
ABSTRACT
Entamoeba histolytica, the causative agent of amebiasis, is one of the top three parasitic causes of mortality worldwide. However, no vaccine exists against amebiasis. Using a lead candidate vaccine containing the LecA fragment of Gal-lectin and GLA-3M-052 liposome adjuvant, we immunized rhesus macaques via intranasal or intramuscular routes. The vaccine elicited high-avidity functional humoral responses as seen by the inhibition of amebic attachment to mammalian target cells by plasma and stool antibodies. Importantly, antigen-specific IFN-γ-secreting peripheral blood mononuclear cells (PBMCs) and IgG/IgA memory B cells (BMEM) were detected in immunized animals. Furthermore, antigen-specific antibody and cellular responses were maintained for at least 8 months after the final immunization as observed by robust LecA-specific BMEM as well as IFN-γ+ PBMC responses. Overall, both intranasal and intramuscular immunizations elicited a durable and functional response in systemic and mucosal compartments, which supports advancing the LecA+GLA-3M-052 liposome vaccine candidate to clinical testing.
Introduction
Amebiasis, caused by Entamoeba histolytica, is the third deadliest parasitic disease, infecting approximately 50 million people worldwide with a resultant 40,000 to 100,000 deaths annually.Citation1,Citation2 E. histolytica is classified as a Category B priority pathogen by the National Institute of Allergy and Infectious Diseases (NIAID). Although most infected individuals remain asymptomatic, invasive amebiasis can clinically manifest as intestinal colitis or extraintestinal amebic liver abscess. Amebiasis can be fatal, particularly in young children.Citation3,Citation4 Other high-risk groups include returning travelers, immigrants, men who have sex with men, and patients with HIV.Citation5,Citation6
Adhesion of E. histolytica to the mucus layer of the intestine, an essential first step in the pathogenesis, is mediated by a surface lectin with high affinity for galactose (Gal) and N-acetyl-D-galactosamine (GalNAc) oligosaccharides on the epithelium and mucin 2 (MUC2).Citation7 Anti-amebic stool IgA and IFN-γ are important components of human adaptive immune responses associated with protection against amebiasis, although specific titers or concentrations for protection have not been established.Citation8,Citation9
In previous work, baboons (Papio sp.) were immunized intranasally using synthetic peptide epitopes spanning amino acids (aa) 758 to 1134 of the lectin heavy chain and cholera toxin as an adjuvant.Citation10 Both serum and stool antibodies elicited by the peptide vaccine exhibited neutralizing activity, as determined by their inhibition of the galactose-specific adherence of E. histolytica trophozoites to Chinese hamster ovary (CHO) cells in vitro. Immunized baboons were then challenged with E. histolytica. All animals cleared the infection by Day 51 and remained free from colitis or parasite invasion.Citation11 However, the choice of adjuvant was problematic for clinical translation, and high doses of peptide antigen were required. Thus, no amebiasis vaccine has progressed to clinical testing.Citation12
LecA is a recombinant protein derived from the Gal/GalNAc lectin (aa 578–1154) and encompasses epitopes necessary for amebic adherence and killing of host cells.Citation13,Citation14 Using LecA and a clinically relevant adjuvant (GLA-3M-052 liposome) delivered by different routes, we successfully characterized and recapitulated the human adaptive immune response to amebiasis in a mouse model, followed by a systematic completion of preclinical testing and stability studies.Citation8,Citation9,Citation15,Citation16 Intranasal immunization with LecA+GLA-3M-052 liposome elicited a concurrent mucosal IgA and systemic IFN-γ response in mice that was antigen specific and protected mice from cecal colitis upon a challenge with E. histolytica.Citation17,Citation18 Moreover, we characterized a clinically relevant intranasal vaccine delivery device (Teleflex MAD) for delivery performance using the LecA+GLA-3M-052 liposome composition.Citation19 Rhesus macaques and humans are both natural hosts for E. histolytica and share important similarities in anatomy and Toll-like receptor (TLR) expression, thus making rhesus macaques the next desirable model to evaluate this amebiasis vaccine candidate containing an adjuvant system that features multiple TLR ligands (GLA and 3M–052).Citation20,Citation21
In this study, we assessed the immunogenicity of intranasally or intramuscularly administered LecA+GLA-3M-052 liposomes in rhesus macaques. Both immunization routes elicited a parasite-adherence inhibitory humoral response in plasma, stool, and saliva. Systemic LecA-specific IFN-γ response was detected along with a strong memory B cell (BMEM) response. This is the first study assessing a pharmaceutically acceptable LecA vaccine formulation in the rhesus macaque primate model.
Materials and methods
Vaccine materials
The LecA antigen was expressed in Escherichia coli and purified by size exclusion chromatography in-house.Citation14 The adjuvant formulation (GLA-3M-052 liposomes) was prepared in-house by high-pressure homogenization and consisted of the synthetic TLR4 ligand glucopyranosyl lipid adjuvant (GLA; Avanti Polar Lipids), the synthetic TLR7/8 ligand 3M–052 (3M Healthcare), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC; Lipoid LLC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DPPE-mPEG2000; CordenPharma), plant-derived cholesterol (PhytoChol, Wilshire Technologies Inc.), ammonium phosphate monobasic and ammonium phosphate dibasic salts (J.T. Baker), and α-tocopherol (Spectrum Chemical). Antigen and adjuvant components were monitored for stability until immunizations had been completed.Citation16
Vaccine preparation
Vaccines were prepared in a laminar flow hood. Sterile saline was first added to the LecA antigen in a sterile glass vial. The adjuvant formulation was then added to the vial containing the antigen-saline mixture, then gently swirled and inverted for several seconds to mix all components. The vials were then placed on wet ice until immunization (see Supplementary Methods), which occurred within 4 h of mixing, an appropriate time frame from our previous mixing stability studies.Citation16 Vaccine formulation was allowed to acclimate to room temperature for at least 20 min prior to administration.
Immunization
The study included four experimental groups designed to assess the impact of antigen dose and route of immunization on vaccine safety and immunogenicity in rhesus macaques. After screening to confirm negative previous or current E. histolytica infection, 27 rhesus macaques were randomly assigned to four experimental groups (see Supplementary Methods): IN10, IN100, IN200, and IM100 (; see Supplementary Table S1 for animal age and weight characteristics). All animal experiments were performed in accordance with national and institutional guidelines for animal care of laboratory animals and approved by the Southwest National Primate Research Center (SNPRC) Institutional Animal Care and Use Committee (Protocol #1770 MM 8).
Table 1. Experimental design.
Each group received three immunizations by intranasal or intramuscular delivery (). Anesthesia was administered prior to each immunization. The intranasal route evaluated three antigen doses (10, 100, and 200 µg), whereas the intramuscular route employed the 100-µg antigen dose (). All animals received the same dose of adjuvant formulation consisting of 25 µg GLA and 10 µg 3M–052. For intramuscular delivery, animals were immunized with a single injection of 200 µL volume in the quadriceps. For intranasal delivery, a 100-µL volume was delivered to each nare for a total of 200 µL (see Supplementary Methods).
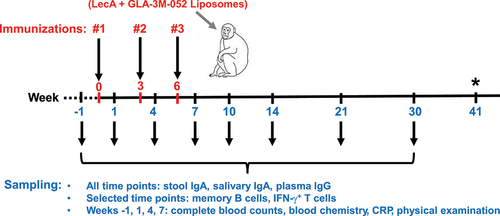
Figure 1. Schematic overview of immunizations and sample collections. A total of 27 rhesus macaques were randomly divided into four groups and assigned to receive either intranasal or intramuscular immunization. The vaccine was formulated with either 10, 100, or 200 µg of LecA plus adjuvant and was administered to designated groups accordingly. Each group received three immunizations with a 3-week interval. Pre-immune samples (blood, saliva, stool, and PBMCs) were collected 1 week prior to the first vaccination, and samples were collected thereafter at the time points shown. Approximately 8-9 months after the third immunization, three animals were euthanized to collect lymph nodes, bone marrow, splenocytes, and PBMCs. Animal health was monitored continuously with regular blood and physical examinations. *Week 41 is an approximate time point representing B and T cell ELISpot assays conducted on tissues harvested from three animals that underwent necropsy 8-9 months after the third immunization, as well as B and T cell ELISpot assays conducted on PBMCs collected 10 months after the third immunization from remaining animals.
Sample collection
Regular clinical observations and weight measurements were recorded for the duration of the study. Plasma, saliva, and stool samples were collected 1 week before the first immunization (pre-immune), 1 week after each immunization (Weeks 1, 4, and 7), and at five additional time points (Weeks 10, 14, 21, 30, and 41) (). Due to the large number of animals, each time point was split for sample collection over two separate days. Analysis of blood chemistry, clinical blood counts (CBC), and C-reactive protein (CRP) was conducted at the pathology lab at SNPRC. Plasma and PBMCs were shipped to the University of Virginia for vaccine-specific immunology analysis. Details of blood collection, processing, and storage have been described under Supplementary Methods.
Mucosal samples consisted of stool and saliva swabs. At least 0.5 g of stool samples were collected per animal at each time point. Details of stool and saliva sample collection, processing, and storage have been detailed under Supplementary Methods. All mucosal samples were shipped on dry ice to the University of Virginia for vaccine-specific immunology analysis.
Plasma or stool samples from three female children aged 1–2 y with known E. histolytica infection were pooled for positive controls. Pooled samples from three uninfected age-matched children were negative controls. Collection of plasma and stool samples from E. histolytica-infected and healthy human individuals was approved by the Ethical and Research Review Committees of the International Centre for Diarrhoeal Disease Research, Bangladesh (PR-13092), and the Institutional Review Board of the University of Virginia (IRB# 20388). The ClinicalTrials.gov identifier is NCT02764918.
Immunogenicity and functional assays
Enzyme-linked immunosorbent assay to detect LecA-specific serum IgG and mucosal IgA
Plasma, stool, and saliva samples were stored at −80°C until assayed for LecA-specific IgG and IgA antibody titers by enzyme-linked immunosorbent assay (ELISA). 96-well ELISA half area plates (Corning Costar, Cat# 3690) were coated with 0.25 µg/well recombinant LecA in 50 mM bicarbonate buffer (pH 9.6) overnight at 4°C and washed three times with 1 × PBS-Tween 20 (PBST; Thermo Scientific, Cat# 28352) using a plate washer (BioTek, Winooski, VT). The plates were blocked with commercial ELISA blocking buffer (Thermo Scientific, Cat# N502) for 1 h at room temperature and air dried for storage until the day of use. All stool samples were processed prior to the mucosal antibody analysis. An aliquot of each stool sample was weighed, and 10 μL of PBS with protease inhibitors (Thermo Scientific, Cat# A32953) was added per mg of stool. Stool samples were mixed with a bead beater (Qiagen, Germantown, MD) at a speed of 250 rev/min for 10 min and spun at 14,000 × g at 4°C for 10 min. Final supernatants were transferred into 1.5-mL microcentrifuge tubes (Axygen), and phenylmethylsulfonyl fluoride was added to a final concentration of 1 mM. Samples were stored at −80°C.
Thawed plasma and saliva samples were used without further processing. Serial dilutions of plasma were prepared with ELISA blocking buffer and were incubated in LecA-coated wells for 3 h at room temperature. Following washes in PBST, horseradish peroxidase (HRP)-conjugated goat anti-human IgG (Jackson ImmunoResearch Laboratories, Cat# 109-035-098) or HRP conjugated goat anti-human IgA (Jackson ImmunoResearch Laboratories, Cat# 109-035-011) was added at 1:1000 or 1:2000, respectively, and incubated for 1 h at room temperature.Citation22 KPL TMB substrate (SeraCare Life Sciences, Cat# 5120–0047) was used to develop the color. The reaction was stopped by addition of 1:100 diluted sulfuric acid (Sigma-Aldrich), and the optical densities (ODs) were measured at 450 nm by an ELISA reader (Synergy, BioTek). Samples from infected humans, infected rhesus macaques, or previously immunized rhesus macaques were positive controls and determined the cutoff. A titration curve with R2 ≥ 0.95 for each plate served as the acceptance criterion. Antibody titers were determined using a sigmoidal dose–response curve generated by the least-squares method. Endpoint titers were determined using an OD cutoff of 0.5 for plasma IgG and 0.1 for mucosal IgA.
ELISpot assays
Enzyme-linked immunospot (ELISpot) assay was utilized to determine the number of LecA-specific memory T cells and BMEM. For the T-cell assay, an IFN-γ kit was purchased from Mabtech (Cat# 3421M-4APW-2). Wells were blocked using complete RPMI medium (1% penicillin-streptomycin [PSN] plus 10% heat-inactivated FBS) for at least 30 min at room temperature. All the animals were bled at approximately Week 41 (see ), and freshly isolated PBMCs were resuspended in complete RPMI medium and incubated in a 5% CO2 incubator at 37°C (three animals that underwent necropsy were bled between Weeks 34 and 38). A total of 250,000 cells were seeded in 200 µL of medium per well and stimulated with either 50 μg/mL recombinant LecA or 1 μg/mL/peptide of a LecA peptide pool (custom synthesis from JPT Peptide Technologies), anti-CD3 control antibody, or left unstimulated for 48 h in a CO2 incubator. Samples were seeded in triplicate. Wells were then washed 5 times with PBS followed by the addition of 100 µL/well biotinylated detection anti-IFN-γ antibody diluted to 1 μg/mL in PBS with 0.5% FBS. After 2 h of incubation at room temperature, the plate was again washed and incubated with 100 µL/well streptavidin diluted 1:1000 in PBS with 0.5% FBS for 1 h at room temperature. BCIP/NBT substrate was filtered through a 0.22-μm syringe filter (Fisher Scientific, Cat# 09-720-511) before use, and the reaction was stopped by tap water after spot development. The plate was dried and stored in the dark until spots were counted on a Biosys (Miami, FL) Bioreader 7000 ELISpot plate reader. The results were then converted to IFN-γ-secreting cells per million cultured PBMCs (antibody-secreting cells [ASCs]/106 PBMCs). Unstimulated wells typically showed low spot counts (<50 per million), and this number was subtracted from the corresponding test sample value.
Long-term B cell ELISpot assay was used to quantitate the BMEM pool that is crucial for generating a secondary response upon antigen re-encounter. Similarly, 96-well multi-screen filter plates (Millipore, Cat# MSIPS4W10) were activated with 35% ethanol prior to use and were briefly washed with sterile PBS. Wells were coated with 2 μg/mL LecA to assess for vaccine-specific responses. For comparison, total IgG- and IgA-secreting cells were also quantitated by coating separate sets of plates with 15 μg/mL anti-human/monkey IgG (Mabtech, Cat# 3850-2A) or anti-human/monkey IgA (Mabtech, Cat# 3860M-2A). Week 14 PBMCs were activated using the B-Poly-S Polyclonal B Cell Activator (R848/IL-2) for 48–72 h prior to developing spots. Roughly 500,000 stimulated cells were seeded per well in 200 µL of medium and incubated for 8–16 h at 37°C in a CO2 incubator. Biotinylated detection anti-human/monkey IgG and IgA were diluted to 1 μg/mL or 0.5 μg/mL, respectively, in PBS with 0.5% FBS, and the same detection procedure above was repeated. Spots were counted and interpreted as above.
IgG avidity assay
The avidity index (AI) was measured as previously described.Citation23,Citation24 In this case, plasma-binding antibody avidities to LecA were measured by a urea disruption ELISA (see Supplementary Methods). An AI value ≥50 was considered high avidity.Citation25
Necropsy analysis of euthanized rhesus macaques
Three rhesus macaques (one each from IN10, IN200, and IM100 groups) were euthanized 6–8 months after the last immunization due to health conditions unrelated to the amebiasis studies (see Supplementary Methods). Fresh whole blood was processed to isolate PBMCs as above. Bone marrow and axillary, iliac, mesenteric, and inguinal lymph nodes were collected. Mononuclear cell suspensions were passed through a 70-μM strainer followed by an RBC lysis step. Lamina propria mononucleated cells (LPMCs) were isolated from the cecum as previously describedCitation26 (see Supplementary Methods). All cells were seeded accordingly in 96-well ELISpot plates as above.
Adherence inhibition assay
Adherence of E. histolytica trophozoites to CHO cells (ATCC, Cat# CCL-61) was assessed in vitro with plasma, stool, and salivary samples as previously describedCitation17 (see Supplementary Methods).
CXCL-13 measurement
Plasma levels of CXCL-13 (chemokine C-X-C motif ligand 13) were measured using a commercial ELISA kit (Bio-Techne, Cat# DCX130) per the instructions. Neat plasma samples were run in triplicate. Concentrations were calculated based on the standard curve.
Analytical and statistical methods
The group size of 6–7 animals was anticipated to provide 80% power to detect a 5-fold increase in cytokine or antibody levels between groups (α = 0.05). Statistically significant differences for adherence inhibition, avidity, ELISpots, and CXCL-13 ELISA were determined by one-way ANOVA with appropriate correction for multiple comparisons using GraphPad Prism version 9.0. Multi-level mixed effects linear regression was used to model the plasma, stool, and salivary antibody titers (IgG or IgA) over time on the treatment groups. The delta method was used to test for differences in the marginal means between treatment groups at each time point. All the regression models used age, sex, and weight as baseline covariates and animal-level random effect to capture the variability due to repeated measurements of the monkeys. To test whether IFN-γ, BMEM response, etc., were different between the male and female monkeys, two-sided t-tests were performed. To measure if antibody titer (IgG or IgA) correlated with IFN-γ, BMEM response, avidity, or CXCL-13 levels, Pearson’s correlation was computed along with p-value to test the null correlation between the antibody levels at all time points and IFN-γ, BMEM response, avidity, or CXCL-13. All the analyses were conducted using R version 4.2.2. Area under the curve (AUC) analysis was performed using GraphPad Prism.
Results
Vaccine safety
Animals were immunized three times by the intranasal or intramuscular route (, ) and monitored for signs of local or systemic reactogenicity. Regardless of immunization route and antigen dose, the vaccine formulations were well tolerated. In general, animal weight was maintained or slightly increased over the course of the study (Supplementary Table S1). For intramuscular immunizations, no local reactogenicity was observed in daily observations conducted for 5 d following vaccine administration. Likewise, no nasal redness was noted following intranasal immunization, and the vaccine spray volume was retained in the nose, except for one animal that exhibited a small amount of vaccine drip back immediately following the first immunization. Although most animals exhibited no nasal discharge after intranasal vaccine administration, a small amount of discharge was noted in one animal after the first immunization and in one animal after the second immunization, where the discharge in both cases occurred within approximately 2 min of vaccine administration. Finally, one animal exhibited vomiting followed by nasal discharge approximately 1 min after the third intranasal immunization; this reaction could be at least partially attributable to the sedation since additional behavioral observations were considered typical of a reaction to sedation. CBC and serum chemistry findings from animals were generally within expected ranges, slightly out of range, transiently out of range, or considered normal. Key indicators including alkaline phosphatase, CRP, white blood cells, lymphocytes, monocytes, eosinophils, hemoglobin, and platelets are summarized in Supplementary Table S1.
Vaccine-specific systemic and mucosal antibody responses
Following the first immunization (Week 1), four of the six animals that were immunized via intramuscular route (IM100) showed plasma IgG response (). Amongst the intranasal groups, one animal in the IN10 and two animals in the IN100 group generated anti-LecA IgG response after the first immunization, whereas none in the IN200 group showed response at this time point. One week after the third and final immunization, all six animals in IM100 showed high levels of anti-LecA IgG titers, whereas only a subset of animals from the other three groups were seropositive at this time point (4 of 7 in IN10, 5 of 7 in IN100, and 4 of 6 in IN200), although all animals turned seropositive at later time points except for one animal from IN200. Peak IgG response for the IM100 and IN100 groups occurred at the Week 10 time point (4 weeks following the final immunization), whereas the response in the IN200 and IN10 groups peaked at later time points (). Although most animals in all four groups remained seropositive at Week 30, the IM100 group tended to elicit more robust anti-LecA IgG titer overall. Indeed, the integrated AUC of IgG titers in the IM100 group was significantly higher compared to each of the intranasally immunized groups (). Regression analysis after adjusting for age, sex, and weight indicated that the IM100 group titers were significantly higher than those of IN10 (all time points), IN100 (Weeks 4, 7, and 10), or IN200 (Weeks 1, 4, 7, 10, and 14), whereas no significant differences were detected amongst the IN groups at any of the time points (Supplementary Table S2).
Figure 2. LecA vaccine elicited a strong systemic antibody response. (a) Anti-LecA plasma IgG endpoint titers. Each group (n = 7 or 6) was immunized three times either intranasally or intramuscularly at a 3-week interval with LecA+GLA-3M-052 liposome formulation. Pre-immune samples collected a week prior to the first immunization served as a negative control. Samples were then collected at the time points noted in (Weeks 1, 4, 7, 10, 14, 21, and 30). All plasma samples were serially diluted and analyzed for LecA-specific antibody levels by ELISA. Any OD value ≤ 0.5 at a 1:8000 dilution of plasma was considered as negative/background based on the pilot runs. Endpoint titers were plotted for all the time points. A plasma sample from an infected human individual collected 28 d after PCR positive detection (human control) as well as a sample from a naturally infected animal (nat. infected) from the same colony identified during the pre-immunization screening were used as positive controls. Plasma from healthy human individuals or naïve animals acted as negative controls and showed background signal (not shown). Data are represented as mean ± SD. (b) Regression analysis. Same data plotted after adjusting for age, sex, and weight. (c) Integrated AUC analysis. Data are represented as mean ± SEM. (d) Avidity index. Plasma dilutions that gave an OD450 nm = 1.0 were determined using Week 10 samples. ELISAs for anti-LecA IgG titers were run as described in two plates in parallel. One plate received standard PBST washes, while the other was washed using 6 M urea. Avidity Index (AI %) was expressed as follows: AI = [(absorbance of urea-treated sample/absorbance of non-urea-treated matched sample) × 100]. All samples were run in triplicate. Data are represented as mean ± SD.
![Figure 2. LecA vaccine elicited a strong systemic antibody response. (a) Anti-LecA plasma IgG endpoint titers. Each group (n = 7 or 6) was immunized three times either intranasally or intramuscularly at a 3-week interval with LecA+GLA-3M-052 liposome formulation. Pre-immune samples collected a week prior to the first immunization served as a negative control. Samples were then collected at the time points noted in Figure 1 (Weeks 1, 4, 7, 10, 14, 21, and 30). All plasma samples were serially diluted and analyzed for LecA-specific antibody levels by ELISA. Any OD value ≤ 0.5 at a 1:8000 dilution of plasma was considered as negative/background based on the pilot runs. Endpoint titers were plotted for all the time points. A plasma sample from an infected human individual collected 28 d after PCR positive detection (human control) as well as a sample from a naturally infected animal (nat. infected) from the same colony identified during the pre-immunization screening were used as positive controls. Plasma from healthy human individuals or naïve animals acted as negative controls and showed background signal (not shown). Data are represented as mean ± SD. (b) Regression analysis. Same data plotted after adjusting for age, sex, and weight. (c) Integrated AUC analysis. Data are represented as mean ± SEM. (d) Avidity index. Plasma dilutions that gave an OD450 nm = 1.0 were determined using Week 10 samples. ELISAs for anti-LecA IgG titers were run as described in two plates in parallel. One plate received standard PBST washes, while the other was washed using 6 M urea. Avidity Index (AI %) was expressed as follows: AI = [(absorbance of urea-treated sample/absorbance of non-urea-treated matched sample) × 100]. All samples were run in triplicate. Data are represented as mean ± SD.](/cms/asset/798b4049-8a6e-471c-8b35-e8577c26c235/khvi_a_2374147_f0002_oc.jpg)
Plasma samples were further tested for avidity of IgG. Avidity is defined as the strength of binding between IgG and its specific target epitope. A higher AI is reflective of affinity maturation and hence potentially better neutralization. Samples from selected animals from all four groups showed a low avidity (AI = 30–50), but most tested animals from the IN100 and IM100 groups showed a high avidity (AI > 50) (). Thus, the avidity results confirmed that the vaccine generated a strong antibody response at the 100-µg antigen dose.
LecA-specific stool IgA response was delayed compared to plasma IgG in all the groups, although peak response was again evident at Week 10 (). All six animals from the IM100 group showed detectable titers even at Week 21. Interestingly, AUC values for all the groups indicated equivalent overall stool IgA responses (). No significant differences were confirmed between groups at any time point except at Week 21, where IM100 titers were significantly higher than those of IN200 (Supplementary Table S3). The positive control human sample showed a stronger response than the immunized animals, which may indicate that the assay is less sensitive for samples from non-human primates (NHPs) or that natural infection elicits a stronger stool IgA response than vaccination.
Figure 3. Mucosal antibody response. Each group (n = 7 or 6) was immunized three times either intranasally or intramuscularly at a 3-week interval with LecA+GLA-3M-052 liposome formulation. Pre-immune samples collected a week prior to the first immunization served as negative controls. Samples were then collected at the time points noted in (Weeks 1, 4, 7, 10, 14, 21, and 30). All samples were serially diluted and analyzed for LecA-specific antibody levels by ELISA. Any OD value ≤ 0.1 at a 1:2 dilution of stool supernatant or an OD value ≤ 0.1 at a 1:5 dilution of saliva was considered as negative/background based on the pilot runs. Endpoint titers were plotted for all the time points. (a) Stool IgA endpoint titers. Stool supernatants were prepared as described under methods. A stool sample from an infected human individual collected 28 d after PCR positive detection (human control) was used as a positive control. Samples from healthy human individuals or naïve animals acted as negative controls and showed background signal (not shown). Data are represented as mean ± SD. (b) Regression analysis for stool IgA. Data plotted after adjusting for age, sex, and weight. (c) Integrated AUC analysis of stool IgA. Data are represented as mean ± SEM. (d) Salivary IgA endpoint titers. Saliva samples were not processed after collection. Data are represented as mean ± SD. (e) Regression analysis for salivary IgA. Data plotted after adjusting for age, sex, and weight. (f) Integrated AUC analysis of salivary IgA. Data are represented as mean ± SEM.
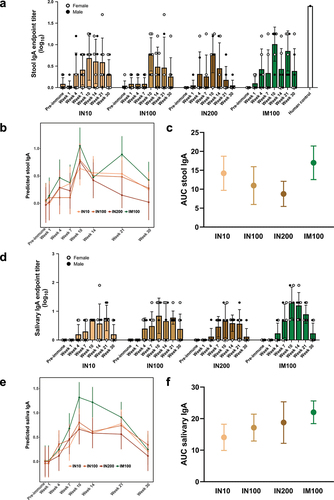
We assessed the ability of the vaccine to elicit antigen-specific salivary IgA response, although its role in protection from amebiasis is not known (). Overall, the titers of salivary IgA were low; however, animals in all the groups were positive at least at one time point. Most animals reached a peak response by Week 10. Similar to stool IgA, fluctuations in titers were observed in a few animals, especially in the intranasal groups. Most animals showed a drop in titers by Week 30. AUC analysis indicated no difference in overall salivary IgA titers between groups (). No statistically significant differences were observed among intranasal groups at any time point, whereas the IM100 group showed higher titers compared to IN10 and IN200, but not IN100, at Week 10. The IM100 group also showed significantly higher titers than all the intranasal groups at Week 14 (Supplementary Table S4).
In summary, both intranasal and intramuscular regimens elicited systemic and mucosal humoral responses. Interestingly, the intramuscular regimen showed mucosal responses that were comparable to those of the intranasal route. Overall, no sex bias was evident for any of the antibody readouts.
Vaccine-generated antibodies inhibited parasite adhesion in vitro
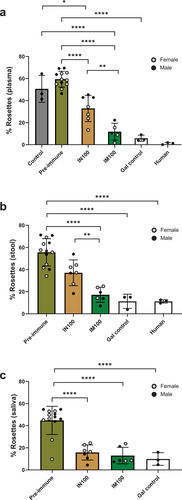
Adherence of E. histolytica trophozoites to target cells is the key initial step to infection.Citation27 Mucosal antibodies are considered particularly important in providing sterilizing immunity against gut pathogens by hindering colonization. While antibody titers measured by ELISA may reflect robustness of the response, it is difficult to define a general protective mucosal IgA titer. The functional capacity of the antibodies to neutralize the parasite reflects a more direct measure of the quality of response. To assess the effectiveness of mucosal IgA and systemic IgG, we tested Week 10 plasma, stool, and saliva samples from the IN100 and IM100 groups for their ability to block adherence of parasites to CHO cells in vitro (). Pre-incubation of trophozoites with plasma, stool supernatants, or saliva from the immunized animals significantly reduced adherence of the trophozoites. Plasma from the IM100 group showed significantly higher adhesion inhibition compared to the IN100 group (), which could be due to higher mean anti-LecA plasma IgG levels in the IM100 group (mean log10 value of 6.6 ± 0.3 in IM100 vs 4.5 ± 0.7 in IN100, p = .016). Nevertheless, the functional activity of Week 10 stool supernatants was also significantly higher in the IM100 group () even though stool IgA titers were similar between these groups (mean log10 value of 1.00 ± 0.39 in IM100 vs 0.77 ± 0.38 in IN100, p > .1). In contrast, the functional activity of salivary samples was equivalent () even though salivary IgA titers were similar between these groups (mean 1.3 ± 0.5 in IM100 vs 0.84 ± 0.6 in IN100, p > .1). As expected, pre-immunization samples failed to show any inhibition. Both galactose (competitive inhibitor) and human specimen controls reduced rosette formation as expected. Overall, vaccine-mediated anti-LecA mucosal and systemic antibodies effectively blocked the adherence step and thus protected mammalian cells in vitro regardless of the immunization route.
Figure 4. Plasma, stool, and salivary antibodies exhibited adherence inhibitory activity. E. histolytica trophozoites were incubated on ice for 1 h with pre-immune or immune plasma, stool supernatant, or saliva samples from the IN100 and IM100 groups collected at Week 10. The mixtures were then incubated on ice with Chinese hamster ovary (CHO) cells at a 1:20 ratio for 1.5 h and centrifuged. Pellets were used to score rosette formation under microscope. (a) Plasma. (b) Stool supernatant. (c) Saliva. Data are represented as mean ± SD. Data were plotted and analyzed using ANOVA in Prism 9.0. Pre-immune samples from both delivery routes were plotted together. Control = amebae + CHO cells only, Gal = galactose control as a competitive inhibitor, human = sample from anti-LecA plasma IgG+/stool IgA+ individual. *p < .05, **p < .01, ****p < .0001.
Vaccine-elicited systemic IFN-γ response
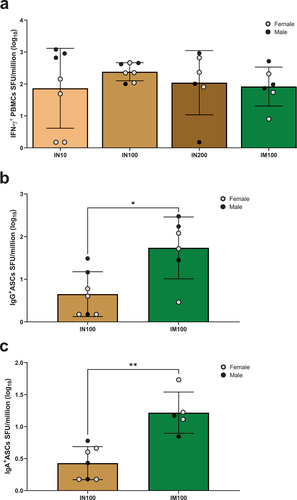
Parasite-specific stool IgA and production of IFN-γ by PBMCs have been associated with protection from E. histolytica re-infection in humans. Therefore, we measured IFN-γ production from PBMCs from the immunized animals using T cell ELISpot assay. Freshly isolated PBMCs were re-stimulated with either recombinant protein () or a LecA peptide pool (Supplementary Figure S1). Animals across the groups showed robust IFN-γ production, and protein stimulation resulted in higher responses compared to peptide pool stimulation (Suplementary Figure S1). Specifically, all the animals from the IN100 group exhibited LecA-specific IFN-γ+ PBMCs (). Most animals in the other groups also showed IFN-γ-secreting cells. The IN100 intranasal regimen tended to sustain the highest number of IFN-γ-secreting cells and the least variation between individual animals, although statistically significant differences were not observed among the groups.
Figure 5. LecA vaccine generated systemic recall responses for T and B cells. (a) LecA-specific IFN-γ+ T cell response. Fresh PBMCs were isolated from blood collected at approximately Week 41 (see ) and stimulated for 48 h with purified recombinant LecA protein in triplicate. Stimulation using anti-CD3 antibodies was used as a positive control, and unstimulated cells were used as a negative control. Spots were counted and expressed as spot forming units (SFU) per million after log-transforming the data. Background signal from unstimulated wells was subtracted and was typically <100 SFU/million. See also Supplementary Figure 1 (b–c) LecA-specific long-term IgG and IgA antibody-secreting cell (ASC) responses in both intranasal and intramuscular regimens. ELISpot assay was used to compare antibody-secreting BMEM between intranasal and intramuscular regimens. PBMCs were isolated from blood collected at the Week 14 time point and activated with B cell activator (R848/IL-2) for 72 h. All cells were then cultured in triplicate on LecA-coated plates for an additional 16–24 h. Pre-immunization PBMCs from the same animals were used to measure baseline response, which was subtracted from the test readings. Spots were developed, counted, and plotted as SFU per million after log-transforming the data. (b) LecA-specific IgG-secreting BMEM, (c) LecA-specific IgA-secreting BMEM. One animal in the IM100 group did not have enough cells. The Mann–Whitney test was used in GraphPad Prism to compare groups. Data are represented as mean ± SD. *p < .05, **p < .01.
Vaccine-generated systemic BMEM
We performed ELISpot assays on PBMCs to evaluate long-term LecA-specific BMEM immune response post-vaccinations. As a direct comparison between delivery routes, thawed and rested PBMCs from only the IN100 and IM100 groups were assayed for both IgG- and IgA-secreting B cells. Pre-immunization PBMCs were used as a negative control. Significant LecA-specific IgG+ () and IgA+ () BMEM responses were detected in the IM100 group compared to the IN100 group. Overall, intramuscular immunization generated a better BMEM response in PBMCs.
Vaccination elicited a strong germinal center (GC) reaction
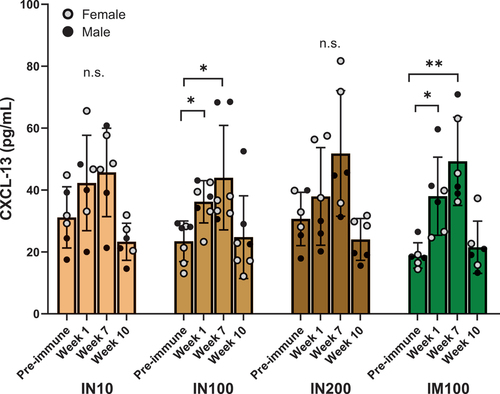
T follicular helper (TFH) cells are essential for the generation of antibody-producing plasma cells and long-lived BMEM. Interaction of B cells with TFH cells is central to GC reaction and somatic hypermutations. Furthermore, plasma levels of CXCL-13, which is expressed by GC TFH cells and follicular dendritic cells (FDCs) in the B cell follicles, correlate with GC activity.Citation28 Therefore, we measured plasma levels of CXCL-13 a week after the first (Week 1) and third (Week 7) immunizations and at Week 10 (). In both IN100 and IM100 groups, CXCL-13 was significantly elevated compared to pre-immunization levels at Weeks 1 and 7. Thus, the adjuvanted LecA vaccine appeared to elicit a robust GC reaction.
Figure 6. CXCL-13 as a plasma biomarker for germinal center activity. Circulating CXCL-13 levels were determined using a commercial ELISA kit (Bio-Techne) according to the instructions provided. Neat plasma samples at the indicated time points from each group were used in triplicate, and CXCL-13 concentrations were extrapolated using the standard curve. All the values were in the linear range of the curve. ANOVA was used to analyze the data in GraphPad Prism. One outlier identified by Grubb’s test was removed from the IN10 group. Data are represented as mean ± SD. *p < .05, **p < .01.
Vaccination stimulated strong T and B cell systemic responses in lymphoid tissues
Lastly, we evaluated immune responses from the necropsy tissues collected from three animals that were euthanized 8-9 months after the third immunization due to unrelated health conditions. Tissues were obtained from one animal each from the IN10, IN200, and IM100 groups. We collected blood, lymph nodes (iliac, mesenteric, inguinal, and axillary), bone marrow, ceca, and spleens. Isolated mononucleated cells were used in ELISpot assays to assess LecA-specific IFN-γ+ T cells and antibody-secreting BMEM. Robust LecA-specific IFN-γ responses were detected in blood, bone marrow, and lymph nodes (Suplementary Figure S2). Multiple tissues analyzed from these three animals (lymph nodes, bone marrow, PBMCs, and lamina propria) showed robust LecA-specific IFN-γ+ T cell and IgA+ and IgG+ BMEM responses. No response was observed in spleens. Overall, these animals generated a LecA-specific response in both T and B cell compartments irrespective of the antigen dose or the immunization route that was durable for at least 8 months post-immunization.
Discussion
The most important conclusions of this study are that 1) the LecA+GLA-3M-052 liposome formulation was well tolerated in rhesus macaques following intranasal or intramuscular immunization and 2) the immunized animals could recapitulate the immune responses observed in humans protected from recurrent amebiasis. This study supports the advancement of LecA+GLA-3M-052 liposomes as a candidate amebiasis vaccine toward first-in-human testing. This study was an extension of our preclinical studies that optimized the antigen and adjuvant dosing and composition for immunogenicity and protective efficacy in mice.Citation16–18 The present work optimized the LecA-specific plasma IgG, stool IgA, and systemic IFN-γ response in rhesus macaques. Both intranasal and intramuscular regimens elicited systemic and mucosal anti-LecA antibody responses that were functional in vitro. Robust and durable IFN-γ and BMEM responses were also evident. Overall, the intramuscular regimen elicited the best qualitative and quantitative antibody and B cell responses, whereas all regimens elicited robust IFN-γ-secreting PBMCs.
Previously, we showed that intranasal immunization elicited better stool IgA and systemic IFN-γ responses in mice compared to subcutaneous immunization, and that antigen dose was critical for optimizing intranasal regimen immunogenicity.Citation16,Citation18 However, due to differences in TLR expression and anatomy, mouse models for intranasal adjuvanted vaccine regimens are not sufficient to predict human responses. For instance, TLR8, one of the TLRs activated by 3M–052, is nonfunctional in mice but functional in NHPs and humans.Citation29 Therefore, NHP models may better approximate the human response to intranasal adjuvanted vaccine candidates. In contrast to our previous results in mice, increasing the antigen dose did not have much impact on immunogenicity responses in the intranasally vaccinated NHP groups, although the IN100 group tended to elicit the best overall immune response profile. Moreover, intramuscular immunization in the rhesus macaques generated an equivalent stool IgA response in magnitude to intranasal immunization, an unexpected finding considering that intranasal immunization of mice with LecA+GLA-3M-052 liposomes elicited significantly higher stool IgA titers compared to subcutaneous immunization.Citation18 On the other hand, other adjuvant systems also induce antigen-specific mucosal antibody responses following intramuscular administration.Citation30 Furthermore, stool supernatant from both regimens inhibited amebic adherence to mammalian cells. We observed fluctuations in stool IgA titers for some animals. These could be due to low response in these animals and a low signal-to-noise ratio resulting from the complexity of stool. Impressively, and consistent with previous mouse data, intranasal vaccination of rhesus macaques with the LecA+GLA-3M-052 liposome vaccine candidate generated a robust antigen-specific IFN-γ response that was equivalent to that from intramuscular administration.
While both intranasal and intramuscular immunizations elicited a high-avidity IgG response that was functional in vitro, the intramuscular group elicited higher IgG titers. Thus, it is unclear whether the better adhesion inhibition by the intramuscular group is due to better quality antibodies, as seen from a higher avidity index, or higher antibody titers. Clinically, over 90% of E. histolytica-infected individuals develop anti-lectin plasma IgG antibodies that show anti-amebic activity in vitro.Citation31 However, anti-amebic plasma IgG generally does not correlate with protection in clinical disease or animal models. The protective or non-protective effects seem to depend on the IgG subclasses induced during the immune response.Citation32,Citation33 Anti-E. histolytica IgG1 subtype has a greater complement fixation ability and was suggested to be protective in humans.Citation34 Lack of protection from IgG antibodies can also be due in part to antibody degradation by parasite cysteine proteinases.Citation35 Nevertheless, the LecA+GLA-3M-052 liposome vaccine candidate elicited a high titer antibody response that blocked amebic adherence. Future studies are needed to determine the IgG subclasses in different immunization regimens and correlate these with disease clearance upon challenge to assess IgG’s role.
Mucosal responses in other compartments were also detected in the present study. Although the role of salivary IgA in protection against E. histolytica is currently unknown, the induction of mucosal responses for protection against other indications, such as respiratory pathogens, would represent an important attribute of adjuvants for mucosal administration. Respiratory tract infections remain the top cause of morbidity and mortality worldwide, and a sudden outbreak like COVID-19 warrants preparedness for a rapid response. The GLA-3M-052 liposome adjuvant also generated SARS-CoV-2 Spike-specific IgA in bronchoalveolar lavage fluid in immunized mice that were subsequently protected against a lethal challenge.Citation36 In the present study, we tested the potential of LecA+GLA-3M-052 liposome to generate a salivary IgA response in NHPs. Both intranasal and intramuscular regimens elicited measurable anti-LecA salivary IgA response, with a higher overall response in the intramuscular group. Importantly, salivary IgA also prevented the attachment of E. histolytica to mammalian cells. Finally, exploratory analysis of other tissues in a limited subset of animals indicated durable antigen-specific immune responses in the gut and lymph nodes. Together, these results emphasize the broader potential of the GLA-3M-052 liposome adjuvant for other vaccine indications requiring mucosal responses.
Recurrent infections are a serious problem with diarrheal infectious diseases, especially in low-resource parts of the world. In a large, multinational, prospective case study of children with moderate-to-severe diarrhea, amebiasis ranked among the top 15 causes of diarrhea in the first 2 y of life in developing countries.Citation37 Thus, the development of immune memory is an important objective for the development of new vaccine candidates. The role of AS03 and MF59 adjuvants in shaping immune memory has been demonstrated for influenza vaccines,Citation38,Citation39 and the adjuvant component 3M–052 has been shown to generate bone marrow-residing antibody-secreting cells.Citation40 In our study, the intramuscular group demonstrated IgG+ and IgA+ BMEM responses. Moreover, intranasally and intramuscularly immunized groups with 100 µg LecA + GLA-3M-052 liposomes demonstrated a significant increase in CXCL-13 following the first and last immunizations. Circulating CXCL-13 a week after vaccination can serve as a useful biomarker to quantify GC TFH cell frequencies in mice and NHPs.Citation28 TFH cells are crucial along with FDCs for productive B cell interactions in the GCs. Thus, the adjuvanted vaccine candidate initiated a strong GC reaction, which likely translated into strong antibody and memory cell responses. These are important findings for developing vaccines toward pathogens that successfully evade the immune response or undergo antigenic variation.
There are several limitations to our study. Due to restrictions on animal availability because of the COVID-19 pandemic, we could not accommodate additional control or experimental groups, and group size was slightly smaller in some groups. For the same reason, we had to accommodate a wide range of animal ages and weights, likely increasing immune response variability, although this was mitigated by adjusting for these factors in our analyses. Unfortunately, though robust IFN-γ-secreting cells were detected in freshly collected PBMCs at the endpoint of the study, we could not detect IFN-γ-secreting cells in thawed PBMCs from earlier time points due to high signals of nonspecific background related to processing, storage, and/or shipping conditions. Finally, a rhesus macaque challenge model for E. histolytica has not been established, so it was not possible to challenge immunized animals with E. histolytica trophozoites to assess protection from colitis in vivo and correlate efficacy to immune responses. As mentioned earlier, only one group has used a non-human primate challenge model, and these studies were not continued. Future vaccine studies may consider development of a challenge model since we know that primates can be infected naturally by E. histolytica variants and that vaccination can recapitulate the human protective immune response.Citation41
Animal models of E. histolytica infections can recapitulate pathogenesis as well as immune responses observed in humans, thus facilitating mechanistic studies, formulation optimization, assessment of memory responses, as well as efficacy trials. However, there is a lack of a complete infection animal model of amebiasis by oral–fecal route involving cyst to trophozoite transition as seen during natural infection of children. Moreover, only a fraction of people colonized with E. histolytica actually go on to develop symptomatic disease, and the factors that influence this are largely unknown. Inability to study the early steps at the parasite–host interface upon natural infection limits our ability to predict the course of the disease in different individuals based solely on the immunogenicity studies in animals.
In conclusion, the reported study represents a significant step toward development of an amebiasis vaccine candidate suitable for first-in-human testing. Considering the similarity between rhesus macaque and human immune responses, this study provides an encouraging demonstration of durable mucosal and systemic antibody and cellular immunogenicity elicited by vaccination using a pharmaceutically acceptable vaccine antigen and adjuvant formulation. Importantly, the desired immune response profile was achievable by intranasal or intramuscular immunization with 100 µg LecA, enabling the potential for two distinct product development pathways. A Phase 1 clinical trial is being planned to assess the vaccine safety and immunogenicity in human participants, using both the intranasal and intramuscular routes to determine the most promising candidate in humans. Moreover, the current study indicates that the GLA-3M-052 liposome adjuvant may be useful in vaccine development for other pathogens. For instance, concurrent mucosal IgA and systemic IFN-γ responses are key readouts for protection from not only amebiasis but also other mucosal pathogens like Cryptosporidium. Thus, advancements in E. histolytica vaccine research as reported here can provide the basis for developing strategies against other priority mucosal pathogens.
Author contributions
MMA, WAP, and CBF conceived and designed the experiments. MMA, FX, DC, AG, EM, CC, DKS, and FV performed the experiments. MMA, FX, DC, WRL, CC, WAP, and CBF analyzed and interpreted the data. SJS, RK, KP, BMM, ARM, MAT, and SG contributed reagents, materials, experimental tools, analysis tools, or data. MMA, SJS, and CBF wrote the paper.
NHP_SupplInfo_HVI_2024_06_14_clean.docx
Download MS Word (479 KB)Acknowledgments
We thank Valerie Soza for scientific editing of the manuscript, Elise Larson for project management assistance, Sudhir Kasturi for scientific discussions and technical advice, and Janet Lathey, Steve Huang, Larry Wolfraim, Nancy Ulbrandt, Lisa Wei, Colleen Sico, Kimberly Taylor, Annie Mo, Clint Florence, and Myron Levine for helpful suggestions and/or programmatic assistance.
Disclosure statement
The authors declare no Competing Non-Financial Interests but the following Competing Financial Interests. MAT is a contract worker for 3M, and 3M-052 is an asset of 3M Healthcare. WAP is a consultant for TechLab, Inc. and in addition receives royalties for amebiasis diagnostics that are donated in their entirety to the American Society of Tropical Medicine and Hygiene. KP is an employee of TechLab, Inc., and amebiasis diagnostics are an asset of TechLab’s. MMA, WAP, and CBF are inventors on patents and/or patent applications involving the vaccine formulation represented in this article. All other authors declare no competing interests.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on request.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website at https://doi.org/10.1080/21645515.2024.2374147.
Additional information
Funding
References
- Ouattara M, N’guéssan NA, Yapi A, N’goran EK. Prevalence and spatial distribution of Entamoeba histolytica/dispar and Giardia lamblia among schoolchildren in Agboville area (Côte d’Ivoire). PLOS Negl Trop Dis. 2010;4:e574. doi:10.1371/journal.pntd.0000574.
- WHO/PAHO/UNESCO report. A consultation with experts on amoebiasis. Epidemiol Bull. Mexico City, Mexico; 1997. 1997 Jan 28–29; 18:13–13.
- Tellevik MG, Moyo SJ, Blomberg B, Hjøllo T, Maselle SY, Langeland N, Hanevik K, Jex AR. Prevalence of Cryptosporidium parvum/hominis, Entamoeba histolytica and Giardia lamblia among young children with and without diarrhea in Dar es Salaam, Tanzania. PLOS Negl Trop Dis. 2015;9(10):e0004125. doi:10.1371/journal.pntd.0004125.
- Mondal D, Petri WA, Sack RB, Kirkpatrick BD, Haque R. Entamoeba histolytica-associated diarrheal illness is negatively associated with the growth of preschool children: evidence from a prospective study. Trans R Soc Trop Med Hyg. 2006;100(11):1032–8. doi:10.1016/j.trstmh.2005.12.012.
- Watanabe K, Gatanaga H, Escueta-de Cadiz A, Tanuma J, Nozaki T, Oka S, Walson JL. Amebiasis in HIV-1-infected Japanese men: clinical features and response to therapy. PLOS Negl Trop Dis. 2011;5(9):e1318. doi:10.1371/journal.pntd.0001318.
- Shirley DT, Farr L, Watanabe K, Moonah S. A review of the global burden, new diagnostics, and current therapeutics for amebiasis. Open Forum Infect Dis. 2018;5(7):ofy161. doi:10.1093/ofid/ofy161.
- Petri WA, Smith RD, Schlesinger PH, Murphy CF, Ravdin JI. Isolation of the galactose-binding lectin that mediates the in vitro adherence of Entamoeba histolytica. J Clin Invest. 1987;80(5):1238–44. doi:10.1172/JCI113198.
- Haque R, Mondal D, Shu J, Roy S, Kabir M, Davis AN, Duggal P, Petri WA. Correlation of interferon-γ production by peripheral blood mononuclear cells with childhood malnutrition and susceptibility to amebiasis. Am J Trop Med Hyg. 2007;76(2):340–4. doi:10.4269/ajtmh.2007.76.340.
- Haque R, Ali IM, Sack RB, Farr BM, Ramakrishnan G, Petri WA Jr. Amebiasis and mucosal IgA antibody against the Entamoeba histolytica adherence lectin in Bangladeshi children. J Infect Dis. 2001;183(12):1787–93. doi:10.1086/320740.
- Abd Alla MD, White GL, Rogers TB, Cary ME, Carey DW, Ravdin JI. Adherence-inhibitory intestinal immunoglobulin A antibody response in baboons elicited by use of a synthetic intranasal lectin-based amebiasis subunit vaccine. Infect Immun. 2007;75(8):3812–22. doi:10.1128/IAI.00341-07.
- Abd Alla MD, Wolf R, White GL, Kosanke SD, Cary D, Verweij JJ, Zhang M-J, Ravdin JI. Efficacy of a Gal-lectin subunit vaccine against experimental Entamoeba histolytica infection and colitis in baboons (Papio sp.). Vaccine. 2012;30(20):3068–75. doi:10.1016/j.vaccine.2012.02.066.
- Quach J, St-Pierre J, Chadee K. The future for vaccine development against Entamoeba histolytica. Hum Vaccin Immunother. 2014;10(6):1514–21. doi:10.4161/hv.27796.
- Mann BJ, Chung CY, Dodson JM, Ashley LS, Braga LL, Snodgrass TL. Neutralizing monoclonal antibody epitopes of the Entamoeba histolytica galactose adhesin map to the cysteine-rich extracellular domain of the 170-kilodalton subunit. Infect Immun. 1993;61(5):1772–8. doi:10.1128/iai.61.5.1772-1778.1993.
- Barroso L, Abhyankar M, Noor Z, Read K, Pedersen K, White R, Fox C, Petri WA Jr, Lyerly D. Expression, purification, and evaluation of recombinant LecA as a candidate for an amebic colitis vaccine. Vaccine. 2014;32(10):1218–24. doi:10.1016/j.vaccine.2013.06.056.
- Guo X, Barroso L, Becker SM, Lyerly DM, Vedvick TS, Reed SG, Petri WA, Houpt ER. Protection against intestinal amebiasis by a recombinant vaccine is transferable by T cells and mediated by gamma interferon. Infect Immun. 2009;77(9):3909–18. doi:10.1128/IAI.00487-09.
- Abhyankar MM, Orr MT, Kinsey R, Sivananthan S, Nafziger AJ, Oakland DN, Young MK, Farr L, Uddin MJ, Leslie JL, et al. Optimizing a multi-component intranasal Entamoeba histolytica vaccine formulation using a design of experiments strategy. Front Immunol. 2021;12:683157. doi:10.3389/fimmu.2021.683157.
- Abhyankar MM, Noor Z, Tomai MA, Elvecrog J, Fox CB, Petri WA. Nanoformulation of synergistic TLR ligands to enhance vaccination against Entamoeba histolytica. Vaccine. 2017;35(6):916–22. doi:10.1016/j.vaccine.2016.12.057.
- Abhyankar MM, Orr MT, Lin S, Suraju MO, Simpson A, Blust M, Pham T, Guderian JA, Tomai MA, Elvecrog J, et al. Adjuvant composition and delivery route shape immune response quality and protective efficacy of a recombinant vaccine for Entamoeba histolytica. NPJ Vaccines. 2018;3(1):22. doi:10.1038/s41541-018-0060-x.
- Murphy BM, Chen JZ, Rolo M, Eldam M, Jordan L, Sivananthan SJ, Kinsey R, Guderian JA, Pedersen K, Abhyankar M, et al. Intranasal delivery of a synthetic Entamoeba histolytica vaccine containing adjuvant (LecA + GLA-3M-052 liposomes): in vitro characterization. Int J Pharm. 2022;626:122141. doi:10.1016/j.ijpharm.2022.122141.
- Hendrickson SM, Thomas A, Prongay K, Haertel AJ, Garzel LM, Gill L, Barr T, Rhoades NS, Reader R, Galan M, et al. Reduced infant rhesus macaque growth rates due to environmental enteric dysfunction and association with histopathology in the large intestine. Nat Commun. 2022;13(1):234. doi:10.1038/s41467-021-27925-x.
- Macaque Genome Sequencing R, Consortium A, Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316(5822):222–34. doi:10.1126/science.1139247.
- Niemuth NA, Rudge TL, Sankovich KA, Anderson MS, Skomrock ND, Badorrek CS, Sabourin CL, Schieffelin J. Method feasibility for cross-species testing, qualification, and validation of the filovirus animal nonclinical group anti-Ebola virus glycoprotein immunoglobulin G enzyme-linked immunosorbent assay for non-human primate serum samples. PLOS ONE. 2020;15(10):e0241016. doi:10.1371/journal.pone.0241016.
- Beghetto E, Buffolano W, Spadoni A, Del Pezzo M, Di Cristina M, Minenkova O, Petersen E, Felici F, Gargano N. Use of an immunoglobulin G avidity assay based on recombinant antigens for diagnosis of primary Toxoplasma gondii infection during pregnancy. J Clin Microbiol. 2003;41(12):5414–8. doi:10.1128/JCM.41.12.5414-5418.2003.
- Bricault CA, Kovacs JM, Badamchi-Zadeh A, McKee K, Shields JL, Gunn BM, Neubauer GH, Ghantous F, Jennings J, Gillis L, et al. Neutralizing Antibody Responses following Long-Term Vaccination with HIV-1 Env gp140 in Guinea Pigs. J Virol. 2018;92(13):e00369–18. doi:10.1128/JVI.00369-18.
- Holec-Gąsior L, Sołowińska K. IgG avidity test as a tool for discrimination between recent and distant Toxoplasma gondii infection—current status of studies. Antibodies. 2022;11(3):52. doi:10.3390/antib11030052.
- Benmeziane K, Delache B, Langlois S, Scarlatti G, Le Grand R, Cavarelli M. Isolation and phenotypic characterization of human and nonhuman primate intestinal lamina propria mononuclear cells. STAR Protocols. 2022;3(4):101815. doi:10.1016/j.xpro.2022.101815.
- Ravdin JI, Guerrant RL. Role of adherence in cytopathogenic mechanisms of Entamoeba histolytica. Study with mammalian tissue culture cells and human erythrocytes. J Clin Invest. 1981;68(5):1305–13. doi:10.1172/JCI110377.
- Havenar-Daughton C, Lindqvist M, Heit A, Wu JE, Reiss SM, Kendric K, Bélanger S, Kasturi SP, Landais E, Akondy RS, et al. CXCL13 is a plasma biomarker of germinal center activity. Proc Natl Acad Sci USA. 2016;113(10):2702–7. doi:10.1073/pnas.1520112113.
- Smirnov D, Schmidt JJ, Capecchi JT, Wightman PD. Vaccine adjuvant activity of 3M-052: an imidazoquinoline designed for local activity without systemic cytokine induction. Vaccine. 2011;29(33):5434–42. doi:10.1016/j.vaccine.2011.05.061.
- Liang H, Poncet D, Seydoux E, Rintala ND, Maciel M, Ruiz S, Orr MT. The TLR4 agonist adjuvant SLA-SE promotes functional mucosal antibodies against a parenterally delivered ETEC vaccine. NPJ Vaccines. 2019;4(1):1–8. doi:10.1038/s41541-019-0116-6.
- Lotter H, Zhang T, Seydel KB, Sl S Jr, Tannich E. Identification of an epitope on the Entamoeba histolytica 170-kD lectin conferring antibody-mediated protection against invasive amebiasis. J Exp Med. 1997;185:1793–801. doi:10.1084/jem.185.10.1793.
- Kaur U, Sharma AK, Sharma M, Vohra H. Distribution of Entamoeba histolytica Gal/GalNAc lectin-specific antibody response in an endemic area. Scand J Immunol. 2004;60(5):524–8. doi:10.1111/j.0300-9475.2004.01512.x.
- Nakada-Tsukui K, Nozaki T. Immune response of amebiasis and immune evasion by Entamoeba histolytica. Front Immunol. 2016;7:175. doi:10.3389/fimmu.2016.00175.
- Bernin H, Marggraff C, Jacobs T, Brattig N, An LV, Blessmann J, Lotter H. Immune markers characteristic for asymptomatically infected and diseased Entamoeba histolytica individuals and their relation to sex. BMC Infect Dis. 2014;14(1):621. doi:10.1186/s12879-014-0621-1.
- Tran VQ, Herdman DS, Torian BE, Reed SL. The neutral cysteine proteinase of Entamoeba histolytica degrades IgG and prevents its binding. J Infect Dis. 1998;177(2):508–11. doi:10.1086/517388.
- Abhyankar MM, Mann BJ, Sturek JM, Brovero S, Moreau GB, Sengar A, Richardson CM, Agah S, Pomés A, Kasson PM, et al. Development of COVID-19 vaccine using a dual toll-like receptor ligand liposome adjuvant. NPJ Vaccines. 2021;6(1):137. doi:10.1038/s41541-021-00399-0.
- Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. The Lancet. 2013;382(9888):209–22. doi:10.1016/S0140-6736(13)60844-2.
- Khurana S, Verma N, Yewdell JW, Hilbert AK, Castellino F, Lattanzi M, Del Giudice G, Rappuoli R, Golding H. MF59 adjuvant enhances diversity and affinity of antibody-mediated immune response to pandemic influenza vaccines. Sci Transl Med. 2011;3(85):8548–8548. doi:10.1126/scitranslmed.3002336.
- Galson JD, Trück J, Kelly DF, van der Most R. Investigating the effect of AS03 adjuvant on the plasma cell repertoire following pH1N1 influenza vaccination. Sci Rep. 2016;6(1):37229. doi:10.1038/srep37229.
- Pino M, Abid T, Pereira Ribeiro S, Vv E, Floyd K, Smith JC, Latif MB, Pacheco-Sanchez G, Dutta D, Wang S, et al. A yeast expressed RBD-based SARS-CoV-2 vaccine formulated with 3M-052-alum adjuvant promotes protective efficacy in non-human primates. Sci Immunol. 2021;6(61):eabh3634. doi:10.1126/sciimmunol.abh3634.
- Levecke B, Dreesen L, Dorny P, Verweij JJ, Vercammen F, Casaert S, Vercruysse J, Geldhof P. Molecular identification of Entamoeba spp. in captive nonhuman primates. J Clin Microbiol. 2010;48(8):2988–90. doi:10.1128/JCM.00013-10.