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90(8):3539–43. doi:10.1073/pnas.90.8.3539.
- Disis ML, Bernhard H, Shiota FM, Hand SL, Gralow JR, Huseby ES, Gillis S, Cheever MA. Granulocyte-macrophage colony-stimulating factor: an effective adjuvant for protein and peptide-based vaccines. Blood. 1996;88(1):202–10. doi:10.1182/blood.V88.1.202.202.
- Warren TL, Weiner GJ. Uses of granulocyte-macrophage colony-stimulating factor in vaccine development. Curr Opin Hematol. 2000;7(3):168–73. doi:10.1097/00062752-200005000-00007.
- Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor–secreting tumor vaccine for pancreatic cancer: a phase i trial of safety and immune activation. J Clin Oncol. 2001;19(1):145–56. doi:10.1200/JCO.2001.19.1.145.
- van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony- stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–66. doi:10.1084/jem.190.3.355.
- Li B, VanRoey M, Wang C, Chen TT, Korman A, Joos K. Anti–programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor–secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res. 2009;15(5):1623–34. doi:10.1158/1078-0432.CCR-08-1825.
- Slingluff CL, Petroni GR, Yamshchikov GV, Barnd DL, Eastham S, Galavotti H, Patterson JW, Deacon DH, Hibbitts S, Teates D, et al. Clinical and immunologic results of a randomized Phase II trial of vaccination using four melanoma peptides either administered in granulocyte-macrophage colony-stimulating factor in adjuvant or pulsed on dendritic cells. J Clin Oncol. 2003;21(21):4016–26. doi:10.1200/JCO.2003.10.005.
- Van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. 2012;110(15):3383–93. doi:10.1182/blood-2011-11-370130.
- Spitler LE, Grossbard ML, Ernstoff MS, Silver G, Jacobs M, Hayes FA, Soong SJ. Adjuvant therapy of stage III and IV malignant melanoma using granulocytes-macrophage colony-stimulating factor. J Clin Oncol. 2000;18(8):1614–21. doi:10.1200/JCO.2000.18.8.1614.
- Spitler LE, Weber RW, Allen RE, Meyer J, Cruickshank S, Garbe E, Lin H, Soong SJ. Recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF, sargramostim) administered for 3 years as adjuvant therapy of states II(T4), III, and IV melanoma. J Immunother. 2009;32(6):632–7. doi:10.1097/CJI.0b013e3181a7d60d.
- Lawson DH, Lee S, Zhao F, Tarhini AA, Margolin KA, Ernstoff MS, Atkins MB, Cohen GI, Whiteside TL, Butterfield LH, et al. Randomized, placebo-controlled, phase III trial of yeast-derived Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) versus peptide vaccination versus GM-CSF plus peptide vaccination versus placebo in patients with no evidence of disease after complete surgical resection of locally advanced and/or stage IV melanoma: a trial of the eastern cooperative oncology group-American college of radiology imaging network cancer research group (E4697). J Clin Oncol. 2015;33(34):4066–74. doi:10.1200/JCO.2015.62.0500.
- Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8. doi:10.1200/JCO.2014.58.3377.
- Andtbacka RH, Collichio F, Harrington KJ, Middleton M, Dosney G, Öhrling K, Kaufman HL. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J ImmunoTher Cancer. 2019;7(1):145. doi:10.1186/s40425-019-0623-z.
- Kaufman HL, Ruby CE, Hughes T, Slingluff CL. Current status of granulocyte–macrophage colony-stimulating factor in the immunotherapy of melanoma. J ImmunoTher Cancer. 2014;2(1):11. doi:10.1186/2051-1426-2-11.
- Dillman RO. An update on GM-CSF and its potential role in melanoma management. Melanoma Manag. 2020;7(3):MMT49. doi:10.2217/mmt-2020-0011.
- Abaid LN, Corr BR, Mason JR, Eskander RN, Cappuccini F, Friedman RL, Lopez KL, Nistor GI, Dillman RO Treatment-related adverse events in a randomized trial in stage 3 and 4 ovarian cancer testing patient-specific dendritic cell vaccines loaded with tumor antigens from self-renewing autologous ovary cancer cells. Poster presented at: the 2023 Society of Gynecologic Oncology Annual Meeting; 2023 Mar 25-28; Tampa Bay, FL.
- Dillman RO, Cornforth AN, Depriest C. Cancer stem cell antigens from autologous tumor cell lines in patient-specific active immunotherapy for metastatic cancer. In: Hayat M editor. Chapter 26 in stem cells and cancer stem cells. Vol. 9. London, England: Springer; 2013. pp. 272–84.
- Selvan SR, Carbonell DJ, Fowler AW, Beatty AR, Ravindranath MH, Dillman RO. Establishment of stable cell lines for personalized melanoma cell vaccine. Melanoma Res. 2010;20(4):280–92. doi:10.1097/CMR.0b013e3283390696.
- Nayak SK, Dillman RO. Establishment of multiple tumor cell lines from a patient with melanoma: a simple method to control fibroblast growth. Clin Biotechnol. 1991;3:237–42.
- Kirkwood JM, Lee S, Moschos SJ, Albertini MR, Michalak JC, Sander C, Whiteside T, Butterfield LH, Weiner L. Immunogenicity and Antitumor effects of vaccination with peptide vaccine +/− granulocyte-monocyte colony-stimulating factor and/or IFN-α2b in advanced metastatic melanoma: Eastern cooperative oncology group phase II trial E1696. Clin Cancer Res. 2009;15(4):1443–51. doi:10.1158/1078-0432.CCR-08-1231.
- Slota M, Lim JB, Dang Y, Disis ML. Elispot for measuring human immune responses to vaccines. Expert Rev Vaccines. 2011;10(3):299–306. doi:10.1586/erv.10.169.
- Butterfield LH, Zhao F, Lee S, Tarhini AA, Margolin KA, White RL, Atkins MB, Cohen GI, Whiteside TL, Kirkwood JM, et al. Immune correlates of GM-CSF and melanoma peptide vaccination in a randomized trial for the adjuvant therapy of resected high-risk melanoma (E4697). Clin Cancer Res. 2017;23(17):5034–43. doi:10.1158/1078-0432.CCR-16-3016.
- Nistor GI, Dillman RO, Robles RM, Langford JL, Poole AJ, Sofro MAU, Nency YM, Jonny J, Yana ML, Karyana M, et al. A personal COVID-19 dendritic cell vaccine made at point-of-care: feasibility, safety, and antigen-specific cellular immune responses. Hum Vaccines Immunother. 2022;18(6):2100189. doi:10.1080/21645515.2022.2100189.
- Nistor GI, Dillman RO. Cytokine network analysis of immune responses before and after therapeutic vaccine immunotherapies in a randomized trial in patients with metastatic melanoma. J Transl Med. 2020;18(1):176. doi:10.1186/s12967-020-02328-6.
- Dillman RO. Unpublished observation NCT02033616.
- Cornforth AN, Lee GJ, Fowler AW, Carbonell DJ, Dillman RO. Increases in serum TARC/CCL17 levels are associated with progression-free survival in advanced melanoma patients in response to dendritic cell-based immunotherapy. Clin Immunol. 2009;29(5):657–64. doi:10.1007/s10875-009-9299-3.
- Vissers JL, Hartgers FC, Lindhout E, Teunissen MB, Figdor CG, Adema GJ. Quantitative analysis of chemokine expression by dendritic cell subsets in vitro and in vivo. J Leukoc Biol. 2001;69(5):785–93. doi:10.1189/jlb.69.5.785.
- Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MW, Louis C, Fleetwood AJ, Lacey DC, Christensen AD, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest. 2016;126(9):3453–66. doi:10.1172/JCI87828.
- Pazdur R. Endpoints for assessing drug activity in clinical trials. Oncologist. 2008;13(Suppl 2):19–21. doi:10.1634/theoncologist.13-S2-19.
- Driscoll JJ, Rixe O. Overall survival: still the gold standard: why overall survival remains the definite endpoint in cancer clinical trials. Cancer J. 2009;15(5):401–5. doi:10.1097/PPO.0b013e3181bdc2e0.
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, et al. New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, national cancer institute of the United States, national cancer institute of Canada. J Natl Cancer Inst. 2000;92(3):205–16. doi:10.1093/jnci/92.3.205.
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47. doi:10.1016/j.ejca.2008.10.026.
- Dillman RO. Why event-free survival is better than tumor response or other measures of survival as an endpoint in cancer trials. Cancer Biother Radiopharm. 1996;11(2):99–104. doi:10.1089/cbr.1996.11.99.
- Korn RL, Crowley JJ. Overview: progression-free survival as an endpoint in clinical trials with solid tumors. Clin Cancer Res. 2013;19(10):2607–12. doi:10.1158/1078-0432.CCR-12-2934.
- Salazar LG, Disis ML. Cancer vaccines: the role of tumor burden in tipping the scale toward vaccine efficacy. J Clin Oncol. 2005;23(30):7397–8. doi:10.1200/JCO.2005.07.020.
- Kim SI, Cassella CR, Byrne KT. Tumor burden and immunotherapy: impact on immune infiltration and therapeutic outcomes. Front Immunol. 2021;11:629722. doi:10.3389/fimmu.2020.629722.
- Dillman RO, Nanci AA, Williams ST, Kim RB, Hafer RL, Coleman CL, Wang PC, Duma CM, Chen PV, Selvan SR, et al. Durable complete response of refractory, progressing metastatic melanoma after treatment with a patient-specific vaccine. Cancer Biother Radiopharm. 2010;25(5):553–7. doi:10.1089/cbr.2010.0819.
- Dillman RO, Nistor GI, Poole AJ. Genomic, proteomic, and immunologic associations with a durable complete remission of measurable metastatic melanoma induced by a patient-specific dendritic cell vaccine. Hum Vaccines Immunother. 2020;16(4):742–55. doi:10.1080/21645515.2019.1680239.
- Piccioni DE, Duma CM, Kesari S, LaRocca RV, Aiken RD, Taylor TH, Carrillo JA, Abedi M, Nistor GI, Keirstead HS, et al. Progression free survival in a phase 2 trial of personal dendritic cell vaccines in patients with newly diagnosed glioblastoma. J Clin Oncol Res Ther. 2022;7:10149.
- Dillman RO. Long-term progression free and overall survival in two melanoma patients treated with patient-specific therapeutic vaccine eltrapuldencel-T after resection of a solitary liver metastasis. Cancer Biother Radiopharm. 2016;31(3):71–4. doi:10.1089/cbr.2016.2003.
- Dillman RO. Patient-specific therapeutic vaccines for metastatic melanoma. Oncol Iss. 2015 Mar-Apr;30(2):48–57. doi:10.1080/10463356.2015.11884014.
- Javed A, Sato S, Sato T. Autologous melanoma cell vaccine using monocyte-derived dendritic cells (Nbs20/eltrapuldencel-T). Future Oncol. 2016;12(6):751–62. doi:10.2217/fon.16.13.
- Dillman RO. Is vaccine research still relevant for metastatic melanoma? Melanoma Manag. 2014;1(2):91–4. doi:10.2217/mmt.14.18.
- Dillman RO. Is there a role for therapeutic cancer vaccines in the age of checkpoint inhibitors? Hum Vaccines Immunother. 2017;13(3):528–32. doi:10.1080/21645515.2016.1244149.
- Collins JM, Redman JM, Gulley JL. Combining vaccines and immune checkpoint inhibitors to prime, expand, and facilitate tumor immunotherapy. Exp Rev Vaccines. 2018;17(8):697–705. doi:10.1080/14760584.2018.1506332.
- Schlom J, Gulley JL. Vaccines as an integral component of cancer immunotherapy. JAMA. 2018;320(21):2195–6. doi:10.1001/jama.2018.9511.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. doi:10.1038/nrc3239.
- Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184(21):5309–37. doi:10.1016/j.cell.2021.09.020.
- Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, Bruneval P, Trajanoski Z, Fridman WH, Pagès F, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29(6):610–8. doi:10.1200/JCO.2010.30.5425.
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. doi:10.1126/scitranslmed.3003689.
- Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012;72(13):3125–30. doi:10.1158/0008-5472.CAN-11-4094.
- Kluger HM, Zito CR, Barr ML, Baine MK, Chiang VL, Sznol M, Rimm DL, Chen L, Jilaveanu LB. Characterization of PD-L1 expression and associated T-cell Infiltrates in metastatic melanoma samples from variable anatomic sites. Clin Cancer Res. 2015;21(13):3052–60. doi:10.1158/1078-0432.CCR-14-3073.
- Das S, Camphausen K, Shankavaram U. Cancer-specific immune prognostic signature in solid tumors and Its relation to immune checkpoint therapies. Cancers (Basel). 2020;12(9):2476. doi:10.3390/cancers12092476.
- Frederico SC, Hancock JC, Brettschneider EES, Ratnam NM, Gilbert MR, Terabe M. Making a cold tumor hot: the role of vaccines in the treatment of glioblastoma. Front Oncol. 2021;11:672508. doi:10.3389/fonc.2021.672508.
- Ullman NA, Burchard PR, Dunne RF, Linehan DC. Immunologic strategies in pancreatic cancer: making cold tumors hot. J Clin Oncol. 2022;40(24):2789–805. doi:10.1200/JCO.21.02616.
- Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–27. doi:10.1056/NEJMoa1012863.
- Dillman RO, Depriest C, McClure SE. High-dose IL-2 in metastatic melanoma: better survival in patients immunized with antigens from autologous tumor cell lines. Cancer Biother Radiopharm. 2014;29(2):53–7. doi:10.1089/cbr.2013.1565.
- Dillman RO, Nistor GI, McLelland BT, Hsieh C, Poole AJ, Cornforth AN, Keirstead HS. Preliminary observations on soluble programmed death protein-1 (sPD1) as a prognostic and predictive biomarker in patients with metastatic melanoma treated with patient-specific autologous vaccines. Oncotarget. 2019;10(51):5359–71. doi:10.18632/oncotarget.27164.
- Dillman RO. Unpublished observation. NCT03400917