
ABSTRACT
Overexpressed tumor-associated antigens (TAAs) are a large group that includes proteins found at increased levels in tumors compared to healthy cells. Universal tumor expression can be defined as overexpression in all cancers examined as has been shown for Tumor Protein D52. TPD52 is an over expressed TAA actively involved in transformation, leading to increased proliferation and metastasis. TPD52 overexpression has been demonstrated in many human adult and pediatric malignancies. The murine orthologue of TPD52 (mD52) parallels normal tissue expression patterns and known functions of human TPD52 (hD52). Here in we present our preclinical studies over the past 15 years which have demonstrated that vaccine induced immunity against mD52 is effective against multiple cancers in murine models, without inducing autoimmunity against healthy tissues and cells.
Introduction
Current statistics reveal an increase in cancer incidence and mortality across all cancers over the past decade, regardless of sex. 1,958,310 new cancer cases are estimated in the USA in 2023, with 609,820 (31%) deaths.Citation1 An increase of 15% incidence over a 10-year period may to some seem relatively small. What stands out is that there is an increase in diagnoses and deaths in an era of remarkable advancement in treatment. Most notable is the improvement of passive antibody therapies referred to as “immune checkpoint treatments” (ICTs). This approach is led by the administration of specific monoclonal antibodies (mAbs) against the T cells inhibitory receptors CTLA-4 and PD-1, referred to as immune checkpoint blockade, and has demonstrated the greatest benefit,Citation2,Citation3 though not without immune-related adverse effect.Citation4 Clearly there is still work to be done and justification for expansion of antigen-specific vaccination research. While ICTs engage the immune system independent of defined tumor antigen targets, active vaccines elicit specific immune responses by targeting defined well-characterized tumor antigens.Citation5 The newest category of specific antigens is the so called “neo-antigens,” which was discovered as a result of successes following ICT.Citation6 Neo-antigens are largely represented by T cell epitope peptides that contain most often a single amino acid change (mutation) in a normally expressed protein within the tumor cells. The point mutation comes about following various unrelated molecular events, so that the target is unique not just to a cancer type but most often to an individual patient.Citation7 Whether a vaccine is therapeutic or preventive, it is clear that future success will depend on the character of the tumor antigen targeted by the vaccine. The current collective of candidate-tumor antigens ranges from specific (non-self) to associated (self) and comprises hundreds, if not thousands of candidates. The group of antigens defined first by their overexpression in tumor cells compared to low but detectable levels in normal cells and second by a role in oncogenesis will be briefly overviewed herein as reference to the inclusion of TPD52 in this catagrory, for more detail see our previous review.Citation8 Tumor protein D52 (TPD52) will be the main focus of this review, specifically to expound on our preclinical work developing it as a vaccine target.
Tumor associated protein antigens (TAAs)
Overexpressed tumor associated antigens (TAAs) are a large and diverse group that includes any protein found at increased levels in tumors compared to normal healthy cells and tissues. When overexpressed antigens that do not play a role in oncogenesis are excluded from discussion there remain roughly half a dozen well characterized overexpressed TAAs that are immunogenic and oncogenic.Citation9 In the following sections, we will briefly highlight those overexpressed oncogenic TAAs closest to the character of TPD52 in shared expression and oncogenic function, followed by focusing intently on our work to develop a vaccine against TPD52 a TAA without mutation that is demonstrably widely shared among cancers, and is oncogenic and immunogenic.
Overexpressed oncogenic TAA: limited to one or a few tumor types
Androgen receptor (AR)
The AR is expressed in normal tissues with the prostate being a major site, and is widely overexpressed in prostate cancer, and in a subset of breast cancers.Citation8,Citation10,Citation11 Pre-existing antibodies and T cells specific for AR peptides have been detected in prostate cancer patients, demonstrating immunogenicity. Peptides capable of eliciting specific CTLs following immunization of transgenic mice and DNA vaccination elicited tumor regression in rat models support the potential for targeting the AR with vaccination. These studies demonstrate that AR is immunogenic in patients and AR-based vaccines are capable of in vivo tumor rejection in pre-clinical animal models.Citation8,Citation12
Her-2/neu
The transmembrane tyrosine kinase Her-2/neu is one of the most studied cancer proteins and therapeutic targets. Her-2/neu is overexpressed in multiple cancers including lung, prostate and most notably breast cancer. Specific CTLs have been demonstrated in the blood of breast cancer patients, driving the majority of vaccine trials targeting the extracellular domain of Her-2 in this patient population.Citation8,Citation13–15
p53
The role played by p53 in cancers of multiple types is widely recognized. Pre-existing p53-specific CTLs have been demonstrated in ovarian, colorectal and lung cancer patients driving ongoing clinical trials evaluating therapeutic vaccines targeting p53 in these patient populations. Though p53-specific immune responses have been observed in vaccinated patients, significant reduction in tumor burdens is beginning to be realized.Citation8,Citation16–22
EphA2
EphA2 is a cell surface-expressed receptor tyrosine kinase and is one of 14 members of the Eph. It is overexpressed in multiple cancers including brain malignancies, which have been the focus of most vaccine trials. Vaccination of mice with EphA2 peptide demonstrated that tolerance could be broken by vaccination without inducing autoimmunity. Additional studies in murine models of colon, liver and brain cancer add support to the potential of EphA2 as a vaccine target. Clinical vaccine trials in adult and pediatric brain cancer patients have shown them to be safe, immunogenic and promising.Citation8,Citation23–26
Overexpressed oncogenic TAA: widely shared among tumor types
hTERT
The activity of hTERT is involved in the proliferative capacity of advanced cancers and differences in normal tissue and tumor expression/function renders it a prototypical universal tumor expression TAA and vaccine target. Demonstration of specific CTLs in patients from approximately 85% of human cancers types led to hTERT peptide(s)/T cell epitope(s) discovery and their pursuit as vaccines in clinical trials. Ongoing clinical trials demonstrate variability of response to hTERT vaccines in patients with several types of cancer. Though specific T cells were commonly induced in the majority of patients, with no adverse toxicities.Citation8,Citation27–32
Survivin
Survivin is an inhibitor of apoptosis and acts upstream of the main effector caspase proteases of apoptosis, thus enhancing survival. Similar to hTERT, it is expressed at low levels in normal tissues but is overexpressed in cancers originating from a variety of tissues including lung, colon, breast, pancreas, prostate cancer and several hematopoietic cancers making it a universal tumor expression TAA. CTLs specific for survivin have been demonstrated in the peripheral blood of a variety of cancer patients, and a peptide vaccine demonstrated clinical benefit for patients with advanced pancreatic cancer, although vaccinated melanoma patients demonstrated contrasting clinical benefits in separate clinical trials.Citation8,Citation33–38
Tumor protein D52 (TPD52)
The tumor protein D52 gene was first identified nearly 30 years ago via its overexpression in human cancer cells or tissues. Early work supporting the significance of TPD52 in cancer demonstrated the gene location on chromosome 8q. This is a region frequently gained in many cancers including as an example prostate carcinoma, and is the location of the MYC oncogene. TPD52’s location at chromosome 8q21 is separate from MYC at chromosome 8q24. It has become clear that TPD52 gains are often independent of MYC, thus TPD52 independently acts as an oncogene. TPD52 is an over expressed tumor self-protein actively involved in transformation, leading to increased proliferation and metastasis.Citation8,Citation39 TPD52 overexpression has been demonstrated in many human malignancies including for example breast,Citation40–42 prostate,Citation43–45 and ovarian carcinomas.Citation46 Expression microarray and other molecular expression analyses predict TPD52 overexpression in most cancers, adding to the list multiple myeloma,Citation47,Citation48 Burkitt’s lymphoma,Citation49,Citation50 pancreatic cancer,Citation51 testicular germ cell tumors,Citation52–54 and melanomas,Citation55,Citation56 as well as multiple other adult and pediatric cancers.Citation39 TPD52 was first discovered in breast and prostate cancers owing to the large body of work in these two cancer types relative to expression and function.
Specific upregulation of TPD52 in human breast cancer is supported by multiple studies focused on chromosome 8q21 the TPD52 locus (reviewed in.Citation39,Citation57 Interestingly, TPD52 expression has also been demonstrated with Her-2/neu expression in human breast cancer cell lines and tissues, and in mammary tissues from Her-2 transgenic mice.Citation58–60 TPD52 has also been reproducibly associated with poorer outcomes in breast cancer patients.Citation61,Citation62
TPD52 overexpression has been reported in human prostate cancer cells and tissues (extensively reviewed elsewhere).Citation8,Citation39,Citation63 TPD52 transcripts have been shown to be increased in tumor and matched tumor adjacent histologically normal tissues, compared with normal, cancer-free prostate tissues.Citation64 In support of these studies, nearly 30 years ago we cloned TPD52 from patient matched malignant and nonmalignant cell culturesCitation65,Citation66 derived from patients undergoing radical prostatectomy, using differential gene expression analysis (unpublished data). TPD52 amplification has been associated with early lethality in prostate cancer patientsCitation67 and may be due to isochromosome 8q formation.Citation68 This suggests that TPD52 copy number may represent a useful prognostic marker in prostate cancer.Citation69
Important for preclinical vaccines studies was the cloning of TPD52 from murine tumor cells. The murine orthologue of TPD52 designated mD52 exhibits normal tissue expression and functions similar tothe human orthologue(hD52). Of note the mouse and human proteins share 86% identity at the amino acid level.Citation70 When mD52 expression is increased in normal, fibroblast type cells it induces anchorage independent growth and spontaneous lung metastasis.Citation71 When hD52 expression is reduced via RNAi resulted in increased apoptosis in human breast cancer cell lines, and increased expression correlated with decreased patient survival.Citation61 These studies demonstrated that TPD52 over-expression is important for initiating and maintaining a transformed and oncogenic phenotype as well as progression to metastasis.
Overview of TPD52 oncogenicity and immunogenicity
It has been demonstrated that TPD52 is involved in initiating and maintaining the malignant state and thus is critical for the cancer cells to survive. Our overexpression studies have examined the effect of increasing TPD52 levels in 3T3 fibroblasts in which TPD52 expression is very low or undetectable. Transfection of 3T3 fibroblasts with a plasmid expressing either hD52 or mD52 resulted in a transformed, metastatic phenotype, increased proliferation and anchorage independent growth.Citation39,Citation61,Citation71 Some TPD52 RNAi transient knockdown studies in human breast cancer cell lines demonstrated increased cell death in cancer cells suggesting a role for TPD52 in regulating cell survival.Citation61,Citation62 Our studies using shRNA for more stable knockdown of TPD52 expression in murine cancer cells resulted in decreased cell proliferation in vitro. Thus, we believe that TPD52 is naturally expressed and involved in tumor formation and metastasis in human cells (hD52) and in mouse cells (mD52), making mD52 a unique and powerful over expressed tumor-self antigen for study as a cancer vaccine target in murine models of cancer.
Why have vaccines against cancers lagged behind the advancement of passive therapies in clinical trials? And will vaccines that target defined tumor-associated antigens emerge as the next generation vaccine targets? To address this, it is necessary to identify the best choice for tumor antigens to target. Given the power with which the immune system is regulated against self-recognition and response, early candidates for vaccine antigen targets were logically those farthest from self-proteins and normal self-expression. This excluded vaccines against the more common cancers like breast and prostate cancer. Proteins that are self, non-mutated with wide tissue expression, but overexpressed or up regulated in malignant cells compared to the normal cell counter parts include, as we have stated above, telomerase, survivin and most relevant to this review TPD52. Tumor-associated antigens such as these have been classified as overexpressed oncogenic tumor-self antigens and may represent the spearhead of the next generation of vaccines against cancer.Citation8,Citation9,Citation72 Our studies that follow are weighted to systems functions as they relate to immunity to TPD52 elicited by vaccination ().
Figure 1. Tumor protein D52 TAA characteristics. Illustrated are the three characteristics of TPD52 defining it as a universal tumor-associated antigen (TAA). Shown are natural cellular expression characteristics (red), oncogenic characteristics (yellow) and immunogenic characteristics as elicited by vaccination with TPD52 (blue). Figure was created using BioRender.
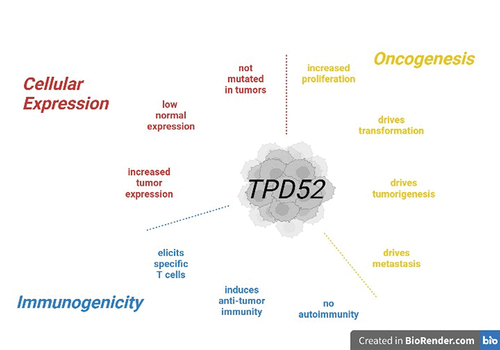
Preclinical studies on TPD52 as a cancer vaccine antigen
In the following sections our preclinical studies have demonstrated that vaccination against TPD52 is effective in mouse models of sarcoma and prostate cancer. Of note, vaccine-induced tumor immunity did not include autoimmunity. It is understood that the T cell immune repertoire is developmentally regulated by negative and positive selection in the thymusCitation73,Citation74 guarding against the generation of highly self-protein-specific T cells. Even so we have elicited CD8 cytotoxic T cells (CTLs) against TPD52 overexpressing tumor cells by vaccination with murine orthologue mD52 in murine cancer models.Citation75–78 We also demonstrated that protective immunity preventing cancer development and metastasis was increased when D52-vaccination was administered with inhibition of conventional CD4 regulatory T cells.Citation77,Citation78 This supports the notion that immunity against a tumor-self protein is also regulated at the cellular level in the periphery of adult mice. CTLs elicited by TPD52 vaccination are capable of recognizing and killing cancer cells yet remain blind to normal cells that also express the TPD52 protein (autoimmunity has not developed following TPD52 vaccination). We have tested 12 different D52 vaccines strategies in three mouse strains against six different tumor cell lines, with tumor protection ranging from 30% to 80%.Citation8 Increased protection was observed with concomitant CD4 Treg depletion. Of note, was the induction of CD8 IL-10 T cells that we believe are a unique subset of CD8 regulatory T cells that arise following TPD52 vaccination. To provide preclinical support for TPD52 as a cancer vaccine antigen and to demonstrate proof of concept to this end, in the following section we will focus on our vaccine studies in two mouse tumor models summarized in .
Table 1. Overview of preclinical TPD52 vaccine studies.
Balb/C sarcoma model for mD52 vaccine studies
Sarcoma vaccine study 1) mD52 protein+ODN vaccination
For the following we asked wether recombinant mD52 protein and CpG/ODN in alum were immunogenic when administered by intramuscular injection to Balb/c mice As a test of tumor immunity vaccinated mice were challenged with syngeneic sarcoma cells that over expressed mD52. Unimmunized or mock immunized sarcoma challenged mice served as controls. 50% of immunized mice challenged with mKSA sarcoma cells and 40% of mice challenged with 3T3.mD52Citation71 spontaneously metastatic sarcoma cells rejected tumor challenge compared to controls. Immunizedmice that were challenged with 3T3.mD52 sarcoma cells also prevented the formation of spontaneous lethal lung metastases. Tumor immunity was mediated by mD52-specific, MHC-I H-2 Kd restricted cytotoxic T lymphocytes (CTLs). Importantly there was no evidence of mice following mD52 immunization.Citation75 These results demonstrated for the first time that TPD52 is immunogenic when administered as a recombinant protein-based vaccine admixed with CpG/ODN as a molecular adjuvant. Of note, mD52 protein recognized by specific CTLs and IgG antibodies from immunized animals usedsubsequently for in vitro experiments was the natural derived from murine tumor cells demonstrating that the recombinant protein delivered as a vaccine induced immunity to the natural form of TPD52. We found measurement of mD52 specific serum IgG titers by ELISA was useful as an indication of protein induced vaccine immunity and involvement of CD4 T cell help for isotype switching to IgG following vaccination with mD52 protein. However, TPD52 is a small intracellular protein that is not expressed on the tumor cell surface as an intact molecule which is needed for antibody binding. Thus, antibodies have not demonstrated a role in tumor immunity and tumor rejection.
There are four points to summarize our findings in the Balb/c sarcoma vaccine model for Study 1. First, Recombinant mD52 protein admixed with CpG/ODN in alum when administered i.m. elicited specific IgG antibody titers useful for monitoring efficacy of vaccination but more importantly elicited specific cell-mediated immunity comprised of CD8 CTLs. Second, D52-specific CTLs were MHC-I H-2 Kd restricted and were responsible for tumor rejection. Third, this vaccine approach resulted in 40–50% protection from lethal sarcoma s.c. tumor challenge and complete rejection of spontaneous sarcoma lung metastases. Finally, since TPD52 is a non-mutated self-antigen that is overexpressed in tumor cells it was critically important that this vaccine formulation did not induce autoimmunity in immunized mice.
Sarcoma vaccine study 2) mD52 protein+ODN vaccine combination treg depletion
In this study we changed the vaccine route and adjuvant to s.c. and IFA. We immunized Balb/c mice s.c. with recombinant mD52 protein mixed with CpG+ODN as an oil in water emulsion using incomplete Freunds adjuvant (IFA). Simply changing the route of injection and the adjuvant was not sufficient for inducing protective immunity in this model (<10% protection). However, 70% protection from sarcoma tumor challenge was observed if this vaccine approach included concomitant administration of mAb specific for CD25 in order to deplete CD4 CD25 Treg cells, compared to < 10% tumor protections of immunized mock-depleted mice. Treg cell depletion did not inhibit the induction of effector T cell memory, as mice rejected a secondary sarcoma tumor challenge administered in the opposite flank more than a month later. CD8 CTLs generated by the vaccine approach combined with CD25 Treg cell depletion demonstrated potent sarcoma tumor cell killing in vitro. This could be due to increasing the frequency of effector CTLs following vaccination, and as a result of diminshed or inhibited CD25 Treg cells. When CD8 CTLs were assayed by ELISA for cytokine production the TH1-type cytokine IFN-γ and IL-10 were detected. The sarcoma tumor cell-specific CD8 CTLs were H-2 Kd restricted. In addition, we demonstrated that this combination approach completely prevented lung mets in the 3T3.mD52 sarcoma spontaneous lung metastasis model, compared to control immunized mice or immunized mock Treg depleted mice.Citation77 These data suggest that mD52 protein+ODN in IFA delivered s.c. requires concomitant CD25 Treg cell depletion to prevent sarcoma tumor growth and progression to spontaneous lethal lung metastases.
There are four points to emphasize our findings in this Balb/c sarcoma vaccine combination Treg depletion model for Study 1. First, mD52 protein+ODN vaccine in IFA administered s.c. with CD25 Treg depletion by specific mAb administered i.p. induced 70% protection from mKSA sarcoma primary tumor challenge, as well as protection from secondary mKSA sarcoma tumor challenge administered in the opposite flank more than a month later. Second, mD52 protein+ODN vaccine in combination with CD25 Treg depletion induced complete rejection of spontaneous 3T3.mD52 sarcoma lung metastases. Third, this mD52 protein+ODN vaccine Treg depletion combination elicited D52 specific CD8 CTLs that were H-2 Kd restricted and produced either IFN-γ or IL-10. Finally, We observed no detectable autoimmunity in this vaccine model.
C57BL/6 TRAMP prostate cancer model for mD52 vaccine studies
Prostate cancer vaccine study 1) mD52 DNA-based vaccination
The above vaccine studies in the Balb/c sarcoma model provided the added opportunity to assess vaccination effects on the 3T3.mD52 spontaneous lung mets. To extend our studies to additional mouse strains we employed the C57BL/6 model of prostate cancer. In this first study using this model we used in place of the recombinant protein-based approach a vaccine comprised of mD52 cDNA plasmid DNA-based vaccination strategy, keeping the s.c. route but adding recombinant murine GM-CSF admixed with the DNA in sterile PBS. To test vaccine efficacy we measured the rejection of s.c. tumor growth following vaccination and inoculation with autochthonous transplantable adenocarcinoma of the mouse prostate (TRAMP) prostate cancer cells.Citation79 In the experimental group 70% of mice were tumor free 85 days post challenge, and all of the mice that survived this initial tumor challenge rejected a second tumor challenge given in the opposite flank >150 days later. These mice remained tumor free from all tumor challenges without autoimmunity. Similar to what we observed in the Balb/c sarcoma model vaccine studies, the mD52 DNA immunized prostate cancer survivor mice generated CD8 CTLs that were restricted for the C57BL/6 MHC-I halplotype H-2 Kb. Also similar to what was observed in the Balb/c sarcoma studies above, the CD8 CTLs elicited by vaccination in the C57BL/6 prostate cancer model were comprised of two distinct populations, TH1-type IFN-γ producers and IL-10 producers.Citation76
There are three key points to emphasize our findings in study 1 using the C57BL/6 prostate cancer model. First, mD52 plasmid DNA admixed with rGM-CSF in PBS and injected s.c. induced 70% protection from s.c. prostate tumor cell challenge, and 100% protection from secondary prostate tumor cell challenge. Second, this vaccine approach elicited prostate tumor cell-specific CD8 CTLs that were MHC-I H-2 Kb restricted and comprised two populations of CD8 T cells that produced either IFN-γ or IL-10. Finally, there was no detectable autoimmunity in vaccinated mice.
For the three independent vaccine studies that follow we used this same C57BL/6 prostate cancer model and further assessed the importance of immune effector cell subsets in tumor protection in vivo following TPD52 vaccination to include; CD4 CD25 Tregs, CD4 T cells, CD8 T cells, as well as a role for IL-10. To increase vaccine potency we also explored variations in this DNA-based vaccine strategy to include, using the human TPD52 ×enoantigen (hD52), with intramuscular injection in place of s.c. injection and in separate studies the employment of cytokine deficient animals.
Prostate cancer vaccine study 2) mD52 DNA vaccine combination treg depletion
In this this study we analyzed mD52 DNA, human TPD52 (hD52) alone or in combination as intramuscular (i.m.) administered DNA-based vaccines. Vaccine efficacy was assessed by s.c. challenge with mD52 positive, autochthonous TRAMP tumor cells. Xenogeneic hD52 plasmid DNA-based vaccine injected i.m. in PBS induced 70% tumor protection, by comparison mD52 plasmid DNA injected i.m. induced maximally 50% protection. Any combination of the two DNA vaccines (priming with hD52 and boosting with mD52; or priming with mD52 and boosting with hD52) tested was less effective and inducing protective tumor immunity. Immunized mice that survived initial tumor challenge with prostate tumor cells rejected a secondary tumor challenge with prostate tumor cells given in the opposite flank more than four months after the first challenge. TH1-type CD8 CTL responses that were H-2 Kb restricted were elicited following i.m. vaccination with either mD52 DNA or hD52 DNA. Of note, CD8 CTLs elicited by vaccination were again comprised of two distinct populations, TH1-type IFN-γ producers and IL-10 producers, the latter suggesting induction of a Treg population. To increase potency of mD52 DNA vaccination, we included injection of CD25-specific mAb i.p. concomitantly with vaccine administration to deplete CD25 Tregs in an attempt to augement tumor immunity. However, unlike what we observed for the Balb/c sarcoma model vaccine studies, depleting CD4 CD25 Tregs in combination with mDNA-based vaccination did not increase tumor protection above 50%.Citation78
A common observation with all our vaccine studies is the induction of CD8 IL-10 T cells following vaccination. Thus, we examined whether the CD8 IL-10 T cells expressed the Treg specific FOXP3 transcription factor, adding support to the possibility that they represent CD8 Treg cells that are elicited by D52 vaccination in our study models. Isolated T cells from immunized mice were analyzed with Realtime RT-PCR for expression of FOXP3, compared to controls. Although FOXp3 expression was not detected in these T cells, we did detect IL-10 as well as transcripts encoding proteins associated with T cell mediated killing; perforin and granzyme-b. Absence of FOXP3 expression suggests they may be a unique subset of CD8 IL-10 Treg elicited by TPD52 vaccination.Citation78
There are three key points to emphasize in C57BL/6 prostate cancer vaccine study 2. First, Vaccination with hD52 DNA induced 70% protection from primary tumor challenge, by comparison mD52 DNA vaccine induced maximally 50% protection from primary tumor challenge. All vaccinated mice that were protected from initial tumor challenge were 100% protected from secondary tumor challenge. Second, CD8 T cells generated by vaccination produced either IFN-γ or IL-10, were MHC-I H-2 Kb restricted, but did not express the Treg transcription factor FOXP3. Finally, no autoimmunity was detected in vaccinated animals.
Prostate cancer vaccine study 3 heterologous prime boost vaccination
As stated above for prostate cancer vaccine study 3, CD25 Treg depetion with mD52 DNA vaccination was no better than mD52 DNA vaccination alone. Interestingly, xenogeneic hD52 DNA vaccine was about 20% more effective at inuducing tumor immunity (70% protection) than mD52 DNA vaccine. We also noted the efficacy of recombinant mD52 protein based vaccine approach (70% protection) in the Balb/c sarcoma model study two. These observations led us to ask whether an heterologous hD52 DNA prime-mD52 protein boost vaccination approach might increase efficacy in the C57BL/6 prostate cancer model. To approach this question we immunized C57BL/6 mice with hD52 DNA i.m. every 10 days for 3 injections followed by a single mD52 protein boost 10 days after the final hD52 DNA injection. Approximately 2 weeks after the boost injections we challenged mice with TRAMP prostate tumor cells s.c. On day 126 post primary tumor challenge 80% of the animals were tumor free compared to controls. The majority of protected mice were capable of rejecting a secondary challenge nearly 240 days from the initial immunization and 200 days from the end of the vaccine schedule, and about 3 months after rejecting the primary challenge. To assess the role of effector CD4 and CD8 T cells in tumor protection following vaccination mice were injected with either CD4 or CD8 specific depleting antibody i.p. on the day of each vaccine injection. Immunized animals depleted of CD4 T cells 80% (8/10) failed to reject primary tumor cell challenge. Similarly, 89% (8/9) of mice depleted of CD8 T cells failed to reject primary tumor cell challenge (). When cytokine production of T cells from immunized and tumor protected mice was analyzed by ELISA, only IL-10 and IFN-γ were detected. This directly corroborates our previously reported findings discussed in the vaccine studies above for both tumor models and mouse strains. Interestingly, these are the only two cytokines present in the supernatants of cultured T cells regardless of the D52 vaccine approach utilized to generate the T cells.
Figure 2. Percent of tumor free animals following heterologous prime-boost immunization: male C57Black/6 mice were vaccinated with human TPD52 (hD52-DNA) i.M. For three injections ending with recombinant mouse TPD52 protein (mD52-protein) + ODN s.C. Tumor challenge was administered 14 days later with mD52+ TRAMP-C2 cells. Mice immunized with control vaccines developed tumors (control). A role for CD4 and CD8 T cells was assessed in response to vaccination and tumor challenge by in vivo depletion of the specific T cell subsets: (a) CD4 or (b) CD8 compared to vaccinated and mock depleted mice.
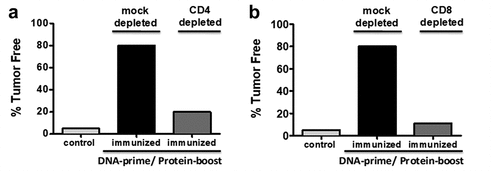
The following four points summarize the important findings from this vaccine approach. First, hD52 DNA prime with mD52 protein boost protected 80% of mice from primary lethal tumor challenge, and the protected from secondary tumor challenge. Second, vaccination along with either CD4 T cell or CD8 T cell depletion eliminated tumor protection demonstrating a critical role for both CD4 T cell help and CD8 CTLs in vaccine induced tumor immunity. Third, specific CD8 CTLs generated by vaccination were H-2 Kb restricted and produced either IFN-γ or IL-10. Finally, no autoimmunity was detected.
Prostate cancer vaccine study 4) mD52 DNA vaccine in IL-10 deficient mice
As we stated above for all our vaccine studies, MHC-I restricted (H-2 Kd for Balb/c mice and H-2 Kb for C57BL/6 mice) effector CD8 CTLs (tumor cell cytotoxic and IFN-γ+) were generated by TPD52 vaccination along with MHC-I restricted CD8 T cells that produce IL-10. We postulated that these IL-10 producing CD8 T cells may have a negative impact on tumor protection. To address this possiblity we vaccinated C57BL/6 IL-10 deficient/knock out(IL-10 KO) mice with mD52 DNA, while vaccinated wild type (wt) mice served as controls for tumor challenge studies. We observed significant delay in tumor growth in vaccinated IL-10 KO mice following primary prostate tumor cell s.c. inoculation. Interestingly, IL-10 KO mice IL-10 had a 100% recall of protective immunity, while wt mice had diminished recall response as measured by secondary prostate tumor cell challenge administered s.c. in the opposite flank. This suggested that IL-10 may play a role in suppressing memory responses following vaccination with TPD52. To explore deeper the role of IL-10 in vaccine induced tumor immunity, mRNA was isolated from bulk T cells or from isolated CD8 T cells generated by mD52 DNA vaccination.
Expression of IL-10 was measured using RT-PCR with specific primers. IL-10 expression was detected in purified CD8 T cells adding further support that CD8 T cells were an active source of IL-10. The detection of IL-10 expression in bulk T cell populations was greatly diminished. Of note, when vaccination was not effective at inhibiting tumor growth, expression of the IL-10 associated transcription factor Blimp-1 was detected by RT-PCR in the same purified CD8 T cells along with detection of IL-10. Together, these data support our hypothesis that CD8 IL-10 T cells elicited by vaccination with the unmutated self-TAA TPD52 may represent a unique CD8 Treg population whose role it is to inhibit TPD52 vaccine induced tumor immunity.Citation80
To summarize this study we offer four key points. First, mD52 DNA vaccination of IL-10 KO mice improved vaccine elicited tumor immunity and augmented cellular memory immune responses demonstrated by rejection of secondary prostate tumor cell challenge compared to controls. Second, mD52 DNA vaccination of IL-10 KO mice elicited tumor specific H-2 Kb restricted effector CTLs with increased tumor cell killing activity in vitro. Third, RT-PCR analyses for IL-10 expression, in CD8 T cells from wt mice induced by mD52 DNA vaccination, demonstrated that a sub-population of CD8 T cells are a key source of vaccine elicited IL-10. Finally, no autoimmunity was detected in vaccinated IL-10 KO mice.
Analysis of CD8 IL10 T cells elicited by mD52 DNA vaccination
As stated above in prostate cancer vaccine study 4, mD52 DNA vaccinated IL-10 KO mice generated a memory immune response against tumor rechallenge compared to w.t. mice, and vaccination with mD52 DNA elicited CD8 IL-10 T cells may represent a distinct regulatory population. To extend these studies in an attempt to better define the vaccine elicited CD8 IL-10 T cell population we administered mD52 DNA vaccination to three distinct categories of C57BL/6 mice: 1) IL-10 KO mice, 2) IFN-γ KO mice, and 3) wild type (wt) mice which served as a control for comparison. Bulk T cells were isolated from all three groups of mD52 DNA vaccinated mice from which mRNA was extracted from purified CD8 T cells for deep RNA sequencing (RNAseq) and RT-PCR expression analysis using specific primers. We identified overexpression of chemokine receptor 8 (CCR8) and inducible co-stimulator (ICOS) in CD8 T cells that produced IL-10 (isolated from vaccinated IFN-γ KO mice) but not in CD8 T cells that produced IFN-γ (isolated from vaccinated IL-10 KO mice). There are reports associating these surface markers with IL-10 producing CD4 T reg cells, adding support to the possibility that mD52 DNA vaccine elicited CD8 IL-10 T cells represent a unique CD8 Treg cell population. As further confirmation of our approachwe demonstrated that mD52 DNA vaccination of IFN-γ KO mice elicited CD8 T cells that make IL-10 only and vaccinated IL-10 KO mice derived CD8 T cells make IFN-γ only, while CD8 T cells from vaccinated w.t. mice contain populations that make either IFN-γ or IL-10. We also observed that expression of ICOS, CTLA-4, PD-1 and PD-L1 on CD8 IL-10 T cells isolated from vaccinated IFN-γ KO mice was measurably different but not statistically significantly different from expression of the same markgers on CD8 IFN-γ T cells isolated from vaccinated IL-10 KO mice. These expression of these markers was confirmed with RT-PCR and were chosen because they represent markers common on activated T cells. RT-PCR confirmed expression of IL-10, CCR8 and the IL-10 transcription factor Blimp-1 differentiated CD8 IL-10 T cells from CD8 INF-γ T cells elicited by mD52 DNA vaccination. The vaccine induced CD8 IL-10 T cells (definable by CCR8, ICOShi) may be specific for TPD52 via pMHC-I peptide epitopes different from those pMHC-I peptide epitopes recognized by vaccine induced effector CD8 CTLs. If so the CD8 IL-10 T cells may be inhibiting vaccine induced tumor immunity by secretion of IL-10, and production of perforin, and granzymes to either suppress or kill effector CD8 CTLs.Citation81 These molecular phenotypic analyses of mD52 DNA vaccine elicited CD8 T cells strengthen our hypothesis that the population of CD8 IL-10 T cells elicited by vaccination likely contribute to the suppression of memory CTL responses and inhibition of vaccine generated tumor immunity.
There are three overarching points that emphasize the results of this study. First, RNAseq analyses identified markers on CD8 IL-10 T cells isolated from vaccinated IFN-γ KO mice that differentiated them from vaccine-induced effector CD8 CTLs isolated from vaccinated IL-10 KO mice and w.t. mice. In addition, multiplex RT-PCR confirmed expression of all common and differentiating T cell markers analyzed for both KO groups of mice and w.t. controls. Second, multiplex RT-PCR confirmed expression of common and unique Treg markers on vaccine induced CD8 IL-10 T cells isolated from vaccinated IFN-γ KO mice compared to CD8 T cells isolated from vaccinated IL-10 KO and w.t. mice. Importantly, differential expression of CCR8 on CD8 IL-10 T cells may be a defining marker for this T cell population. Finally, no autoimmunity was detected in vaccinated mice.
Conclusions
TPD52 tumor overexpression, involvement in transformation, in increased proliferation, in tumorigenesis and metastasis all support its classification as on overexpressed oncogenic TAA. In addition, evidence suggests TPD52 may be useful as prognostic biomarker. This was first and most convincingly demonstrated for human prostate cancer, where it was included in a twelve gene molecular profile of aggressive cancer.Citation82 Our TPD52 preclinical vaccine studies focused on using the entire non-mutated protein or cDNA as vaccine to enable the involvement of multiple potential T cell peptide epitopes restricted by more than one MHC-I and II molecules. We have preliminary data that suggests that human HLA-A2.1 does present epitopes to autologous CD8 T cells, however given that we have not studied and know of no other studies involving additional HLA-A, B, C haplotypes in TPD52-specific CD8 T cell recognition we cannot say for certain if there is a bias for a specific HLA haplotype. We are unaware of studies on the involvement of HLA-DP, DQ, DR and TPD52-specific CD4 T cells. However, we have data demonstrating the critical role of both CD4 and CD8 T cells in TPD52 vaccinated mice and describe above. While we have not yet explored CD4 T cell epitopes, we have ongoing studies that identify MHC-I CD8 T cell epitopes that appear to be responsible for vaccine induced responses we observe. These peptide epitopes are restricted by H-2 Kd and H-2 Kb (preliminary unpublished data).
In this review we demonstrated support for TPD52 as a shared TAA that is widely expressed in numerous human cancers as well as mouse tumor models, is oncogenic and likely indispensable for tumor cell survival and is immunogenic in preclinical studies. Moreover, preclinical vaccine studies against the TPD52 self-protein TAA by our group (reviewed above) and othersCitation83 do not elicit autoimmune side effects yet do elicit anti-tumor immunity.
Our preclinical vaccine data demonstrate efficacy of targeting TPD52 as a preventive vaccine using multiple vaccine formulations in murine models and have characterized the T cell response to include involvement of type I CD4 helper T cells, CD8 cytoxic T cells, classic CD4 CD25 Tregs and the identification and characterization of a TPD52 vaccine elicited unique CD8 Treg population. In addition to targeting classic CD4 Tregs (discussed above) our data suggests that administration of specific monoclonal antibody targeting CCR8 with the combination of TPD52 vaccine therapy may enhance tumor rejectionby inhibiting CCR8 CD8 IL-10 Tregs during an optimum window of efficacy during vaccine administration, thus augmenting productive and effective anti-tumor T cells responses specific for TPD52 while minimizing or avoiding autoimmunity.
Abbreviations
| TPD52 | = | tumor protein D52 |
| mD52 | = | murine D52 |
| hD52 | = | human D52 |
| IL-10 | = | interleukin 10 |
| IFN-γ | = | interferon gamma |
| Treg | = | T regulatory cell |
| ICOS | = | inducible co-stimulatory |
| CCR8 | = | chemokine receptor 8 |
| wt | = | wild type |
| im | = | intra muscular |
| sc | = | subcutaneous |
| KO | = | knock out (gene deficient mice) |
| CTL | = | cytotoxic T lymphocyte |
Acknowledgments
This work was supported by Funds from the TTUHSC School of Medicine. We would like to thank those who specifically contributed to this work over the years: JD (Lewis) Bright, LA (Payton) Sullivan, CR Elizondo, JA Byrne, HN Shultz, JF Aldrich, JG Whitford and K Korac. We thank BioRender.com for use of their figure software, the image for was generated using Biorender.com with permission.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- American Cancer Society. Cancer facts & figures 2023. Atlanta: American Cancer Society; 2023.
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy come of age. Nature. 2011;480(7378):480–11. doi:10.1038/nature10673.
- Couzin-Frankel J. Cancer immunotherapy. Sci. 2013;342(6165):1432–3. doi:10.1126/science.342.6165.1432.
- Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–7. doi:10.1200/JCO.2012.41.6750.
- Lewis JD, Reilly BD, Bright RK. Tumor-associated antigens: from discovery to immunity. Int Rev Immunol. 2003;22(2):81–112. doi:10.1080/08830180305221.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Sci. 2015;348(6230):69–74. doi:10.1126/science.aaa4971.
- Schumacher TN, Scheper W, Kvistborg P. Cancer neoantigens. Annu Rev Immunol. 2019;37(1):173–200. doi:10.1146/annurev-immunol-042617-053402.
- Bright RK, Bright JD, Byrne JA. Overexpressed oncogenic tumor-self antigens: new vaccine targets. Hum Vaccin Immunother. 2014;10(11):3297–305. doi:10.4161/hv.29475.
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15(17):5323–37. doi:10.1158/1078-0432.CCR-09-0737.
- Augello MA, Den RB, Knudsen KE. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev. 2014;33(2–3):399–411. doi:10.1007/s10555-013-9471-3.
- McNamara KM, Yoda T, Nurani AM, Shibahara Y, Miki Y, Wang L, Nakamura Y, Suzuki K, Yang Y, Abe E, et al. Androgenic pathways in the progression of triple-negative breast carcinoma: a comparison between aggressive and non-aggressive subtypes. Breast Cancer Res Treat. 2014;145(2):281–93. doi:10.1007/s10549-014-2942-6.
- Olson BM, Johnson LE, McNeel DG. The androgen receptor: a biologically relevant vaccine target for the treatment of prostate cancer. Cancer Immunol Immunother. 2013;62(3):585–96. doi:10.1007/s00262-012-1363-9.
- Wiedman U, Davis AB, Zielinski CC. Vaccination for the prevention and treatment of breast cancer with special focus on Her-2/neu peptide vaccines. Breast Cancer Res Treat. 2013;138(1):1–12. doi:10.1007/s10549-013-2410-8.
- Baxevanis CN, Voutsas IF, Gritzapis AD, Perez SA, Papamichail M. Her-2/neu as a target for cancer vaccines. Immuotherapy. 2010;2(2):213–26. doi:10.2217/imt.09.89.
- Tobias J, Garner-Spitzer E, Drinić M, Wiedermann U. Vaccination against Her-2/neu, with focus on peptide-based vaccines. ESMO Open. 2022;7(1):100361. doi:10.1016/j.esmoop.2021.100361.
- Leffers N, Vermeij R, Hoogeboom BN, Schulze UR, Wolf R, Hamming IE, van der Zee AG, Melief KJ, van der Burg SH, Daemen T, et al. Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int J Cancer. 2012;130(1):10–12. doi:10.1002/ijc.25980.
- Rahma OE, Ashtar E, Czystowska M, Szajnik ME, Wieckowski E, Bernstein S, Herrin VE, Shams MA, Steinberg SM, Merino M, et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol Immunother. 2012;61(3):373–84. doi:10.1007/s00262-011-1100-9.
- Zeestraten EC, Speetjens FM, Welters MJ, Saadatmand S, Stynenbosch LF, Jongen R, Kapiteijn E, Gelderblom H, Nijman HW, Valentijn AR, et al. Addition of interferon-α to the p53-SLP® vaccine results in increased production of interferon-γ in vaccinated colorectal cancerpatients: a phase I/II clinical trial. Int J Cancer. 2013;132(7):1581–91. doi:10.1002/ijc.27819.
- Hall RD, Gray JE, Chiappori AA. Beyond the standard of care: a review of novel immunotherapy trials for the treatment of lung cancer. Cancer Control. 2013;20(1):22–31. doi:10.1177/107327481302000105.
- Zhou S, Fan C, Zeng Z, Young KH, Li Y. Clinical and immunological effects of p53-targeting vaccines. Front Cell Dev Biol. 2021;9:762796. doi:10.3389/fcell.2021.762796.
- Chiappori AA, Williams CC, Gray JE, Tanvetyanon T, Haura EB, Creelan BC, Thapa R, Chen DT, Simon GR, Bepler G, et al. Randomized-controlled phase II trial of salvage chemotherapy after immunization with a TP53-transfected dendritic cell-based vaccine (Ad.P53-DC) in patients with recurrent small cell lung cancer. Cancer Immunol Immunother. 2019;68(3):517–27. doi:10.1007/s00262-018-2287-9.
- Carlsen L, Zhang S, Tian X, De La Cruz A, George A, Arnoff TE, El-Deiry WS. The role of p53 in anti-tumor immunity and response to immunotherapy. Front Mol Biosci. 2023;10:1–13. doi:10.3389/fmolb.2023.1148389.
- Park JE, Alexander IS, Zhou R. Roles of EphA2 in development and disease. Genes. 2013;4(3):334–57. doi:10.3390/genes4030334.
- Yeung JT, Hamilton RL, Okada H, Jakacki RI, Pollack IF. Increased expression of tumor-associated antigens in pediatric and adult ependymomas: implication for vaccine therapy. J Neurooncol. 2013;111(2):103–11. doi:10.1007/s11060-012-0998-x.
- Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, Mintz AH, Engh JA, Bartlett DL, Brown CK, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–6. doi:10.1200/JCO.2010.30.7744.
- Ladomersky E, Genet M, Zhai L, Gritsina G, Lauing KL, Lulla RR, Fangusaro J, Lenzen A, Kumthekar P, Raizer JJ, et al. Improving vaccine efficacy against malignant glioma. Oncoimmunology. 2016;5(8):e1196311. doi:10.1080/2162402X.2016.1196311.
- Beatty GL, Vonderheide RH. Telomerase as a universal tumor antigen for cancer vaccines. Expert Rev Vaccines. 2008;7(7):881–7. doi:10.1586/14760584.7.7.881.
- Carpenter EL, Vonderheide RH. Telomerase-based immunotherapy of cancer. Expert Opin Biol Ther. 2006;6(10):1031–9. doi:10.1517/14712598.6.10.1031.
- Vonderheide RH, Domchek SM, Schultze JL, George DJ, Hoar KM, Chen DY, Stephans KF, Masutomi K, Loda M, Xia Z, et al. Vaccination of cancer patients against telomerase induces functional antitumor CD8+ T lymphocytes. Clin Cancer Res. 2004;10(3):828–39. doi:10.1158/1078-0432.CCR-0620-3.
- Brunsvig PF, Kyte JA, Kersten C, Sundstrøm S, Møller M, Nyakas M, Hansen GL, Gaudernack G, Aamdal S. Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Ca Res. 2011;17(21):6847–57. doi:10.1158/1078-0432.CCR-11-1385.
- Vetsika EK, Konsolakis G, Aggouraki D, Kotsakis A, Papadimitraki E, Christou S, Menez-Jamet J, Kosmatopoulos K, Georgoulias V, Mavroudis D. Immunological responses in cancer patients after vaccination with the therapeutic telomerase-specific vaccine Vx-001. Cancer Immunol Immunother. 2012;61(2):157–68. doi:10.1007/s00262-011-1093-4.
- Ellingsen EB, Mangsbo SM, Hovig E, Gaudernack G. Telomerase as a target for therapeutic cancer vaccines and considerations for optimizing their clinical potential. Front Immunol. 2021;12:682492. doi:10.3389/fimmu.2021.682492.
- Stauber RH, Mann W, Knauer SK. Nuclear and cytoplasmic survivin: molecular mechanism, prognostic, and therapeutic potential. Cancer Res. 2007;67(13):5999–6002. doi:10.1158/0008-5472.CAN-07-0494.
- Andersen MH, Svane IM, Becker JC, Sraten PT. The universal character of the tumor-associated antigen survivin. Clinical Cancer Research. 2007;13(20):5991–4. doi:10.1158/1078-0432.CCR-07-0686.
- Kameshima H, Tsuruma T, Kutomi G, Shima H, Iwayama Y, Kimura Y, Imamura M, Torigoe T, Takahashi A, Hirohashi Y, et al. Immunotherapeutic benefit of α-interferon (IFNα) in survivin2B-derived peptide vaccination for advanced pancreatic cancer patients. Cancer Sci. 2013;104(1):124–9. doi:10.1111/cas.12046.
- Filipazzi P1, Pilla L, Mariani L, Patuzzo R, Castelli C, Camisaschi C, Maurichi A, Cova A, Rigamonti G, Giardino F, et al. Limited induction of tumor cross-reactive T cells without a measurable clinical benefit in early melanoma patients vaccinated with human leukocyte antigen class I–modified peptides. Clinical Cancer Research. 2012;18(23):6485–96. doi:10.1158/1078-0432.CCR-12-1516.
- Becker JC1, Andersen MH, Hofmeister-Müller V, Wobser M, Frey L, Sandig C, Walter S, Singh-Jasuja H, Kämpgen E, Opitz A, et al. Survivin-specific T-cell reactivity correlates with tumor response and patient survival: a phase-II peptide vaccination trial in metastatic melanoma. Cancer Immunol Immunother. 2012;61(11):2091–103. doi:10.1007/s00262-012-1266-9.
- Lee J, Uy BR, Liau LM. Brain tumor vaccines. Neurosurg Clin N Am. 2021;32(2):225–34. doi:10.1016/j.nec.2021.01.003.
- Byrne JA, Frost S, Chen Y, Bright RK. Tumor protein D52 (TPD52) and cancer—oncogene understudy or understudied oncogene? Tumor Biol. 2014;35(8):7369–82. doi:10.1007/s13277-014-2006-x.
- Balleine RL, Schoenberg Fejzo M, Sathasivam P, Basset P, Clarke CL, Byrne JA. The D52 (TPD52) gene is a candidate target gene for events resulting in increased 8q21 copy number in human breast carcinoma. Genes Chromosom Cancer. 2000;29(1):48–57. doi:10.1002/1098-2264(2000)9999:9999<:AID-GCC1005>3.0.CO;2-O.
- Byrne JA, Tomasetto C, Garnier JM, Rouyer N, Mattei MG, Bellocq JP, Rio MC, Basset PA. A screening method to identify genes commonly overexpressed in carcinomas and the identification of a novel complementary DNA sequence. Cancer Res. 1995;55:2896–903.
- Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Børresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002;99(20):12963–8. doi:10.1073/pnas.162471999.
- Wang R, Xu J, Saramaki O, Visakorpi T, Sutherland WM, Zhou J, Sen B, Lim SD, Mabjeesh N, Amin M, et al. PrLZ, a novel prostate-specific and androgen-responsive gene of the TPD52 family, amplified in chromosome 8q21.1 and overexpressed in human prostate cancer. Cancer Research. 2004;64(5):1589–94. doi:10.1158/0008-5472.CAN-03-3331.
- Wang R, Xu J, Mabjeesh N, Zhu G, Zhou J, Amin M, He D, Marshall FF, Zhau HE, Chung LW. PrLZ is expressed in normal prostate development and in human prostate cancer progression. Clin Cancer Res. 2007;13(20):6040–8. doi:10.1158/1078-0432.CCR-07-0640.
- Rubin MA, Varambally S, Beroukhim R, Tomlins SA, Rhodes DR, Paris PL, Hofer MD, Storz-Schweizer M, Kuefer R, Fletcher JA, et al. Overexpression, amplification, and androgen regulation of TPD52 in prostate cancer. Cancer Res. 2004;64(11):3814–22. doi:10.1158/0008-5472.CAN-03-3881.
- Byrne JA, Balleine RL, Schoenberg Fejzo M, Mercieca J, Chiew YE, Livnat Y, St Heaps L, Peters GB, Byth K, Karlan BY, et al. Tumor protein D52 (TPD52) is overexpressed and a gene amplification target in ovarian cancer. Int J Cancer. 2005;117(6):1049–54. doi:10.1002/ijc.21250.
- Largo C, Alvarez S, Saez B, Blesa D, Martin-Subero JI, González-García I, Brieva JA, Dopazo J, Siebert R, Calasanz MJ, et al. Identification of overexpressed genes in frequently gained/amplified chromosome regions in multiple myeloma. Haematologica. 2006;91:184–91.
- Tiacci E, Orvietani PL, Bigerna B, Pucciarini A, Corthals GL, Pettirossi V, Martelli MP, Liso A, Benedetti R, Pacini R, et al. Tumor protein D52 (TPD52): a novel B-cell/plasma-cell molecule with unique expression pattern and Ca(2+)-dependent association with annexin VI. Blood. 2005;105:2812–20. doi:10.1182/blood-2004-07-2630.
- Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, et al. Molecular diagnosis of Burkitts lymphoma. N Engl J Med. 2006;354(23):2431–42. doi:10.1056/NEJMoa055759.
- Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354(23):2419–30. doi:10.1056/NEJMoa055351.
- Loukopoulos P, Shibata T, Katoh H, Kokubu A, Sakamoto M, Yamazaki K, Kosuge T, Kanai Y, Hosoda F, Imoto I, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98(3):392–400. doi:10.1111/j.1349-7006.2007.00395.x.
- Skotheim RI, Autio R, Lind GE, Kraggerud SM, Andrews PW, Monni O, Kallioniemi O, Lothe RA. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol. 2006;28(5–6):315–26. doi:10.1155/2006/219786.
- Korkola JE, Heck S, Olshen AB, Reuter VE, Bosl GJ, Houldsworth J, Chaganti RS. In vivo differentiation and genomic evolution in adult male germ cell tumors. Genes Chromosomes Cancer. 2008;47(1):43–55. doi:10.1002/gcc.20504.
- McIntyre A, Summersgill B, Lu YJ, Missiaglia E, Kitazawa S, Oosterhuis JW, Looijenga LH, Shipley J. Genomic copy number and expression patterns in testicular germ cell tumours. Br J Cancer. 2007;97(12):1707–12. doi:10.1038/sj.bjc.6604079.
- Hoek KS. DNA microarray analyses of melanoma gene expression: a decade in the mines. Pigment Cell Res. 2007;20(6):466–84. doi:10.1111/j.1600-0749.2007.00412.x.
- Roesch A, Becker B, Bentink S, Spang R, Vogl A, Hagen I, Landthaler M, Vogt T. Ataxia telangiectasia-mutated gene is a possible biomarker for discrimination of infiltrative deep penetrating nevi and metastatic vertical growth phase melanoma. Cancer Epidemiology, Biomarkers & Prevention. 2007;11(11):2486–90. doi:10.1158/1055-9965.EPI-07-0224.
- Byrne JA, Chen Y, Martin La Rotta N, Peters GB. Challenges in identifying candidate amplification targets in human cancers: chromosome 8q21 as a case study. Genes Cancer. 2012;3:87–101. doi:10.1177/1947601912456287.
- Wilson KS, Roberts H, Leck R, Harris AL, Geradts J. Differential gene expression patterns in Her-2/neu-positive and -negative breast cancer cell lines and tissues. The American Journal Of Pathology. 2002;161(4):1171–85. doi:10.1016/S0002-9440(10)64394-5.
- Landis MD, Seachrist DD, Abdul-Karim FW, Keri RA. Sustained trophism of the mammary gland is sufficient to accelerate and synchronize development of ErB2/Neu-induced tumors. Oncogene. 2006;25(23):3325–34. doi:10.1038/sj.onc.1209365.
- Chen H, Pimienta G, Gu Y, Sun X, Hu J, Kim M-S, Chaerkady R, Gucek M, Cole RN, Sukumar S, et al. Proteomic characterization of Her2/neu-overexpressing breast cancer cell lines. Proteomics. 2010;10(21):3800–10. doi:10.1002/pmic.201000297.
- Shehata M, Bieche I, Boutros R, Weidenhofer J, Fanayan S, Spalding L, Zeps N, Byth K, Bright RK, Lidereau R, et al. Non-redundant functions for tumor protein D52-like proteins support specific targeting of TPD52. Clin Cancer Res. 2008;14(16):5050–60. doi:10.1158/1078-0432.CCR-07-4994.
- Roslan N, Bieche I, Bright RK, Lidereau R, Chen Y, Byrne JA. TPD52 represents a survival factor in ERBB2-amplified breast cancer cells. Mol Carcinog. 2014;53(10):807–19. doi:10.1002/mc.22038.
- Shehata M, Weidenhofer J, Thamotharampillai K, Hardy JR, Byrne JA. Tumor protein D52 overexpression and gene amplification in cancers from a mosaic of microarrays. Crit Rev Oncol. 2008;14(1):33–5. doi:10.1615/CritRevOncog.v14.i1.30.
- Haaland CM, Heaphy CM, Butler KS, Fischer EG, Griffith JK, Bisoffi M. Differential gene expression in tumor adjacent histologically normal prostatic tissue indicates field cancerization. Int J Oncol. 2009;35:537–46. doi:10.3892/ijo_00000365.
- Bright RK, Lewis JD. Long-term culture of normal and malignant human prostate epithelial cells. In: Pfragner R Frehsney R, editors. Culture of specialized cells, culture of human tumor cells. Hoboken, NJ: John Wiley & Sons, Inc; 2003. pp. 125–44.
- Bright RK, Vocke CD, Emmert-Buck MR, Duray PH, Solomon D, Fetsch P, Rhim JS, Linehan WM, Topalian SL. Generation and genetic characterization of immortal human prostate epithelial cell lines derived from primary cancer specimens. Cancer Res. 1997;57:995–1002.
- Liu W, Xie CC, Thomas CY, Kim ST, Lindberg J, Egevad L, Wang Z, Zhang Z, Sun J, Sun J, et al. Genetic markers associated with early cancer-specific mortality following prostatectomy. Cancer. 2013;119(13):2405–12. doi:10.1002/cncr.27954.
- Virgin JB, Hurley PM, Nahhas F, Bebchuk KG, Mohamed AN, Sakr WA, Bright RK, Cher ML. Isochromosome 8q formation is associated with 8p loss of heterozygosity in a prostate cancer cell line. Prostate. 1999;41(1):49–57. doi:10.1002/(SICI)1097-0045(19990915)41:1<49:AID-PROS7>3.0.CO;2-8.
- Bismar TA, Demichelis F, Riva A, Kim R, Varambally S, He L, Kutok J, Aster JC, Tang J, Kuefer R, et al. Defining aggressive prostate cancer using a 12-gene model. Neoplasia. 2006;8(1):59–68. doi:10.1593/neo.05664.
- Byrne JA, Mattei MG, Basset P. Definition of the D52 gene/protein family through cloning of D52 homologues in human (hD53) and mouse (mD52). Genomics. 1996;35(3):523–32. doi:10.1006/geno.1996.0393.
- Lewis JD, Payton LA, Whitford JG, Byrne JA, Smith DI, Yang L, Bright RK. Induction of tumorigenesis and metastasis by the murine orthologue of tumor protein D52. Mol Cancer Res. 2007;5(2):133–44. doi:10.1158/1541-7786.MCR-06-0245.
- Bright RK. Cancer vaccines: the next generation immunotherapy. Int J Vaccine Immun. 2015;2(1):1–2. doi:10.16966/2470-9948.105.
- Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol. 2012;30(1):95–114. doi:10.1146/annurev-immunol-020711-075035.
- Bright RK, Mamula MJ. Regulation of adaptive immune responses to self-antigens in cancer and autoimmunity. Curr Trends Immunol. 2014;15:47–57.
- Payton LA, Lewis JD, Byrne JA, Bright RK. Vaccination with the metastasis-related tumor associated antigen TPD52 and CpG/ODN induces protective tumor immunity. Cancer Immunol Immunother. 2008;57(6):799–811. doi:10.1007/s00262-007-0416-y.
- Lewis JD, Sullivan LA, Byrne JA, DeRiese W, Bright RK. Memory and cellular immunity induced by a DNA vaccine encoding self antigen TPD52 administered with soluble GM-CSF. Cancer Immunol Immunother. 2009;58(8):1337–49. doi:10.1007/s00262-009-0659-x.
- Bright JD, Schultz HN, Byrne JA, Bright RK. Injection site and regulatory T cells influence durable vaccine-induced tumor immunity to an over-expressed self tumor associated antigen. OncoImmunol. 2013;2(7):e25049-1–11. doi:10.4161/onci.25049.
- Bright JD, Aldrich JF, Byrne JA, Bright RK. Vaccination with the prostate cancer over-expressed tumor-self protein TPD52 elicits protective tumor immunity and a potentially unique subset of CD8+ T cells. Au J Clin Immunol. 2014;1:1–13. http://www.austinpublishinggroup.com/clinical-immunology/fulltext/ajci-v1-id1007.php.
- Gingrich JR, Barrios RJ, Morton RA, Boyce BF, DeMayo FJ, Finegold MJ, Angelopoulou R, Rosen JM, Greenberg NM. Metastatic prostate cancer in a transgenic mouse. Cancer Res. 1996;56:4096–102.
- Elizondo CR, Bright JD, Byrne JA, Bright RK. Analysis of the CD8+ IL-10+ T cell response elicited by vaccination with the oncogenic tumor-self protein D52. Hum Vaccin Immunother. 2020;16(6):1413–23. doi:10.1080/21645515.2019.1689746.
- Elizondo CR, Bright JD, Bright RK. Vaccination with a shared oncogenic tumor-self antigen elicits a population of CD8+ T cells with a regulatory phenotype. Hum Vaccin Immunother. 2022;18(6):e2108656-1–14. doi:10.1080/21645515.2022.2108656.
- Bismar TA, Demichelis F, Riva A, Kim R, Varambally S, He L, Kutok J, Aster JC, Tang J, Kuefer R, et al. Defining aggressive prostate cancer using a 12-gene model. Neoplasia. 2006;8(1):59–68. doi:10.1593/neo.05664.
- Mirshahidi S, Kramer VG, Whitney JB, Essono S, Lee S, Dranoff G, Anderson KS, Ruprecht RM. Overlapping synthetic peptides encoding TPD52 as breast cancer vaccine in mice: prolonged survival. Vaccine. 2009 Mar 13;27(12):1825–33. doi:10.1016/j.vaccine.2009.01.089.