
ABSTRACT
DNA vaccines containing only antigenic components have limited efficacy and may fail to induce effective immune responses. Consequently, adjuvant molecules are often added to enhance immunogenicity. In this study, we generated a tumor vaccine using a plasmid encoding NMM (NY-ESO-1/MAGE-A3/MUC1) target antigens and immune-associated molecules. The products of the vaccine were analyzed in 293 T cells by western blotting, flow cytometry, and meso-scale discovery electrochemiluminescence. To assess the immunogenicity obtained, C57BL/6 mice were immunized using the DNA vaccine. The results revealed that following immunization, this DNA vaccine induced cellular immune responses in C57BL/6 mice, as evaluated by the release of IFN-γ, and we also detected increases in the percentages of nonspecific lymphocytes, as well as those of antigen-specific T cells. Furthermore, immunization with the pNMM vaccine was found to significantly inhibit tumor growth and prolonged the survival of mice with B16-NMM+-tumors. Our data revealed that pNMM DNA vaccines not only confer enhanced immunity against tumors but also provide a potentially novel approach for vaccine design. Moreover, our findings provide a basis for further studies on vaccine pharmacodynamics and pharmacology, and lay a solid foundation for clinical application.
Introduction
Global cancer epidemiological statistics (GLOBOCAN) indicate that the number of new cancer cases per year will increase to 20 million by 2025.Citation1 In this regard, considerable efforts have been expended in areas such as mechanism identification, cancer prevention, and therapy development. Along with surgery, radiotherapy, and chemotherapy, immunotherapy has been identified as the fourth major type of tumor treatment. Immunotherapy can contribute to activating the immune system and prime immune cells to recognize and clear tumor cells.Citation2,Citation3 However, although checkpoint-like monoclonal antibodies and CAR-T are recognized as representative approaches of tumor immunotherapy, the efficacy of these treatments has been questioned in some cases, and their application can be hindered in patients with abnormal baseline immune status.Citation4,Citation5
DNA vaccination has been shown to induce CD4+ (Th1) and CD8+ cytotoxic T lymphocyte (Tc) immune responses,Citation6,Citation7 and recombinant DNA vaccines have been established offer advantages and novel avenues for cancer therapy. In contrast to recombinant bacterial/viral vaccines, DNA vaccines consist exclusively of plasmids. When transfected into cells, these plasmids generate an immune response by expressing the antigen genes they carry. DNA plasmids are safe, readily manufactured, stored, and delivered, and conducive to the rapid evaluation of a range of target antigens.Citation8–10 In addition, DNA vaccines can be readily modified and their immunogenicity can be enhanced by adopting various approaches.Citation11
However, although compared with other treatments, DNA vaccines have potentially superior anti-tumor specificity and immune memory,Citation12 the immunogenicity conferred by vaccines containing only tumor antigens is often limited and generally induces only a weak immune response. Consequently, adjuvant molecules are often used to boost the immunogenicity and therapeutic efficacy of these vaccines. Nevertheless, adjuvants generally provide only limited improvements, and the immunogenicity obtained using DNA vaccines remains poor in large animals and humans, which can probably be attributed to the low uptake of plasmids by antigen-presenting cells (APCs), thereby resulting in low levels of antigen expression.Citation13
In recent years, interest in therapeutic tumor DNA vaccines has undergone something of a resurgence, with promising results being obtained.Citation14,Citation15 Tumor vaccines can deliver tumor-targeting antigens into the human body to stimulate specific immune responses against tumor cells. Given that tumor immunotherapy is primarily dependent on the cytotoxic T lymphocyte (CTL) reaction, which does not significantly affect normal tissue and cells, it is generally considered relatively safe.
On the basis of long-term research, we have constructed a technical platform for the development of efficient replication and expression of DNA vectors, which involves transforming the pSFV1 vector into the replicon expression vector pSFVK1, and using the internal ribosomal entry site (IRES) sequence to design a double cis-trans recombinant expression plasmid based on the IRES sequence. In this system, the sequence upstream of the IRES used to express a fusion antigen and the sequence downstream is used to express the adjuvant. This facilitates the efficient expression of multiple target genes under the control of a single promoter, and the antigen and immune adjuvant work in conjunction to enhance the immunogenicity of DNA vaccines. Using this technical platform, we have conducted studies on prostate cancerCitation16,Citation17 and melanoma (Patent No. Publication No. CN103215216A),Citation18,Citation19 and achieved good results in animal experiments.
To date, low delivery efficiency has been important factor hampering the development of DNA vaccines; however, with its ease of operation and high delivery efficiency, the emerging technology of electric pulse-mediated gene delivery has largely contributed to solving this problem. Accordingly, the inclusion of electric pulse-mediated gene delivery during immunization enables the effective introduction and enhanced expression of exogenous genes in vivo.Citation20
A further prominent problem associated with immunotherapy is that of immune tolerance caused by homogeneity. To avoid this phenomenon, we plan to use chimeric genes that express heterogeneous tumor-associated antigens. With regards to target gene selection, NY-ESO-1, MUC1, and MAGE-A3 are currently the focus of considerable interest in cancer immunology research. NY-ESO-1 (New York esophageal squamous cell carcinoma-1) is a cancer-testis antigen (CTA) that is among the strongest tumor antigens identified to date. It is expressed in a range of tumor tissues, including malignant melanoma, lymphoma, synovial sarcoma, and lung cancer, in which it has been established to trigger spontaneous humoral and cellular immune responses. Given its limited expression pattern, it is generally considered a good candidate target for tumor immunotherapy.Citation21 MUC1 (cancer-associated mucin 1), a member of the mucin family, is closely associated with the protective function of the mucosal epithelium and also plays an important role in cell signaling. The overexpression, aberrant cellular localization, and altered glycosylation modification of MUC1 have been demonstrated to be associated with tumorigenesis. In addition, it has been established that MUC1 masks tumor cell antigens, is involved in immune escape, and is strongly expressed in patients with a diverse range of cancer types, including breast cancer, lymphoma, lung cancer, and colon cancer.Citation22,Citation23 Similar to NY-ESO-1, MAGE-A3 (melanoma-associated antigen 3) is also a CTA that, with the exception of testicular cells, is not expressed in normal human cells, although is widely expressed on the surface of multiple types of tumor cells, including melanoma, breast cancer, skin cancer, and lung cancer cells. Moreover, MAGE-A3 is the most immunogenic CTA identified to date.Citation24,Citation25 Given that these three tumor antigens are highly expressed in cancers such as breast cancer, lymphoma, skin cancer, thyroid cancer, and melanoma,Citation26–30 we designed and constructed a novel recombinant plasmid DNA (pNMM) vaccine carrying these tumor antigens.
To further enhance the immunogenicity of the developed vaccine, in addition to the aforementioned antigenic genes we also introduced adjuvants with cellular co-stimulatory molecules. Relevant studies have confirmed that the tyrosine kinase receptor 3 ligand (FLT3L) can promote the differentiation and maturation of dendritic, natural killer, and CTL cells.Citation31 Interactions between tumor necrosis factor-associated activator proteins CD40L and CD40 can directly inhibit the growth of numerous types of cancers, including breast, ovarian, bladder, and non-small cell lung cancers,Citation32 whereas GM-CSF can activate dendritic cells, as well as promoting the proliferation of Th cells and effector cells,Citation33,Citation34 and the binding of CD80 to its ligand, CD28, can promote T cell proliferation and the secretion of chemokines.Citation35,Citation36 To date, recombinant DNA vaccines encoding FLT3L,Citation37 CD40L,Citation38 GM-CSF,Citation39 and costimulatory molecules CD80Citation40 have also have been studied and clinically applied, and in our previous study, we demonstrated that the application of these molecules could enhance the efficacy of prostate cancer DNA vaccines.Citation41
On the basis of findings reported in the literature, we constructed a pNMM recombinant DNA vaccine with the objective of enhancing the efficacy of therapeutic cancer vaccines. This recombinant pNMM vaccine carries fusion inserts comprising tumor-associated genes (NY-ESO-1, MUC1, and MAGE-A3) and inserts of cytokine and co-stimulatory molecule gene sequences (FLT3L, CD40L, GM-CSF, CD80), and was constructed as a double cis-trans DNA vaccine based on IRES sequences. To assess the efficacy of adopting this novel approach, we used an immunologically healthy mouse model to evaluate the immunogenic response following DNA vaccine administration and constructed a mouse melanoma cell line stably expressing human NMM (NY-ESO-1/Muc-1/MAGE-A3) to investigate the protective and therapeutic efficacy of the pNMM DNA vaccine in B16-NMM+-tumor-bearing mice.
Materials and methods
Ethics statement
Female C57BL/6 mice (6–8 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). All mice were maintained in accordance with the appropriate Laboratory animal guidelines (GB/T 35,892–2018 National Institutes of Health Publication No. 85–23, Revised 1996). The experimental procedures conformed to international guidelines for the care and use of laboratory animals and the study was approved by the Animal Ethics Committee of Gu’an Dingtai Haigui Biotechnology Co., Ltd.
Cell lines and a recombinant NMM protein
293T and B16 cell lines were purchased from the Cell Bank NICR. The cell line B16-NMM+ expressing NY-ESO-1, MUC1, and MAGE-A3 was previously constructed in our laboratory. Briefly, NY-ESO-1 (amino acids 157–165), MUC1 (amino acids 1159–1181), and MAGE-A3 (amino acids 1–314) fusion genes with a 6× his tag fused to the C termini were synthesized and inserted into the eukaryotic expression vector pIRES-neo via DNA recombination. B16 cells were transfected with cationic polymers, and cells stably expressing these three antigens were obtained based on pressure screening using G418, and expression of the fusion antigens in B16-NMM+ cells was confirmed based on western blotting.
Recombinant NMM protein was obtained from Nanjing Mingyan Biotechnology Co., Ltd. Briefly, NY-ESO-1 (amino acids 157–165), MUC1 (amino acids 1159–1181), and MAGE-A3 (amino acids 1–314) fusion genes with a 6× his tag fused to the C termini were synthesized and inserted into the prokaryotic expression vector pET-30a via DNA recombination. Successful construction of the recombinant NMM protein was verified based on western blot analysis and endotoxin testing.
The pNMM plasmid construct
In this study, we used the replicon-based vector pSFVK1 to construct the pNMM plasmid (). The replication complex of pSFVK1 consisting of the Alphavirus nsp1–4 protein was employed to enhance the efficiency of RNA replication. The nsp1–4 replicase gene promoter sequence incorporated the CMV early promoter and the target gene promoter from the 26S subgenome promoter. In addition, the transcription termination/polyadenylation sequence incorporated the SV40 poly (A) tail, and kanamycin was used as the antibiotic resistance marker.
Figure 1. Map of the recombinant plasmid pNMM vaccine and expression pattern of control plasmids.
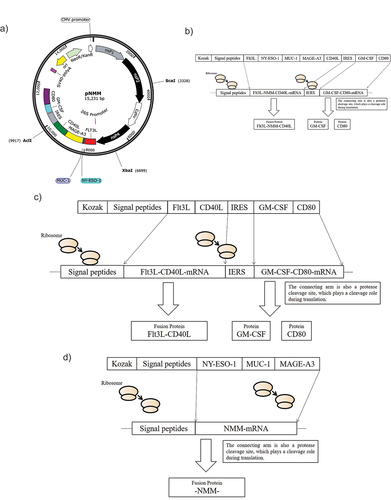
A bicistronic pNMM DNA tumor vaccine, consisting of tumor-associated antigens (NY-ESO-1, MUC1, and MAGE-A3) and adjuvant co-stimulators (FLT3L, CD40L, GM-CSF, and CD80), was constructed based on the IRES sequence. The upstream transcription product of the IRES was an FLT3L-NY-ESO-1-MUC1-MAGE-A3-CD40L fusion protein (referred to as FLT3L-NMM-CD40L), which was mainly expressed on cell membranes, whereas downstream transcription mainly transcribed GM-CSF-CD80. A designed cleavage protease sequence was inserted between GM-CSF and CD80 sequences, such that both proteins could retain their natural structure (GM-CSF for secretion and CD80 for membrane expression) (). For details of the recombinant plasmid construction procedure, please refer to the published patent (201810908222.1).Citation42 In addition to the pNMM plasmid, we also constructed control adjuvant plasmids (pSFVK1- FLT3L -CD40L-IRES-GM-CSF-CD80) () and an antigen plasmid (pSFVK1-NMM) ().
In vitro expression of the construct
293T cells were transfected with the pNMM plasmid using jetOPTIMUS® DNA Transfection Reagent (Lot# 0000000672; Polyplus) following the manufacturer’s instructions. After 6 h, the medium was replenished with fresh DMEM medium and zVAD-FMK (InvivoGen, Cat. No. 6307-43-02, a pan caspase inhibitor that blocks apoptosis). Forty-eight hours later, the culture medium and cells were harvested for further protein analysis. For western blotting, cells were treated with RIPA cell lysis buffer and equal amounts of total protein were separated on 10% SDS-PAGE gels and thereafter transferred to PVDF membranes. Membranes were blocked with PBST containing 5% skim milk for 1 h at room temperature (RT), then rinsed and incubated overnight with mouse anti-human MAGE-A3 antibodies at 4°C. The following day, the membranes were rinsed and incubated with horseradish peroxidase-labeled goat anti-mouse IgG for 1 h at RT. The membranes were visualized using an ECL western blotting system (Tanon). For flow cytometry analysis, transfected cells were pelleted and rinsed twice with pre-chilled buffer (1 × PBS+2% fetal bovine serum) prior to resuspending in 0.5 mL pre-chilled buffer. Following incubation with PE-labeled anti-human CD40L monoclonal antibodies (Cat. No. 310806; BioLegend,) and PE-labeled anti-human CD80 monoclonal antibodies (305208; BioLegend,) on ice for 30 min in the dark, the expression of CD40L and CD80 was measured by flow cytometry. For meso-scale discovery electrochemiluminescence (MSD-ECL), culture medium from the transfected cells was added to a 96 well-plate using an MSD kit (K151RID–1) according to manufacturer’s protocol. Following incubation and rinsing at RT, the GM-CSF signals of samples in the plate were measured using an MSD device (Meso QuickPlex SQ 120).
Immunization
Female C57BL/6 mice (6 to 8 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. The mice were vaccinated three times at 7-day intervals via intramuscular injection into the quadriceps femoris. Immediately after injection, multiple electrical pulse stimulation was performed at the injection site using an in vivo gene transducer operated using following parameters: voltage, 60 V; pulse time, 50 ms; and pulse frequency, 1 Hz. Each stimulation was repeated six times.
Interferon (IFN)-γ ELISPOT assay
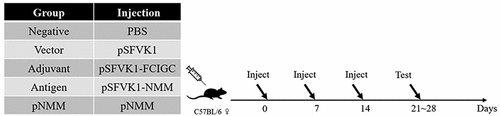
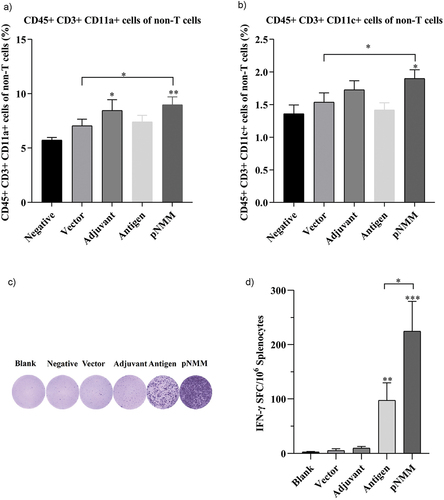
For immunogenicity experiments, mice were divided into five groups, namely, negative control, empty vector, adjuvant, antigen, and antigen plus pNMM groups (), each of which comprised 8 to 10 mice. One week following the final vaccination, spleens were extracted from the immunized mice and macerated with grinder in a Petri dish. Splenocytes were collected by centrifugation using a lymphocyte separation medium (Dakewe Biotech Co., Ltd.) following the manufacturer’s manual. An IFN-γ ELISPOT assay was performed according to the manufacturer’s protocol (Mabtech, Sweden). Briefly, freshly isolated splenocytes (4 × 105 cells/well) were added to plate wells and incubated with 2 μg/well recombinant NMM protein for 20 h. Each assay was performed in triplicate. The plates were counted and analyzed using an ELISPOT Reader (CTL, USA).
Figure 2. A schematic diagram of the experimental animal groups used in DNA vaccine immunogenicity experiments.
Evaluation of the non-T cell response
One week following the final vaccination, splenocyte suspensions were transferred to tubes. To tubes containing 1 × 106 cells splenocytes, we added an antibody cocktail (CD45 + CD3+ CD11a/CD11c; CD45 (56-0451-82; Thermofisher), CD3 (47-0031-82; Thermofisher), CD11a (101120; Biolegend), and CD11c (117349; Biolegend)). Following incubation at RT in the dark for 20–30 min, the cells were washed and analyzed by flow cytometry.
Protective and therapeutic vaccinations
For an evaluation of protective vaccination, mice were divided into the following three groups: blank control, negative control, and pNMM groups. B16-NMM+ cells (3 × 104) were subcutaneously inoculated into the backs of mice on day 7 following the final immunization (please refer to the immunization regimen shown in ). When palpable, the length and vertical short diameter of the developed tumors were measured at 2-day intervals using a vernier caliper. Tumor volumes were calculated using the following formula:
Tumor volume (mm3) = 0.5 × length diameter × short diameter2
On the basis of the values thus obtained, we constructed growth curves for an assessment of tumor development. Tumor tissues were also weighed and the rate of growth inhibition was calculated using the following formula:
Tumor growth inhibition rate (%) = (average tumor weight in blank group - average tumor weight in experimental group)/average tumor weight in blank group × 100%.
All tumor-bearing mice were euthanized when the tumor volume had reached 2000 mm3, or if ulceration was present or the mice showed any signs of discomfort.
For an evaluation of therapeutic vaccination, mice were divided into two groups, namely, a blank control and a pNMM group. B16-NMM+ cells (3 × 104) were subcutaneously inoculated into the backs of mice and 3 day post-inoculation, the mice were immunized with 1 µg of pNMM plasmid (). The length and short vertical diameter of the developing tumor were measured at 2-day intervals using a vernier caliper. Values for tumor volume and growth inhibition were obtained as described for our evaluation of protective vaccination. All tumor-bearing mice were euthanized when the volume of tumors reached 2000 mm3, or if ulceration was present or the mice showed signs of discomfort.
Statistical analysis
For statistical analyses, we used GraphPad Prism 8 software. The results are expressed as the means ± standard deviation. Differences between two groups were analyzed using Student’s t-test, and differences among three groups were evaluated based on single-factor analysis of variance. Survival curves were constructed using the Kaplan – Meier method, and differences were assessed using a Log rank test. Levels of statistical significance were set at the following p values: *p < .05; **p < .01; and ***p < .001.
Results
In vitro expression of the pNMM construct
In this study, we constructed a novel plasmid DNA (pNMM) recombinant carrying three tumor antigens (NY-ESO-1, MUC1, and MAGE-A3) and co-stimulatory molecules. To confirm expression, we transiently transfected 293T cells with the pNMM plasmid. Cells were harvested 48 h later and protein expression was assessed by western blotting. As shown in , the expression of the fusion protein (FLT3L-NMM-CD40L) was detected using specific anti-human MAGE-A3 antibodies.
Figure 3. Expression of the pNMM vaccine in 293T cells.
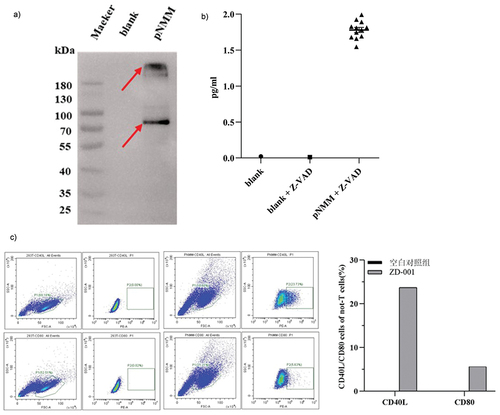
To detect the secretion of GM-CSF in culture media, we used the MSD method, which revealed that only the culture supernatant of 293T cells transfected with the pNMM plasmid contained GM-CSF (). These findings accordingly indicated that the GM-CSF sequence of IRES can be successfully expressed in the pNMM plasmid.
To confirm the expression of CD40L and CD80, 293T cells were harvested 48 h post-transfection and analyzed by flow cytometry. In cells transfected with the pNMM plasmid we detected percentage CD40L and CD80 expression of 23.73% and 5.63%, respectively, as shown in , thereby verifying successful transfection of the plasmid.
Changes in nonspecific lymphocytes and antigen-specific T cells
Splenocytes isolated from the vaccinated mice and labeled with antibodies in vitro were subsequently analyzed using flow cytometry. Observations revealed significant changes in the number of CD45+ CD3+ CD11a+ and CD45+ CD3+ CD11c+ non-T cells (macrophage and dendritic cells) among the different groups. More specifically, in the treatment naive, vector control, adjuvant only, antigen only, and pNMM groups, we detected CD45+ CD3+ CD11a+ percentages of 5.74%, 7.05%, 8.47%, 7.4%, and 8.99% respectively. Compared with the treatment naive group, there were significant increases in the percentages of CD45+ CD3+ CD11a+ cells in the adjuvant only and the pNMM groups (). Comparatively, we detected CD45+ CD3+ CD11c+ percentages of 1.36%, 1.54%, 1.73%, 1.42% and 1.9% in the treatment naive, vector control, adjuvant only, antigen only and pNMM groups respectively. Compared with the treatment naive group, the percentage of CD45+ CD3+ CD11c+ cells was significantly higher in the pNMM group (). In subsequent analyses of T cells, we found that compared with the treatment naive, vector control, and adjuvant only groups, there were significant increases in the percentages of T cells in the antigen only and pNMM groups ().
Figure 4. Evaluation of non-T cell response and specific IFN-γ-producing T cells.
Protective efficacy of the pNMM vaccine
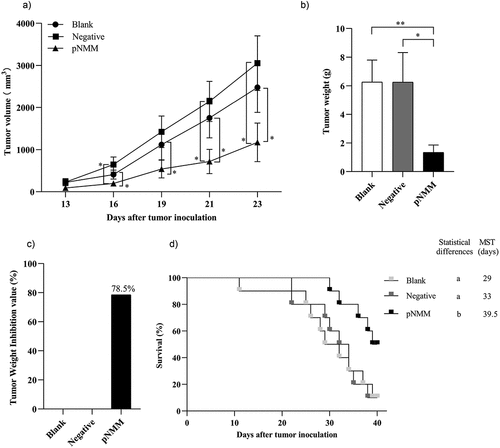
To evaluate the efficacy of the pNMM vaccine against tumor growth, we immunized a group of five C57BL/6 female mice with 1 µg of the pNMM plasmid three times at days 0, 7 and 14. After completing immunization, B16-NMM+ tumor cells were subcutaneously transplanted into the mice on day 21, and we subsequently monitored tumor growth (). We accordingly found that on day 28, those mice receiving the pNMM vaccine were characterized by a tumor inhibition rate of 78.5% (). Moreover, we recorded survival rates of 10%, 10%, and 50% among mice in the blank, negative control, and pNMM groups respectively, with corresponding median survival times of 30.5, 33, and 39.5 days ().
Figure 5. Protective efficacy of the DNA vaccine in a B16-NMM+-tumor model.
Therapeutic efficacy of the pNMM vaccine
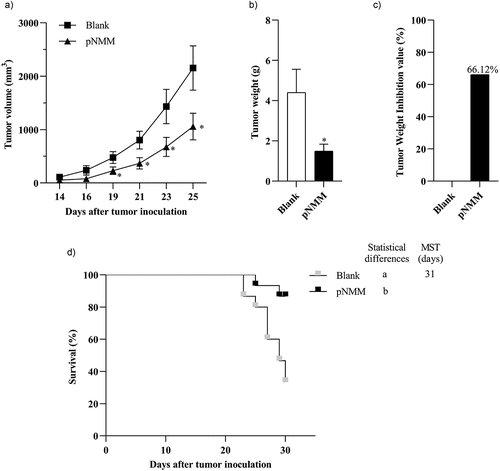
To evaluate the therapeutic effect of pNMM vaccination against preexisting tumors, C57BL/6 female mice were inoculated with B16-NMM+ tumor cells and 3 day thereafter, we commenced immunization treatment using 1 µg of the pNMM plasmid. Tumor growth in immunized mice was monitored and is presented in . Compared with the blank control group, we detected a significant reduction in the volumes of tumors in the pNMM group mice as early as the 19th day post-inoculation (). On the basis of measurements of changes in tumor weight among the mice in each group, we detected a tumor inhibition rate of 66.12% in the pNMM group mice (). Furthermore, at the end of the assessment period (31 days after tumor cell inoculation), we recorded survivals of 86.6% and 33.3% among mice in the pNMM and control groups, respectively ().
Figure 6. Therapeutic efficacy of the DNA vaccine in a B16-NMM+-tumor model.
Discussion
Although multiple types of tumor vaccine have shown encouraging results in animal experiments, these vaccines have generally proved to be less effective when assessed in clinical trials. Accordingly, there is a necessity to further study and identify effective tumor antigens, as well as co-stimulatory molecules, to develop more potent vaccines. In this regard, genetically engineered vaccines have been established to have a number of disadvantages, among which are their instability and weak immunogenicity. Consequently, a range of alternative methods of vaccine development have been proposed, including the incorporation of adjuvants and cytokines within the vaccine construct. In addition to weak immunogenicity and difficulties associated with delivery, it has often been questioned as to whether conventional DNA vaccines would integrate into host cell genomes. However, given that the self-replication and transcription of replicon DNA vaccines occur within the cytoplasm, this effectively eliminates the risk of integration into the host cell genome, and thereby markedly enhances vaccine safety.Citation43 In addition, DNA vaccines have structural advantages that facilitate fusion expression of multiple epitope peptides.Citation11,Citation44 Furthermore, the use of electrical pulses for the introduction of plasmid DNA overcomes the problem of low transfection efficiency associated with the inoculation of DNA vaccines, all of which highlights the promise of DNA vaccines for therapeutic vaccine development.
To take into account the factors of antigen recognition, antigen presentation, and full activation of the immune response in vaccine design, in this study, we used a strategy based on the co-expression of tumor-specific antigen genes and adjuvant genes in a double cis-trans plasmid. Initially, we fused three tumor-associated antigens with the aim of enhancing the rate of tumor cell binding. Given that it has been established that vaccine adjuvants can enhance specific immune responses and boost immunogenicity, in addition to tumor antigens, the pNMM plasmid also expresses intramolecular components such as cytokines and co-stimulatory molecules. Among these, upstream of the IRES sequence, we expressed a tumor antigen fused with FLT3L and CD40L. Along with the two immunostimulatory molecules, the NMM antigen expressed in this way has an enhanced capacity to synergistically target antigen-presenting cells, thereby facilitating antigen processing and presentation by dendritic cells. Furthermore, GM-CSF is expressed with CD80 molecules downstream of the IRES sequence. The expression of these molecules promotes the proliferation or maturation of immune cells such as dendritic cells, macrophages, and T cells, thereby enhancing antigen-specific immune responses.
In this study, to assess the activation of immune cells, we used splenocytes isolated from C57BL/6 mice that had been immunized with pNMM plasmid along with other plasmids (empty vector, antigen expression only, and adjuvant expression only), and accordingly detected increases in the numbers of CD45+ CD3+ CD11a+ and CD45+ CD3+ CD11c+ expressing cells in the splenocytes of mice vaccinated with pNMM. Such elevation of the populations of CD45+ CD3+ CD11a+ cells (macrophages) and CD45+ CD3+ CD11c+ cells (dendritic cells) can potentially contribute to the induction of antigen-specific immune cells. In addition, we detected increases in the numbers of IFN-γ-producing T-cells in pNMM-immunized mice. Collectively, these findings provide convincing evidence to indicate that the pNMM vaccine developed in this is study is effective in activating an antigen-specific cellular immune response.
In the tumor prevention model evaluated in this study, we found that vaccination with pNMM inhibited the growth of implanted B16-NMM+ tumor cells and enhanced the total survival of treated mice. In the tumor treatment model, we detected a retardation of the rate of tumor growth and an overall increase in the survival of mice receiving the pNMM vaccination. Our findings accordingly revealed that those mice receiving the pNMM were characterized the strongest T lymphocyte activity and the best antitumor effect, thereby indicating that the expression of antigens from a fusion construct incorporating immunostimulatory molecules and cytokines could represent a potentially viable strategy for enhancing the efficacy of tumor vaccines.
Although on the basis of our comparison between the pNMM plasmid and the blank and negative control groups, we succeeded in confirming the efficacy of the pNMM vaccine, both with respect to tumor suppression and enhanced survival, we regrettably obtained no direct evidence to indicate that adjuvant genes can contribute to enhancing the efficacy of the DNA vaccine. Accordingly, in follow-up animal studies, we will conduct more comprehensive comparative evaluations, in which we will assess the effects of antigen alone and adjuvant alone plasmids as controls, thereby enabling us to further determine whether the adjuvant genes harbored in the pNMM plasmid can contribute to enhancing DNA vaccine efficacy. Moreover, we also intend to expand the range of assessed relevant cell populations, particularly regulatory T cells (Tregs).
Among the DNA vaccine currently available on the market worldwide is ZyCoV-D, a DNA vaccine against COVID-19 developed in India by Zydus Cadila, which represents a milestone in the history of DNA vaccine development.Citation45 DNA vaccines have the capacity to store vast amounts of information, and thereby potentially encode large, complex proteins, or even multiple proteins.Citation46 We accordingly believe that such vaccines hold considerable promise as anti-cancer vaccines, and in the near future, we would anticipate a notable increase in the development and application DNA vaccines.
Acknowledgments
This work was financially supported by the Key projects of Research and Development of Hebei Provincial Department of Science and Technology (contract No. 20372404D). We are grateful for the support received from the Support Fund for Young Talents in Langfang.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Steven A, Fisher SA, Robinson BW. Immunotherapy for lung cancer. Respirology. 2016;21(5):821–11. doi:10.1111/resp.12789.
- Asmar R, Rizvi NA. Immunotherapy for advanced lung cancer. Cancer J. 2015;21(5):383–91. doi:10.1097/PPO.0000000000000151.
- Yang B, Jeang J, Yang A, Wu TC, Hung CF. DNA vaccine for cancer immunotherapy. Hum Vacc Immun. 2014;10(11):3153–64. doi:10.4161/21645515.2014.980686.
- Esfandiary A, Ghafouri-Fard S. MAGE-A3: an immunogenic target used in clinical practice. Immunotherapy. 2015;7(6):683–704. doi:10.2217/imt.15.29.
- Batchu RB, Gruzdyn O, Potti RB, Weaver DW, Gruber SA. MAGE-A3 with cell-penetrating domain as an efficient therapeutic cancer vaccine. JAMA Surg. 2014;149(5):451–7. doi:10.1001/jamasurg.2013.4113.
- Franck CO, Fanslau L, Bistrovic Popov A, Tyagi P, Fruk L. Biopolymer-based carriers for DNA vaccine design. Angew Chem Int Ed Engl. 2021;60(24):13225–43. doi:10.1002/anie.202010282.
- Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451(7179):725–9. doi:10.1038/nature06537.
- Signori E, Iurescia S, Massi E, Fioretti D, Chiarella P, De Robertis M, Rinaldi M, Tonon G, Fazio VM. DNA vaccination strategies for anti-tumour effective gene therapy protocols. Cancer Immunol Immun. 2010;59:1583–91. doi:10.1007/s00262-010-0853-x.
- Alam S, McNeel DG. DNA vaccines for the treatment of prostate cancer. Expert Rev Vacc. 2010;9:731–45. doi:10.1586/erv.10.64.
- Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9:776–88. doi:10.1038/nrg2432.
- Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008;8:108–20. doi:10.1038/nrc2326.
- Xu Q, Zhu YF, Wang HC, Gong ZW, Yu YZ. Enhanced efficacy of DNA vaccination against botulinum neurotoxin serotype a by co-administration of plasmids encoding DC-stimulating Flt3L and MIP-3α cytokines. Biologicals. 2016;44(5):441–7. doi:10.1016/j.biologicals.2016.04.008.
- Demoulin S, Herfs M, Delvenne P, Hubert P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: insight into the molecular mechanisms. J Leuk Biol. 2013;93:343–52. doi:10.1189/jlb.0812397.
- Zurkova K, Babiarova K, Hainz P, Krystofova J, Kutinova L, Otahal P, Nemeckova S. The expression of the soluble isoform of hFlt3 ligand by recombinant vaccinia virus enhances immunogenicity of the vector. Oncol Rep. 2009;21(5):1335–43. doi:10.3892/or_00000359.
- Lee SH, Danishmalik SN, Sin JI. DNA vaccines, electroporation, and their applications in cancer treatment. Hum Vacc Immun. 2015;11(8):1889–900. doi:10.1080/21645515.2015.1035502.
- Han G, Gao JP, Yan JQ, Jia R, Zhang L, Dong JK. Construction and eukaryotic expression of a heterologous PSCA fusion gene DNA vaccine against prostate cancer. J Chin PLA Postgrad Med Sch. 2009;30(1):88–90.
- Dong JK, Chen LJ, Yu JY. Preliminary study on the tumor suppressive activity of immune potentiated anti-prostate cancer DNA vaccine. In: Proceedings of the 2019 China Oncology Conference. Chongqing (China); 2019. p. 5201.
- Cheng SH, Ma XH, Chen X, Yu JY. Study on the tumor suppressive effect of anchor-expressing GM-CSF vaccine. Chin J Cancer Bio. 2004;11(1):27–30.
- Tian R, Cheng SH, Yu JY, Li W, Liu YF. Inhibitory effect of mGM-CSF gene transfection on lung metastasis of mouse melanoma cells. J Fourth Military Med Uni. 2006;27(6):484–6.
- Zhang L, Wang Y, Xiao Y, Wang Y, Dong J, Gao K, Gao Y, Wang X, Zhang W, Xu Y, et al. Enhancement of antitumor immunity using a DNA-based replicon vaccine derived from Semliki forest virus. Plos One. 2014;9(3):e90551. doi:10.1371/journal.pone.0090551.
- Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, Decock J. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol. 2018;9:947. doi:10.3389/fimmu.2018.00947.
- Asín A, García-Martín F, Busto JH, Avenoza A, Peregrina JM, Corzana F. Structure-based design of anti-cancer vaccines: the significance of antigen presentation to boost the immune response. Curr Med Chem. 2022;29(7):1258–70. doi:10.2174/0929867328666210810152917.
- Zhu H, Wang K, Wang Z, Wang D, Yin X, Liu Y, Yu F, Zhao W. An efficient and safe MUC1-dendritic cell-derived exosome conjugate vaccine elicits potent cellular and humoral immunity and tumor inhibition in vivo. Acta Biomater. 2022;138:491–504. doi:10.1016/j.actbio.2021.10.041.
- Schäfer P, Paraschiakos T, Windhorst S. Oncogenic activity and cellular functionality of melanoma associated antigen A3. Biochem Pharmacol. 2021;192:114700. doi:10.1016/j.bcp.2021.114700.
- Das B, Senapati S. Immunological and functional aspects of MAGEA3 cancer/testis antigen. Adv Protein Chem Struct Biol. 2021;125:121–47.
- Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011;239(1):62–84. doi:10.1111/j.1600-065X.2010.00980.x.
- Ligtenberg MA, Rojas-Colonelli N, Kiessling R, Lladser A. NF-κB activation during intradermal DNA vaccination is essential for eliciting tumor protective antigen-specific CTL responses. Hum Vacc Immun. 2013;9(10):2189–95. doi:10.4161/hv.25699.
- Li L, Petrovsky N. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert Rev Vacc. 2016;15(3):313–29. doi:10.1586/14760584.2016.1124762.
- Iurescia S, Fioretti D, Rinaldi M. Strategies for improving DNA vaccine performance. Meth Mol Biol. 2014;1143:21–31.
- Anandasabapathy N, Feder R, Mollah S, Tse SW, Longhi MP, Mehandru S, Matos I, Cheong C, Ruane D, Brane L, et al. Classical Flt3L-dependent dendritic cells control immunity to protein vaccine. J Exp Med. 2014;211(9):1875–91. doi:10.1084/jem.20131397.
- Xu ZX. Advances in the study of Flt3 ligands. For Med Sci. 2000;23:4.
- Tong A, Stone M. Prospects for CD40-directed experimental therapy of human cancer. Cancer Gene Ther. 2003;10(1):1–13. doi:10.1038/sj.cgt.7700527.
- Nakazaki Y, Tani K, Lin ZT, Sumimoto H, Hibino H, Tanabe T, Wu M-S, Izawa K, Hase H, Takahashi S, et al. Vaccine effect of granulocyte–macrophage colony-stimulating factor or CD80 gene-transduced murine hematopoietic tumor cells and their cooperative enhancement of antitumor immunity. Gene Ther. 1998;5:1355–62. doi:10.1038/sj.gt.3300726.
- Kass E, Parker J, Schlom J, Greiner JW. Comparative studies of the effects of recombinant GM-CSF and GM-CSF administered via a poxvirus to enhance the concentration of antigen- presenting cells in regional lymph nodes. Cytokine. 2000;12:960–71. doi:10.1006/cyto.2000.0684.
- Boussiotis VA, Freeman GJ, Gribben JG, Nadler LM. The role of B7-1/B7-2: CD28/CLTA-4 pathways in the prevention of anergy, induction of productive immunity and down-regulation of the immune response. Immunol Rev. 1996;153:5–26. doi:10.1111/j.1600-065X.1996.tb00918.x.
- Herold KC, Lu J, Rulifson I, Vezys V, Taub D, Grusby MJ, Bluestone JA. Regulation of C-C chemokine production by murine T cells by CD28/B7 costimulation. J Immunol. 1997;159(9):4150–3. PMID: 9379007. doi:10.4049/jimmunol.159.9.4150.
- Li Y, Mention JJ, Court N, Masse-Ranson G, Toubert A, Spits H, Legrand N, Corcuff E, Strick-Marchand H, Di Santo JP. A novel Flt3-deficient HIS mouse model with selective enhancement of human DC development. Eur J Immunol. 2016;46(5):1291–9. doi:10.1002/eji.201546132.
- Mwangi DM, Honda Y, Graham SP, Pelle R, Taracha EL, Gachanja J, Nyanjui JK, Bray J, Palmer GH, Brown WC, et al. Treatment of cattle with DNA-encoded Flt3L and GM-CSF prior to immunization with Theileria parva candidate vaccine antigens induces CD4 and CD8 T cell IFN-γ responses but not CTL responses. Vet Immunol Immunopathol. 2011;140(3–4):244–51. doi:10.1016/j.vetimm.2010.12.013.
- Iwabuchi R, Ikeno S, Kobayashi-Ishihara M, Takeyama H, Ato M, Tsunetsugu-Yokota Y, Terahara K. Introduction of human Flt3-L and GM-CSF into humanized mice enhances the reconstitution and maturation of myeloid dendritic cells and the development of Foxp3+CD4+ T cells. Front Immunol. 2018;9:1042. doi:10.3389/fimmu.2018.01042.
- Wang H, Yu J, Li L. A DNA vaccine encoding mutated HPV58 mE6E7-Fc-GPI fusion antigen and GM-CSF and CD80. Onco Targ Ther. 2015;8:3067–77. doi:10.2147/OTT.S84888.
- Wang Y, Li ZM. A nucleic acid vaccine for prostate cancer: China patent no. 2015107650912.
- Yu JY, Wang Y, Xu Q, Ke ZY, Song WW, Guo RZ. An anti-tumor recombinant plasmid DNA vaccine carrying NMM fusion antigen: China patent no. 2018109082221.
- Polo JM, Gardner JP, Ji Y, Belli BA, Driver DA, Sherrill S, Perri S, Liu MA, Dubensky TW. Alphavirus DNA and particle replicons for vaccines and gene therapy. Dev Biol. 2000;104:181–5.
- Loisel-Meyer S, Foley R, Medin JA. Immuno-gene therapy approaches for cancer: from in vitro studies to clinical trials. Front Biosci. 2008;13:3202–14. doi:10.2741/2921.
- Momin T, Kansagra K, Patel H, Sharma S, Sharma B, Patel J, Mittal R, Sanmukhani J, Maithal K, Dey A, et al. Safety and immunogenicity of a DNA SARS-CoV-2 vaccine (ZyCoV-D): results of an open-label, non-randomized phase I part of phase I/II clinical study by intradermal route in healthy subjects in India. EClin Med. 2021;38:101020. doi:10.1016/j.eclinm.2021.101020.
- Smriti M. India’s DNA COVID vaccine is a world first–more are coming. Nature. 2021;597:161–2. https://www.nature.com/articles/d41586-021-02385-x