
ABSTRACT
To overcome challenges associated with adoptive cell therapy (ACT), we developed a personalized autologous T-cell therapy program. Patients with advanced cancer with HLA-A *02:01 allele and tumor expression of PRAME, MAGEA1, MAGEA4, MAGEA8, NY-ESO-1, COL6A3 exon 6, MXRA5, and/or MMP1 underwent leukapheresis and T-cell product manufacturing. Patients received lymphodepletion, IMA101 infusion and interleukin 2 for 14 days. Of 214 screened patients, 14 were treated (6, IMA101; 8, IMA101 and atezolizumab). The most common adverse events were cytokine release syndrome (G1, n = 6; G2, n = 4) and cytopenia. At 6 weeks, 12 (85.7%) patients had stable disease. Three patients had prolonged disease stabilization for 12.9, 7.3, and 13.7 months, respectively. The median progression-free survival and overall survival were 3.4 months and 9.4 months, respectively. Target-specific T cells expanded to constitute up to 78.7% of CD8+ cells. In conclusion, IMA101 was feasible and well tolerated, leveraging the potential of multi-targeted ACT that warrants further investigation.
Introduction
Adoptive cell therapy (ACT) is a treatment strategy that uses genetically modified human lymphocytes to fight diseases. It is currently being studied for its potential to treat hematologic malignancies and solid tumors, and it is already approved for the treatment of several hematologic malignancies.Citation1 The process involves using T cells that are derived from either the patient or a donor, activating them outside of the body, and then infusing them into the patient along with other agents to help them fight the cancer. ACT has the potential to be more effective than traditional immunotherapies and vaccines because it allows for the selection and expansion of T cells that specifically target cancer cells.
There are two primary methods being used to develop ACT. One involves introducing an artificial receptor called a chimeric antigen receptor (CAR) into immune cells to help them recognize and target cancer cells. The other method involves using T-cell receptor (TCR)-engineered cells that can identify tumor-specific epitopes presented by major histocompatibility complex (MHC) molecules on the tumor cell surface.
Early studies have focused on targeting lineage-specific antigens such as gp100 or Melan-A/MART-1. However, these antigens may also be present in normal tissues; therefore, other studies have focused on using well-established cancer germline antigens like MAGE-A3 and NY-ESO-1, which are only present in tumors.Citation2,Citation3 ACT is a promising new approach to cancer treatment that may provide better outcomes for patients.
Although ACT has demonstrated substantial clinical activity in hematologic cancers, only a small proportion of patients with solid tumors have benefited from these advances due to the lack of suitable immunotherapy targets with high specificity in solid tumors and the high frequency of relapse following immunotherapy to single targets, which is often associated with loss of target expression in the tumor.Citation1 To overcome these challenges, we conducted a clinical trial of IMA101 (NCT02876510), an ACT that uses autologous T-cell products against multiple defined tumor-associated, peptide-human leukocyte antigen (pHLA) targets. T-cell products were infused after administration of an optimized lymphodepleting regimen. Low-dose interleukin 2 (IL-2) was also given, and the study had an additional cohort in which a checkpoint inhibitor was added to the above treatments. In this article, we discuss our experience with the development of autologous T-cell products and their specific characteristics as a therapy for patients with advanced metastatic cancer, as recently publishedCitation1 (NCT02876510). We will also share the lessons we have learned, as well as the challenges and opportunities that come with this approach.
Scientific rationale
The scientific rationale for developing IMA101 was based on previous studies that demonstrated clinical benefit and a favorable toxicity profile in patients with melanoma and leukemia who had undergone endogenous ACT.Citation4–7 Specifically, in a phase I trial, the safety, effectiveness, and persistence of CD8+ T cell clones that targeted MART1/MelanA and gp100 were investigated in patients with metastatic melanoma.Citation4 This treatment was associated with in vivo persistence, migration, and antitumor effect of transferred T cells.Citation4 Ten patients were treated with four autologous T-cell clone infusions each, with the initial infusion lacking IL-2 and the following infusions containing low-dose IL-2. The adopted T-cell clones survived and reacted to the low IL-2 levels. Disease stabilization was noted in 8 of 10 patients, lasting up to 21 months.Citation4 In another study, the use of autologous, non-engineered NY-ESO-1-specific CD4+ T cells in metastatic melanoma was associated with lasting clinical remissions and activated natural responses against other melanoma antigens.Citation5 Following the infusion, the clone cells proliferated and were detectable in patients’ blood for >80 days.Citation5 In another study, 11 patients with metastatic melanoma received cyclophosphamide followed by an infusion of antigen-specific CD8+ cytotoxic T-lymphocyte clones and IL-2.Citation6 One patient had a complete response for >3 years, and five patients had disease stabilization at 8 weeks. Treatment was well tolerated. The melanoma-specific CD8+ T cells persisted, mediated tumor regression, and acquired a central memory phenotype, thus allowing for sustained clinical benefit.Citation6 Investigators have published results of other studies in which they utilized donor-derived CD8+ cytotoxic T-cell clones that targeted WT1.Citation7 These clones were specific to HLA A × 02:01 and were used to treat 11 patients who either had relapsed leukemia or were at high risk of leukemia after receiving allogeneic hematopoietic cell transplantation. To increase the survival of cytotoxic T-lymphocytes (CTLs) in vivo, the last four patients were given CTL clones that were exposed to IL-21 during their generation to restrict the terminal differentiation of antigen-specific T cells produced in vitro. One patient had a transient response and another with minimal residual disease achieved prolonged remission.Citation7
These encouraging results prompted us to design and initiate a clinical trial of IMA101 autologous T-cell therapy for patients with advanced cancer.Citation1 In our study, the use of antigen-specific T cells against predefined targets (PRAME, MAGEA1, MAGEA4, MAGEA8, NY-ESO-1, COL6A3 exon 6, MXRA5, and/or MMP1) was intended to overcome the limitations associated with ACT in solid tumors by addressing multiple novel relevant tumor antigens per patient. Specifically, autologous T-cell products were manufactured against the most relevant tumor target peptides for individual patients whose tumors expressed any of the eight aforementioned targets.
Endpoints
The primary objective of the study was to assess safety and tolerability, and the secondary objectives were to evaluate the in vivo persistence of transferred T cells and to assess tumor response. The exploratory endpoints included progression-free survival (PFS), overall survival (OS), feasibility, and assessment of biomarkers.
Biomarker characterization
Prior to all procedures, patients signed an informed consent form, stating that they were aware of the investigational nature of the study. Patients had HLA typing, and those with HLA-A *02:01 positivity underwent a tumor biopsy and were screened for the following tumor target antigen peptides: PRAME, MAGEA1, MAGEA4, MAGEA8, NY-ESO-1, COL6A3 exon 6, MXRA5, and MMP1. These peptides are expressed in tumors and have very limited, if any, expression in normal tissues. Patients with expression of any of the above biomarkers underwent leukapheresis. Subsequently, the antigen-specific T cells were primed, expanded, and sorted in vitro with IL-21. Details of the cell manufacturing process were previously published.Citation8 A product of T cells with a less terminally differentiated phenotype was produced after approximately 9 weeks, and it was cryopreserved until patients met the eligibility criteria for treatment. During the T-cell product manufacturing period, patients received “bridging” therapy if deemed necessary by their physician.
When the eligibility criteria for treatment were met, patients were admitted to a specialized unit to receive a lymphodepleting regimen, followed by infusion of up to four T-cell products and low-dose IL-2 administered twice daily subcutaneously for 14 days. The lymphodepleting regimen consisted of fludarabine (FLU) and cyclophosphamide (CY), and it was optimized in 2016 at MD Anderson on the basis of mechanistic studies of drug interactions performed by Valdez and Andersson.Citation9 IMA101 was investigated as a monotherapy (Cohort 1) or combined with atezolizumab (anti – PD-L1; Cohort 2). The rationale for combining FLU and CY is based on data showing that lymphodepleting regimens promote the successful engraftment and continued presence of T-cells.Citation10–12 Τhe checkpoint inhibitor was added to improve T-cell therapy by triggering antigen spreading and maintaining transferred T cells with a central memory phenotype, leading to positive clinical outcomes.Citation13
FLU and CY were administered from day −6 to day −3.Citation14 FLU was given as a 1-hour intravenous (IV) infusion of 40 mg/m2; this was followed by a 2-hour IV infusion of 500 mg/m2 CY 1 hour later. On day 0, patients received the IMA101 products (up to 1.75 × 1010 cells per product, not exceeding 4 × 1010 cells in total). Subsequently, the study participants were given subcutaneous IL-2 treatment of 1.0 × 106 IU twice daily for 14 consecutive days, which amounted to a total of 28 doses. This treatment was initiated within 6 hours of receiving the initial IMA101 treatment. The patients in Cohort 1 were given only IMA101, and once its safety was established, Cohort 2 was initiated, adding atezolizumab (Tecentriq, Genentech, F. Hoffman-La Roche) to IMA101. Atezolizumab was administered intravenously at a dosage of 1,200 mg over a period of 30–60 minutes, starting 3 weeks after Τ cell infusion (after hematologic recovery) and then every 3 weeks for up to a year. The patients remained on the trial until they experienced disease progression or unacceptable toxicity, withdrew their consent, or discontinued the treatment at the discretion of their physician. Standard institutional guidelines were followed for prophylaxis against bacterial, viral, and fungal infections, and Mesna was given as renal prophylaxis to prevent toxicity from cyclophosphamide. Growth colony stimulating factors were administered starting on day 5 after T-cell infusion.
To assess the efficacy of the treatment, all patients underwent computed tomography (CT)/magnetic resonance imaging (ΜRI) studies, including brain imaging, at baseline and approximately every 6 weeks. The National Cancer Institute Common Terminology Criteria of Adverse Events version 5.0 were used to monitor the patients for any toxicity, while cytokine release syndrome (CRS)Citation15,Citation16 and immune effector cell-associated neurotoxicity syndrome were assessed using standard institutional CARTOX guidelines.Citation17
Results
Demographics
From July 2017 to January 2020, we screened 214 patients with advanced metastatic cancer; 99 (46.3%) patients were HLA-A *02:01 positive, and of those 61 (28.5%) had a tumor biopsy (). Overall, 57 products were released for 36 patients (16.8%). The remaining products were not released because the peripheral blood mononuclear cells lacked antigen-specific T-cell precursors (52.5%), did not fulfill the release conditions (42.5%), or were contaminated (5.0%). A total of 15 patients underwent lymphodepletion; among them, 14 received T-cell products. Their baseline characteristics are shown in . Regarding the 14 treated patients, their median age was 43 years (range, 20–63 years), 12 (85.71%) patients were women, and they were heavily treated and had received a median of 6 prior therapies (range, 3–12). Bridging therapy was administered to all patients in the study because of the prolonged time required to manufacture the T-cell products.
Figure 1. Workflow of patient screening and treatment with IMA101 autologous engineered T cell receptor therapy. This flowchart illustrates the stepwise process involved in screening patients for IMA101 T-cell therapy. Of 214 patients who started screening for the study and underwent HLA typing, 14 patients received autologous T cell receptor therapy. Twelve of these 14 patients had stable disease at 6 weeks. The patient’s journey on the trial encompasses HLA typing, tumor biopsy, biomarker profiling, leukapheresis, product preparation, and finally, lymphodepletion and infusion of T cell therapy for eligible patients, showcasing the steps from screening to therapeutic efficacy assessment using RECIST 1.1. criteria.
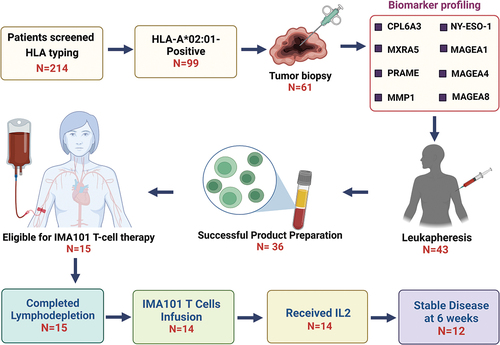
Table 1. Demographic and clinical profile of 14 patients treated with IMA101.
Treatment
The median number of T-cell products administered was two (range, 1–3). The median dose of target-specific T cells was 1.19 × 1010 (range, 0.1 × 1010 to 3.02 × 1010). Cohort 1 included four treated patients, and Cohort 2 was planned to include 10 patients, but two patients in Cohort 2 did not receive atezolizumab because they did not meet the criteria for treatment. Twelve patients received 28 doses of IL-2. Οne patient who underwent lymphodepletion did not receive ΙΜΑ101 because the T-cell product was damaged during thawing.
Safety
The most common treatment-emergent adverse events (TEAEs) were Grade 1–2 CRS and cytopenia.Citation8 No immune effector cell-associated neurotoxicity syndrome was noted. T-cell products were administered to 14 patients, among whom 10 (71.4%) developed CRS. Six patients encountered Grade 1 CRS, whereas four patients had Grade 2 CRS. All patients exhibited Grade 4 lymphopenia, with 13 patients encountering Grade 4 lymphopenia, and one patient experiencing Grade 3 neutropenia, which persisted for a duration of 1–2 weeks following T-cell infusion. Notably, 50% of the patients (n = 7) developed Grade 3–4 thrombocytopenia. Infectious complications were noted in 35.71% (n = 5) of patients, which included Grade 3 bacteremia in two patients, port catheter-related infections in two patients, and Grade 3 appendicitis in one patient. Additionally, some patients experienced adverse events such as nausea, vomiting, and anorexia. One patient experienced Grade 3 sinus bradycardia on Day 9 after T-cell therapy, following which IL2 treatment was discontinued. The patient’s heart rate normalized on day 15. Notably, his cardiac workup was normal, and testing for hemochromatosis revealed that he was a carrier of an H63D hemochromatosis gene mutation. Another patient encountered Grade 3 QTc interval prolongation on Day 100 after T-cell therapy, which was associated with hypokalemia. Grade 3 orthostatic hypotension was noted in another patient.
Response
Fourteen patients were evaluable for response. Six weeks after T-cell infusion, 12 (85.7%) of 14 patients had stable disease by RECIST 1.1. Three patients had prolonged disease stabilization for 12.9, 7.3, and 13.7 months, respectively. An additional patient with squamous cell carcinoma of the anus treated with two T-cell products (COL6A3, PRAME) had a decrease in tumor measurements by 26% after therapy at 6 weeks.
Progression-free and overall survival
Of the 14 patients who underwent treatment, 13 had disease progression (PD). The median PFS was 3.4 months (range, 1.8–13.7 months) (). The median OS was 9.4 months (). There were no deaths related to treatment. Four patients were alive for over 20 months after Τ-cell therapy. All patients died from PD.
Immuno-monitoring studies and correlation with progression-free survival
Immuno-monitoring using flow cytometry was used to characterize the infused T cells in the patients’ blood. After IMA101 T-cell treatment, target-specific T-cells increased to a peak frequency of up to 78.74% of CD8+ T-cells within 1–3 weeks (median 25.58%, range 0.39–78.74%) and were detected up to 79 weeks. Patients with higher frequency of T-cells in the blood over time had longer PFS, but the small number of patients precludes any statistically significant conclusions.
Detection of target-specific T cells in tumor tissue
The assessment of overall T-cell infiltration in tumor tissue, conducted through TCRβ sequencing, exhibited variations among patients. No significant correlation was identified between the overall T-cell infiltration in tumor tissue before treatment and the infiltration of target-specific T-cells after treatment.
Discussion
The IMA101 treatment modality incorporates the theoretical framework of ACT, specifically targeting endogenous T cells.Citation18 It shows that an actively personalized, multitargeted, multi–T-cell product approach for treating solid cancers is feasible and safe, based on confirmed target expression of endogenous tumor antigens in biopsies from patients’ tumor tissue. Targeting tumor biomarkers that were expressed in 90% of HLA-A *02:01-positive patients with cancer helped overcome the challenge of identifying suitable targets for ACT. T-cell product(s) were successfully generated for 36 (83.7%) of 43 patients who underwent leukapheresis. Overall, 14 patients received the T-cell product(s), and this therapeutic approach was feasible and safe.
The ideal lymphodepleting treatment for T-cell-based therapy in solid tumors is currently unknown. For IMA101, a modified lymphodepleting regimen was designed to optimize the potential for synergy between FLU and CY when used in this setting. More commonly, CY is given before FLU, which yields antagonistic rather than synergistic effects between the two drugs.Citation9 The timing of administration of FLU and CY was based on previously published data in cell line experiments.Citation9,Citation19 The FLU/CY treatment in this protocol is unique in its dosage, timing, and sequencing, important elements for achieving effective lymphodepletion while minimizing normal organ toxicity effects.Citation9,Citation19 The combination of FLU and CY potentially facilitated the expansion of T-cells and ensured the prolonged persistence of the infused T-cells. The infusion of T cells took place two days after the 4-day lymphodepletion regimen, ensuring an adequate timeframe for the clearance of FLU.Citation20,Citation21 A low-dose IL-2 regimen was implemented to reduce toxicities (compared to a higher dose) and maintain T-cell persistence, replacing the commonly used high-dose regimen with ACT FLU.Citation20–23 The potential concern of preferential expansion of regulatory T cells through low-dose IL-2Citation24–26 may be less significant in patients who are lymphodepleted.
In 2019, when the atezolizumab cohort was initiated, this monoclonal antibody that binds to the PD-L1 protein and prevents its interaction with the PD-1 and B7.1 receptors, was expected to activate T-cells via inhibition of PD-L1. However, anti-PD1 antibodies may constitute a valid alternative and could have been used.
The safety profile of IMA101 treatment was favorable in patients with relapsed or refractory solid tumors, compared to CAR T-cell trials for diffuse large B-cell lymphoma.Citation1 Among the 14 treated patients, Grade 1–2 CRS manifested in 10 patients, accounting for 71.4% of the cohort. Notably, there were no reported instances of neurotoxicity among the patients. Following lymphodepletion, Grade 4 lymphopenia was observed in all patients, and 13 patients experienced Grade 4 neutropenia, while only one patient presented with Grade 1 neutropenia. Infectious complications occurred in five patients and were successfully treated.
Notably, the TIL- and TCR-T trials initially contained no lymphodepletion regimens, and when lymphodepletion was added, the tumor responses improved, particularly when IL-2 was introduced to maintain the persistence of the T-cells post-infusion. The use of high-dose cyclophosphamide for lymphodepletion (e.g., 3–4 g/m2) is a confounding factor, where the cyclophosphamide dose may cause direct normal organ toxicity, myelotoxicity, and immunosuppression that can lead to serious opportunistic bacterial and fungal infections. We strived to combine cyclophosphamide and fludarabine at dose levels that would yield significant lymphodepletion while limiting the duration of neutropenia to 3–5 days. We also aimed for an absolute neutrophil count no lower than 0.5 × 109/L, which was accomplished in all patients on the IMA101 trial. With the use of a low dose of post-infusion IL2, no serious adverse events were noted that could be attributed to the T-cell preparation, which was similar to the experience of other investigators. In contrast, the CAR-T cell infusions after lymphodepletion often yield serious clinical adverse events related to the cell preparation rather than to the lymphodepletion regimen.
In the study cohort, Grade 3–4 thrombocytopenia was noted in eight patients, constituting 53.3% of the participants. Importantly, no instances of treatment-related mortality were observed during the 100-day period following T-cell infusion. Of note, the occurrence and intensity of adverse effects appeared to be milder and less frequent when compared to prior investigations employing distinct lymphodepletion protocols for CAR T-cell therapy.Citation27,Citation28 It is essential to highlight that our patients received a comprehensive prophylactic regimen, encompassing measures to mitigate allergic reactions, as well as prophylaxis against bacterial, viral, and fungal infections. Additionally, they were treated with Mesna to prevent renal toxicity owing to cyclophosphamide. These prophylactic measures were aimed at reducing potential complications.
Previous studies have shown that targeting multiple antigens through ACT could be possible. The combination of autologous, non-engineered, multi-target T cells with chemotherapy has been associated with extended tumor control in multiple myeloma,Citation29 melanomaCitation30 and pancreatic cancer.Citation31 In our trial, the engraftment and persistence of T cells exhibited a favorable profile compared to previously reported data with similar ACT approaches.Citation32 Importantly, T-cell persistence is necessary for achieving prolonged PFS. Several factors contributed to the robust T cell persistence, including the modified lymphodepletion and IL-2 regimen, the characteristics of the targets, the manufacturing process, and, ultimately, the phenotype of the infused T cells.
A beneficial outcome of in vitro expanded T-cells can be attributed to both an (optimized) lymphodepletion regimen and the use of post-infusion IL-2 for the following reasons: a) we noticed prolonged disease stabilization in some patients in the IMA101 trial that was associated with detection of circulating engineered T-cells, and b) in our trial and in other trials of both expanded TILs and engineered antigen-specific T-cells, the clinical tumor responses were associated with the use of a lymphodepletion regimen, and the responses were even greater when post-infusion IL 2 was used to maintain the T-cells after infusion.Citation6,Citation33 In contrast, Chandran et al.Citation34 treated a cohort of patients with melanoma, targeting either gp100 (n = 10 patients) or MART-1 (n = 5 patients), and while these investigators used lymphodepletion with cyclophosphamide and fludarabine, they did not use IL-2 for the MART-1 group and achieved no objective tumor responses among those patients. Further, in a T-cell trial in HER-2+ breast cancer patients, the objective responses were long-lasting in 43% of the patients and included increased HER2-specific T-cell activity in vivo compared with pre-infusion levels that persisted for a prolonged period of time.Citation35 Although the number of treated patients overall is limited, we believe that when taken together with the observations in the IMA101 trial, the data indicate that both an optimized lymphodepletion regimen and the use of post-infusion IL-2 contribute to the persistence of circulating T-cells and the observed disease stabilization.
The T-cell products demonstrated a multifunctional effector memory profile characterized by CD45RO, CD127, and CD27 surface markers. The sustained or even induced expression of CD28, CD27, and CD127 following infusion, without the use of immune checkpoints, suggests the presence of a durable effector memory phenotype. This phenotype retains the ability to signal through these molecules, slowly transitioning toward terminal differentiation and less polyfunctional T cells post-infusion. Studies with similar manufacturing processes, including IL-21, have shown improved T-cell persistence and responses.Citation36
Target-specific T cells were rarely detected prior to the infusion, indicating T-cell priming from the naïve repertoire during manufacturing.Citation32 Post-treatment biopsies of all tested patients revealed the presence of IMA101 T cells, demonstrating their ability to infiltrate the tumor, an essential requirement for antitumor activity. While the T-cell products showed the capability to eliminate target-positive cell lines, their effectiveness varied and was comparatively lower than that of TCR-engineered T cells. The analysis of individual TCRs within the native T-cell products showed variable avidity, reflecting the natural immune repertoire directed against self-epitopes.
We investigated the significance of the T-cell products before and after infusion in relation to clinical outcomes. One patient suffering from nasopharyngeal squamous cell carcinoma (SCC) was given T-cell product targeting COL6A3 and experienced high engraftment, persistence, and tumor infiltration. A dominant T-cell clone was identified with low functional avidity, resulting in disease stabilization for 12.9 months. Another patient diagnosed with anal SCC had a 26% decrease in tumor measurements six weeks after receiving two T-cell products targeting PRAME and COL6A3. Both products exhibited high-avidity clones and multifunctional characteristics. A patient with ovarian cancer had high T-cell infiltration within the tumor. This patient was administered three T-cell products targeting COL6A3, MXRA5, and MMP1, resulting in disease stabilization for 7.3 months. Despite the promising biological data and exciting case studies, no objective responses were observed. The primary challenge faced in achieving clinical effectiveness with this approach seems to be the limited occurrence of highly potent target-specific TCRs in the native T-cell products. The adoptive transfer of autologous TCR-engineered T cells provides a potential solution to overcome this limitation. This therapeutic strategy aims to enhance antitumor activity by utilizing thoroughly characterized, potent, and high-avidity target-specific TCRs, which will lead to reduced manufacturing time, faster treatment availability, and potentially improved patient outcomes. Currently, trials of this strategy are underway (NCT03686124, NCT03441100, and NCT03247309).
Conclusion
Our findings demonstrate the feasibility and overall tolerability of employing autologous T-cells targeted against multiple well-defined pHLA (peptide-human leukocyte antigen) targets (PRAME, MAGEA1, MAGEA4, MAGEA8, NY-ESO-1, COL6A3 exon 6, MXRA5, and/or MMP1) in individuals with HLA-A *02:01 allele with advanced metastatic solid tumors. The observation of sustained T-cell presence detected in patients’ blood and their infiltration into tumor sites has provided valuable insights into the mechanism of action of IMA101. However, it is noteworthy that mere attributes such as extended and robust T-cell persistence, a favorable T-cell phenotype, elevated T-cell receptor (TCR) avidity, or tumor infiltration in isolation may be insufficient for effective tumor eradication. Our findings indicate that adopting multi-target ACT strategies, leveraging potent, high-affinity autologous TCR-engineered T-cells to target specific pHLA targets, hold promise for enhancing the clinical outcomes of patients with solid tumors.
Author contributions
A.M.T. contributed substantially to the development and discussion of the content. All authors researched data for the article, wrote the article, and reviewed and/or edited the manuscript before submission.
Acknowledgments
The authors would like to thank the patients who participated in this study and their families.
Disclosure statement
A.M.T. declares receipt of clinical trial research funding (to The University of Texas MD Anderson Cancer Center) from Agenus, OBI Pharma, Parker Institute for Cancer Immunotherapy, Tachyon, Orionis, Tempus and Tvardi, Novocure and IMMATICS; fees for consulting or advisory roles for Avstera Therapeutics, Bioeclipse, BrYet, Diaccurate, Macrogenics, NEX-I and VinceRx; and travel expenses from ASCO, Cancer Care Crossroads, GenomeWeb conference and Precision Medicine World Conference.
The other authors declare no relevant competing interests.
Additional information
Funding
References
- Tsimberidou AM, Van Morris K, Vo HH, Eck S, Lin YF, Rivas JM, Andersson BS. T-cell receptor-based therapy: an innovative therapeutic approach for solid tumors. J Hematol Oncol. 2021;14(1):102. doi:10.1186/s13045-021-01115-0.
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15(17):5323–8. doi:10.1158/1078-0432.CCR-09-0737.
- Debets R, Donnadieu E, Chouaib S, Coukos G. TCR-engineered T cells to treat tumors: seeing but not touching? Semin Immunol. 2016;28(1):10–21. doi:10.1016/j.smim.2016.03.002.
- Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99(25):16168–73. doi:10.1073/pnas.242600099.
- Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358(25):2698–703. doi:10.1056/NEJMoa0800251.
- Chapuis AG, Thompson JA, Margolin KA, Rodmyre R, Lai IP, Dowdy K, Farrar EA, Bhatia S, Sabath DE, Cao J, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci USA. 2012;109(12):4592–7. doi:10.1073/pnas.1113748109.
- Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;5(174):174ra27. doi:10.1126/scitranslmed.3004916.
- Tsimberidou AM, Guenther K, Andersson BS, Mendrzyk R, Alpert A, Wagner C, Nowak A, Aslan K, Satelli A, Richter F, et al. Feasibility and safety of personalized, multi-target, adoptive cell therapy (IMA101): first-in-human clinical trial in patients with advanced metastatic cancer. Cancer Immunol Res. 2023;11(7):925–45. doi:10.1158/2326-6066.CIR-22-0444.
- Valdez BC, Andersson BS. Interstrand crosslink inducing agents in pretransplant conditioning therapy for hematologic malignancies. Environ Mol Mutagen. 2010;51(6):659–68. doi:10.1002/em.20603.
- Yee C, Lizee G, Schueneman AJ. Endogenous T-cell therapy: clinical experience. Cancer J. 2015;21(6):492–500. doi:10.1097/PPO.0000000000000158.
- Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26(2):111–17. doi:10.1016/j.it.2004.12.003.
- Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–9. doi:10.1200/JCO.2008.16.5449.
- Chapuis AG, Roberts IM, Thompson JA, Margolin KA, Bhatia S, Lee SM, Sloan HL, Lai IP, Farrar EA, Wagener F, et al. T-cell therapy using interleukin-21–primed cytotoxic T-cell lymphocytes combined with cytotoxic T-cell lymphocyte antigen-4 blockade results in long-term cell persistence and durable tumor regression. J Clin Oncol. 2016;34(31):3787–95. doi:10.1200/JCO.2015.65.5142.
- Zhou D, Byers LA, Sable B, Smit MD, Sadraei NH, Dutta S, Upreti VV. Clinical pharmacology profile of AMG 119, the first chimeric antigen receptor T (CAR-T) cell therapy targeting delta-like ligand 3 (DLL3), in patients with relapsed/refractory small cell lung cancer (SCLC). J Clin Pharmacol. 2023. doi:10.1002/jcph.2346.
- Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, Maus MV, Park JH, Mead E, Pavletic S, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–38. doi:10.1016/j.bbmt.2018.12.758.
- Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci USA. 1992;89(8):3429–33. doi:10.1073/pnas.89.8.3429.
- Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, Steen MS, LaMadrid-Herrmannsfeldt MA, Williamson DW, Livingston RJ, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4(1):2680. doi:10.1038/ncomms3680.
- Britten CM, Singh-Jasuja H, Flamion B, Hoos A, Huber C, Kallen KJ, Khleif SN, Kreiter S, Nielsen M, Rammensee H-G, et al. The regulatory landscape for actively personalized cancer immunotherapies. Nat Biotechnol. 2013;31(10):880–2. doi:10.1038/nbt.2708.
- Yamauchi T, Nowak BJ, Keating MJ, Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4-hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res. 2001;7:3580–9.
- Long-Boyle JR, Green KG, Brunstein CG, Cao Q, Rogosheske J, Weisdorf DJ, Miller JS, Wagner JE, McGlave PB, Jacobson PA, et al. High fludarabine exposure and relationship with treatment-related mortality after nonmyeloablative hematopoietic cell transplantation. Bone Marrow Transplant. 2011;46(1):20–6. doi:10.1038/bmt.2010.53.
- Sanghavi K, Wiseman A, Kirstein MN, Cao Q, Brundage R, Jensen K, Rogosheske J, Kurtzweil A, Long-Boyle J, Wagner J, et al. Personalized fludarabine dosing to reduce nonrelapse mortality in hematopoietic stem-cell transplant recipients receiving reduced intensity conditioning. Transl Res. 2016;175:103–15 e4. doi:10.1016/j.trsl.2016.03.017.
- Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6:S11–4.
- Rosenberg SA. Interleukin-2 and the development of immunotherapy for the treatment of patients with cancer. Cancer J Sci Am. 2000;6:S2–7.
- Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, Ghobadi A, Budde LE, Bot A, Rossi JM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285–95. doi:10.1016/j.ymthe.2016.10.020.
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N Engl J Med. 2017;377(26):2531–44. doi:10.1056/NEJMoa1707447.
- Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi:10.1056/NEJMoa1804980.
- Houot R, Schultz LM, Marabelle A, Kohrt H. T-cell–based immunotherapy: adoptive cell transfer and checkpoint inhibition. Cancer Immunol Res. 2015;3(10):1115–22. doi:10.1158/2326-6066.CIR-15-0190.
- Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-in CAR-T. Int J Mol Sci. 2018;19(2):340. doi:10.3390/ijms19020340.
- Lulla PD, Tzannou I, Vasileiou S, Carrum G, Ramos CA, Kamble R, Wang T, Wu M, Bilgi M, Gee AP, et al. The safety and clinical effects of administering a multiantigen-targeted T cell therapy to patients with multiple myeloma. Sci Transl Med. 2020;12(554). doi:10.1126/scitranslmed.aaz3339.
- Puig-Saus C, Sennino B, Peng S, Wang CL, Pan Z, Yuen B, Purandare B, An D, Quach BB, Nguyen D, et al. Neoantigen-targeted CD8(+) T cell responses with PD-1 blockade therapy. Nature. 2023;615:697–704. doi:10.1038/s41586-023-05787-1.
- Smaglo BG, Musher BL, Vasileiou S, Kuvalekar M, Watanabe A, Robertson CS, Wang T, Francois M, Ramos CA, Hill L, et al. A phase I trial targeting advanced or metastatic pancreatic cancer using a combination of standard chemotherapy and adoptively transferred nonengineered, multiantigen specific T cells in the first-line setting (TACTOPS). J Clin Oncol. 2020;38:4622–. doi:10.1200/JCO.2020.38.15_suppl.4622.
- Chapuis AG, Desmarais C, Emerson R, Schmitt TM, Shibuya K, Lai I, Wagener F, Chou J, Roberts IM, Coffey DG, et al. Tracking the fate and origin of clinically relevant adoptively transferred CD8 + T cells in vivo. Sci Immunol. 2017;2(8). doi:10.1126/sciimmunol.aal2568.
- Khammari A, Labarriere N, Vignard V, Nguyen JM, Pandolfino MC, Knol AC, Quéreux G, Saiagh S, Brocard A, Jotereau F, et al. Treatment of metastatic melanoma with autologous melan-A/MART-1-specific cytotoxic T lymphocyte clones. J Invest Dermatol. 2009;129(12):2835–42. doi:10.1038/jid.2009.144.
- Chandran SS, Paria BC, Srivastava AK, Rothermel LD, Stephens DJ, Dudley ME, Somerville R, Wunderlich JR, Sherry RM, Yang JC, et al. Persistence of CTL clones targeting melanocyte differentiation antigens was insufficient to mediate significant melanoma regression in humans. Clin Cancer Res. 2015;21(3):534–43. doi:10.1158/1078-0432.CCR-14-2208.
- Disis ML, Dang Y, Coveler AL, Marzbani E, Kou ZC, Childs JS, Fintak P, Higgins DM, Reichow J, Waisman J, et al. HER-2/neu vaccine-primed autologous T-cell infusions for the treatment of advanced stage HER-2/neu expressing cancers. Cancer Immunol Immunother. 2014;63(2):101–9. doi:10.1007/s00262-013-1489-4.
- D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, Grupp S, Tap WD, Chagin K, Binder GK, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 (c259)T cells in synovial sarcoma. Cancer Discovery. 2018;8(8):944–57. doi:10.1158/2159-8290.CD-17-1417.