
ABSTRACT
This paper reviews recent research relating to the application of bioinformatics approaches to determining HIV-1 protease specificity, outlines outstanding issues, and presents a new approach to addressing these issues. Leading machine learning theory for the problem currently suggests that the direct encoding of the physicochemical properties of the amino acid substrates is not required for optimal performance. A number of amino acid encoding approaches which incorporate potentially relevant physicochemical properties of the substrate are identified, and are evaluated using a nonlinear task decomposition based neuroevolution algorithm. The results are evaluated, and compared against a recent benchmark set on a nonlinear classifier using only amino acid sequence and identity information. Ensembles of these nonlinear classifiers using the physicochemical properties of the substrate are demonstrated to consistently outperform the recently published state-of-the-art linear support vector machine based approach in out-of-sample evaluations.
Introduction
Human immunodeficiency virus (HIV) is the causative agent of AIDS (acquired immunodeficiency syndrome).Citation1,2 For the HIV virus to become infectious, it must mature to its virion stage, allowing it to travel between cells. HIV encodes many of the proteins required for its lifecycle in polypeptide chains which must be cleaved to produce several different structural and functional peptides. To achieve this, HIV also encodes the HIV-1 protease.Citation3 The specificity of HIV-1 protease allows it to cleave the viral Gag and Gag-Pol precursor polyproteins.Citation4
According to figures released by the World Health Organization (WHO), the AIDS epidemic was responsible for between 1.4 and 1.7 million deaths globally in 2013.Citation5 Although no cure for HIV or AIDS has been found, this number has decreased since 2005. In the United States of America, HIV incidence is stable at 50,000 per year.Citation6 One approach which has contributed to the reduction in AIDS deaths is the use of a protease inhibitor, which binds to the active site of the HIV-1 protease preventing it from functioning correctly. This interrupts an essential part of the HIV maturation process, rendering it noninfectious. HIV is however a highly robust virus, where it is estimated that, in an infected individual, every possible single-point mutation can occur between 104 and 105 times per day.Citation7 For a given protease inhibitor, it is likely that resistant variations of the strain will eventually evolve.Citation8 Therefore, to design efficient inhibitors or cocktails of inhibitors that are able to combat the robustness of HIV, a thorough understanding of the protease specificity is required.Citation9
The dataset
The activity of the HIV-1 protease-peptide interaction is dictated by the 4 amino acids at either side of a scissile bond, forming the substrate of the protease.Citation10 The AAs in this 8 amino acid area (8-mer/octamer) are labeled { P4, P3, P2, P1, P1′, P2′, P3′, P4′} under the typically used nomenclature introduced by Schechter and Berger,Citation11 with the protease active site, if it exists, located between positions P1 and P1′.
As each letter in the octamer has 20 different possible identities, there are 20Citation8 (25,600,000,000) possible 8 character amino acid sequences. With numbers such as this, it is unrealistic to define the specificity through brute force laboratory work. Focus has therefore turned to the use of machine learning approaches to generalize classifications for octamers as matching or not matching HIV-1 protease specificity, given a subset of octamers for which the specificity of HIV-1 protease has been defined.Citation12
The data used in this case study was taken from the UCI machine learning data repository.Citation13 Each exemplar has 2 attributes: an 8 letter string representing the 8 amino acids in the P4 to P4′ locations, and a label, “1” or “−1,” representing whether this octamer would be cleaved (case) or not cleaved (control) respectively by the HIV-1 protease at the P1-P1′ site. The allowed alphabet for the character string representing the octamer is {A,R,N,D,C,Q,E,G,H,I,L,K,M,F,P,S,T,W,Y,V}, each representing a different common amino acid. The dataset is comprised of 4 separately published datasets:
746: 746 exemplars (401 cleaved, 345 noncleaved)Citation14
1625: 1,625 exemplars (374 cleaved, 1251 noncleaved)Citation8
Schilling: 3,272 exemplars (434 cleaved, 2,838 noncleaved)Citation15
Impens: 947 exemplars (149 cleaved, 798 noncleaved)collected from 4 publicationsCitation16-19
This corresponds to a total of 6,590 exemplars, of which 1,358 represent HIV-1 protease cleavages. These 4 data sets contain 740 repeated exemplars, and 10 octamers with different classifications in different sets. Removing the conflicting exemplars and reducing the repeated exemplars each to a single instance, lowers the count to 5830 exemplars, of which 991 represent octamers cleavable by the HIV-1 protease. The available data represents roughly 0.00002% of the possible combinations.
Literature review
Much of machine learning research identified on this topic relates to the definition and encoding the octamer sequences in a manner suitable for interpretation by a machine learning algorithm. Given a limited number of exemplars, the training algorithm can learn patterns in the training data (typically a sample of the full distribution) which are not reflected in the full distribution, i.e., the learning algorithm will overfit the training data.Citation20 Removing irrelevant and redundant features can produce more robust classifiers which are more resilient to overfitting,Citation21 as well as reducing the complexity and computation time of the solutions.Citation22 Therefore, dimensionality reduction has been a very active topic in this domain.
A critical review of work in this field up to 2007 is presented in the paper “Bioinformatic approaches for modeling the substrate specificity of HIV-1 protease: an overview,” by Rögnvaldsson, You and Garwicz.Citation23 Much of the work carried out up to this point uses only a very limited number of exemplars. Additionally, as discussed in the review, many of the exemplars were generated using a single point mutation on known cleavages, representing a very biased sampling and likely the introduction of artificial patterns in the dataset.
The 2004 paper “Why neural networks should not be used for HIV-1 protease cleavage site prediction,” by Rögnvaldsson and You, noted that nonlinear classifiers such as the Multilayer Perceptron offered no advantage for this problem over simple linear classifiers (typically considered less powerful) such as the perceptron or SVM using a linear kernel (LSVM), when orthogonal encoding is employed.Citation24 In orthogonal encoding, each of the 20 amino acids is represented by a unique 20 bit binary vector comprising 19 0′s and a single 1. This approach encodes only the identities and sequence of the amino acids, without any direct encoding of their physicochemical properties. However, given sufficient data, this encoding can be used to indirectly learn relevant properties of the amino acids. Recently (March 2015), a paper entitled “State of the art prediction of HIV-1 protease cleavage sites,” by Rögnvaldsson, You and Garwicz, was published in the journal Bioinformatics, which purports that, in the context of increased data availability, the “state of the art” for this problem is achieved using an LSVM and orthogonal encoding, which outperforms a number of direct physicochemical encodings combined with an SVM using a (nonlinear) radial basis function kernel (RSVM).Citation25 The good performance for orthogonal encoding combined with linear classifiers on this problem has been corroborated by a number of sources.
In contrast to this, other recent work on the topic purports good performance when the physicochemical properties of the amino acids are directly encoded, although much of this work has been carried out on much smaller data sets. Under this encoding, the use of nonlinear classifiers also appears more relevant. A recently published example by Niu et al. evaluated a set of 30 physicochemical properties of the amino acids at specific offsets from the scissile bond, e.g. the “normalized frequency of coil” for the amino acid at position P1.Citation26 The feature set was identified by carrying out a filtering operation on the AAindex database, which defines numerous scales relating to a range of properties of the 20 coding amino acids.Citation27 An AdaBoost (Adaptive Boosting) approach was applied to classify the reduced feature set.Citation28 The results achieved were positive, and the relevance of many of the features identified by their algorithm could be rationalized a posteriori in the context of HIV-1 protease, suggesting credibility to the power of the filtering algorithm used, and the relevance of numerous physicochemical properties to defining specificity of HIV-1 protease.
The work of Nanni and Lumini also suggests that physicochemical properties can be used to increase performance over orthogonal encoding.Citation29 Under their MppS (multiple physicochemical properties and support vector machines) algorithm, relevant physicochemical properties are selected from the AAindex database using a sequential forward floating selection (SFFS) algorithm. Individual LSVMs are trained using a singular different physicochemical property. The SVMs were combined into an ensemble and their collective output selected using the “max rule.”
Öztürk et al.Citation30 present a novel hybrid algorithm to reduce the dimensionality of orthogonal encoding which looks at the overlap between 2 separate feature selection algorithms; consistency based, and an SVM based method of Recursive Feature Elimination (RFE). Öztürk et al. also highlight combining orthogonal encoding and physicochemical properties as an approach to improving performance.
Song et al. developed the PROSPER (Protease specificity prediction server) web server for the identification of target cleavage sites for 24 different proteases, including the HIV-1 protease, using a combination of sequence and structure characteristics.Citation31 The features used are orthogonal encoding, secondary structure (using PSIPREDCitation32), solvent accessibility (using SCRATCHCitation33), and flexible areas of peptide which are not static in conformation (using DISOPRED2Citation34). An SVM with an RBF kernel is used to classify the exemplars.
Gök and Özcerit evaluated a feature set, denoted OETMAP, combining orthogonal encoding with physicochemical properties of the amino acids.Citation35 Physicochemical properties are encoded for each amino acid using a binary vector representing membership of 10 respective groups: {small, tiny, proline, charged, negative, positive, hydrophobic, polar, aromatic, aliphatic}, as defined by Taylor et al..Citation36 Although Gök and Özcerit found the use of OETMAP encoding to offer an improvement over orthogonal encoding, later evaluations by Rögnvaldsson et al. on a larger dataset found it to in fact be inferior to orthogonal encoding.Citation25
Later work by Gök further evaluated the use of physicochemical properties.Citation37 Each of the 544 physicochemical properties in the AAindex was evaluated individually using an encoding where the non-zero values in an orthogonal encoding are replaced by the corresponding value from the scale.Citation38 The performance of the top 10, 20, and 30 most relevant physicochemical scales identified using this approach were evaluated. The use of 20 features per amino acid was shown to outperform orthogonal encoding, but performance was reduced when the best 10 or 30 features were evaluated. An LSVM outperformed an RSVM when both were trained on this physicochemical feature set.
Newell evaluated a number of typical feature selection algorithms on synthetic active substrates and found that even the best algorithms identified mostly incorrect features, and were, in general, only able to detect simple or extremely strong features with confidence.Citation39 Following this observation, Newell introduces a new algorithm, which uses the background probability of observing each amino acid to adjust the significance placed on localized sequence features (sets of amino acids at particular positions) identified in the training data. The approach is capable of detecting first order features that are over or under-represented, or higher order features that are over-represented.
Li et al. employ a nonlinear dimensionality reduction, to reduce the features in orthogonally encoded octamers.Citation40 An SVM is used to classify the reduced feature set. Their results show that the information relevant to the specificity of the HIV-1 protease in orthogonal encoding can be maintained in a reduced dimensionality.
Similarly, Kim et al. introduced the FS-MLP feature selection algorithm to reduce the orthogonal encoding from 160 bits to 14 key features.Citation41 FS-MLP is a 2 stage process in which a trained MLP is evaluated using a heuristic approach to test combinations of input vectors to evaluate which produce an activation value above a pre-selected threshold. A decision tree, a simple perceptron and an LSVM were evaluated on the reduced feature set, and all were shown to offer an improvement over the full feature set. The 14 feature vector suggested by Kim et al. was evaluated briefly by us, by training an LSVM on the {746, 1625, Schilling} data set, and testing it on the Impens dataset. The performance in terms of area under the curve for the receiver operating characteristic (ROC-AUC)Citation42 was observed to drop from 0.900 to 0.834 compared to standard orthogonal encoding.
Jaeger and ChenCitation43 suggest a new reduced feature set in which each amino acid is represented by 4 real valued scales; hydropathy index, molecular mass, polarity and occurrence percentage. The occurrence percentage describes the average occurrence of each particular amino acid calculated from a set of more than 1150 proteins. This feature set is used to train a multiple classifier systems (MCS) comprising neural networks, SVMs, and decision trees. An RSVM was shown to out-perform an LSVM in these experiments.
Nanni and LuminiCitation44 evaluate the use of ensembles using different feature encodings and demonstrate that ensembles can outperform stand-alone methods. The best performance was achieved using an ensemble with 3 different feature sets; 1) a variation of the quasi-residue couple model, 2) a selection of physicochemical properties, and 3) a method for reducing the alphabet from 20 amino acids to a set value. Orthogonal encoding was also shown to be inferior to both the quasi-residue couple model and the physicochemical properties individually.
Later work by Nanni and Lumini evaluated a number of different encoding methods.Citation45 By applying principal component analysis to the AAIndex, they were able to identify a set of 19 physicochemical properties (denoted PC19) that best describe the variance of the database. The feature set was evaluated using an RSVM. Notably, performance was further increased when the orthogonal and PC19 encodings are combined. The suitability of the PC19 data set was evaluated for the research carried out here by training an RSVM using the parameters provided by Nanni and Lumini on the {746, 1625, Schilling} dataset, and evaluating it on the Impens data set. The classifier achieved an accuracy of 88.49%, and an ROC-AUC of 0.899.
Yuan et al.Citation46 present an approach which builds heavily on previous research of Nanni and Lumini,Citation45 by combining features extracted from the AAindex database using PCA, with features extracted by Nonlinear Fisher transform, and orthogonal encoding. The work of Yuan et al. goes further by reducing the resultant feature vectors from 160, 152 and 144 values, to 120, 124 and 106 values for orthogonal, PCA and Fisher transform encoding respectively. Both the PCA and Fisher feature sets are demonstrated to outperform orthogonal encoding in a number of experiments.
The performance of the Fisher feature set was evaluated by us, by training an RSVM on the {746, 1625, Schilling} dataset using this encoding, and evaluating it on the Impens data set. The RSVM performed well, achieving an accuracy of 88.7% and an ROC-AUC of 0.898. An RSVM trained on a 456 feature vector combining orthogonal encoding with the PCA and Fisher feature sets, and using parameters suggested by Yuan et al. achieved an accuracy of 85.22% and an ROC-AUC of 0.881 when tested on the Impens dataset, much lower than the performance rates suggested by the experiments of Yuan et al.
Recent work by Nanni and Lumini in this field relates to representing the octamer as an 8×8 matrix instead of the typical vector based encoding.Citation47 The matrix represents both the sequence information and a selected physicochemical scale.Citation48 The matrix is then treated as an image, and a texture descriptor taken from image processing theory is applied to characterize the key features, which in turn form the inputs to a standard classifier. Using the matrix based representation was shown to give lower performance than the standard vector based encoding, but offered improved performance when combined with the standard vector encoding for HIV-1 protease specificity.Citation49
Oğul noted that existing approaches tended to use the identities or properties of the amino acids at specific sites, but did not directly consider potential interactions between amino acids in the substrate.Citation50 Oğul used a modified version of the variable order Markov model to represent this information. Very positive results were reported, but the evaluations were limited to the 1625 data set.
It is typical for the physicochemical properties of the substrate to play a role in the specificity of a protease. Proteases preferentially cleave substrates within extended loop regions,Citation31 while properties such as hydrophobicity can affect the solvent accessible area of the substrate.Citation51 Additionally, previous wet lab research has suggested that HIV-1 protease specificity depends on the conformation of the substrate, rather than the recognition of specific amino acids.Citation20 Orthogonal encoding, as was employed in the recently published state of the art for detecting HIV-1 protease cleavage points in a peptide,Citation25 can be considered only an indirect encoding of such properties of the amino acids from which the learning algorithm must derive its own interpretation and estimates of values. However, it is not apparent if there is currently sufficient available training data (and of sufficient quality) to allow the learning algorithms to infer these properties optimally from an orthogonal encoding. Indeed, a number of the papers highlighted in the literature review purport improved performance through the use of the direct encoding of relevant physicochemical properties. The previous definition of the problem as linear relates to when orthogonal encoding is employed, but the use of physiochemical scales are a lossy lower dimensional encoding of the substrate, under which the problem does not necessarily maintain its linear separability.
A diversity matrix is presented in summarizing the different approaches stated in the reviewed papers to addressing the problem. There is a large amount of support in the literature for both the relevance and irrelevance of physicochemical properties to HIV-1 protease specificity.
Table 1. Diversity matrix of the different approaches taken to defining HIV-1 specificity, as noted in the literature reviewed. [*] denotes future planned work.
MFF-NEAT
The literature reviewed supports the assumption that orthogonal encoding is preferably handled with a linear classifier, but direct physicochemical encoding, which is also likely beneficial, typically requires a nonlinear classifier. If physicochemical properties are used in conjunction with the orthogonal encoding, it is unclear if standard learning algorithms would be able to handle both these sets of information optimally in conjunction with each other, or indeed if the 2 sets of data are best handled independently. Therefore, the classifier selected for evaluating combinations of disparate encodings should be capable of automatically identifying and handling multiple distinct patterns appropriately from a dataset. For this reason, the MFF-NEAT (modular feed forward neuroevolution of augmenting topologies) classifier was selected for this evaluation.Citation52 MFF-NEAT is a topology and weight evolving algorithm for artificial neural networks. It is a modification to the standard NEAT algorithm which adds the potential for automatic task decomposition to build and train solution models with a mixture-of-experts architecture.Citation53
NEAT is a robust neuroevolution algorithm based on 3 key concepts: complexification, speciation, and principled crossover.Citation54 In complexification, the initial solutions are minimalist, comprising only the input and output neurons connected by synapses. Over time new neurons and synapses are added through mutation to increase the complexity of the solution architectures. Architectural additions which don't improve performance are not expected to propagate through the population. Speciation identifies how similar solutions are by tracking the lineage of the architectural additions, and is used to maintain diversity in the population. Principled crossover uses the lineage tracking to limit crossover to common genes across solutions when producing children to minimize the “competing conventions problem.”Citation55
A solution produced by MFF-NEAT here is referred to as a “system” comprising a “gating network” and zero or more “expert networks.” The number of expert networks and the individual network architectures are dictated by the algorithm given the problem. To evaluate an input vector, it is first applied to each expert network. The original input vector and the outputs of the expert networks form the input vector to the gating network, which generates the overall output of the system. The expert networks do not work as an ensemble; none of the networks individually may work as a classifier for the problem. The expert networks are trained to deal with patterns in the data which may not directly or independently corresponding to classifier outputs. MFF-NEAT harnesses the complexification, speciation, and principled crossover of the NEAT algorithm, co-evolves a population of expert networks, and employs a form of negative correlation to assemble the expert and gating networks into complete systems. An example of a simple MFF-NEAT system is presented in .
Figure 1. Sample architecture produced by MFF-NEAT for a feature set with 4 inputs and 3 outputs.
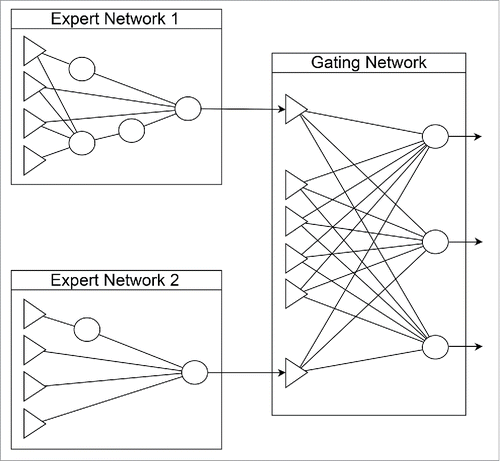
Initial systems comprise only a gating network. Both the expert and gating networks are evolved using standard NEAT operators, but are speciated independently. Performance is recorded on each individual training exemplar in a structure referred to as a “coverage vector.” The coverage vector of a system is simply the performance of the system on each exemplar. The coverage vector of an expert species is calculated by sampling the performance of different systems that employ that expert network species. Each new expert species identified is recorded in an “expert archive” with the associated coverage vector and a holotype of that species. The negative correlation uses the coverage vectors of the systems and the coverage vector recorded for each species to determine the species of expert network to add to a system. An expert of that species is then spawned from the population or the holotype, and added to the system. Expert networks are connected to a system by adding an additional input neuron to the gating network. When a maximum number of expert networks allowed for a system is reached, mutation allows for swapping of the expert networks.
Although MFF-NEAT was designed to increase coverage of the search space by maintaining diversity of the solutions, the population members can settle on suboptimal solutions known as local minima due to the greediness of genetic algorithms. If functionality desirable for the improvement of a system is not being evolved elsewhere in the population, the advantages of the MFF-NEAT system are limited. The previously published algorithm was modified here to address this short coming, through the spawning of an island which runs another instance of the algorithm,Citation56 but which focuses training only on the exemplars handled poorly by the best performing system. The island starts with a completely new population of solutions and its own empty expert archive. After a number of generations, useful expert network species identified in the island can be made available to systems in the main population through adding them to the original expert archive.
The task decomposition provided by MFF-NEAT should allow it to optimally combine features of different encodings, or handle them independently as relevant, but also be able to isolate and handle relevant subsets of features without the need for an external dimensionality reduction algorithm. The MFF-NEAT software is available by e-mail.
Encodings
Based on the literature review, a number of different encodings of the physicochemical properties of the amino acid substrate were identified which were considered to potentially provide information relevant to defining the specificity of HIV-1 protease. An overview of the encodings is provided in .
Table 2. Selection of amino acid encoding formats identified in the literature review.
The z-Scales
The z-Scales are a set of 5 real valued principal properties for describing the variance of amino acids. The scales were derived using PCA from a larger set of physicochemical properties by Hellberg et al. and Sandberg et al.Citation57,58 The scales used in this research correspond to hydrophobicity, steric properties, polarizability, polarity, and electronic effects.
Physicochemical group
The physicochemical group corresponds to a set of 7 real valued scales taken from the AAindex database. The 7 values selected relate to the volume, mass, hydrophobicity, surface area, and the propensity to form an α-helix, β-strand and turn.Citation59,60
Hydrophobicity group
This encoding places each amino acid into 1 of 3 groups, {{D,E,N,Q,R,K}, {C,S,T,P,G,H,Y}, {A,M,I,L,V,F,W}}, corresponding to whether the amino acid is considered hydrophobic, hydrophilic or neutral.Citation59 Membership of the groups is encoded as a 3 bit orthogonal vector per amino acid.Citation60
BLOMAP
BLOMAP uses the BLOSUM62 substitution matrix and the nonlinear Sammon projection to map the information in the matrix to a lower dimensionality (5 real values per amino acid) while retaining relevant information about the relationships between the amino acids.Citation60
Exchange (substitution) group
This encoding represents the groupings of amino acids which have a propensity to be substituted in homologous sequences through evolution, as derived from a PAM (Point accepted mutation) matrix.Citation59 The groups are as follows: {{H,R,K}, {D,E,N,Q}, {C}, {S,T,P,A,G}, {M,I,L,V}, {F,Y,W}}. Each amino acid is encoded as a 6 bit binary vector.Citation60
Orthogonal, PC19, Fisher, and Niu encoding are described in the literature review.
Results
The importance of out of sample testing has been well defined for the HIV-1 protease specificity problem.Citation8,25 For this reason, the {746, 1625, Schilling} data sets were used for training the classifiers and the Impens dataset used to evaluate the classifiers. The Impens data set was selected for generating the performance measures as it has not been used in the definition or evaluation of any of the physicochemical encodings we wish to evaluate, removing the possibility of a resubstitution error. Additionally, a benchmark has been published for this experiment using the current state of the art approach.Citation25
Individual classifiers
Several instances were identified in the literature review where it was noted that superior performance was achieved by combining the use of orthogonal or physicochemical encoding, over the performance of either individually.Citation30,31,35,46 Given this information, it was decided that each physicochemical encoding should be evaluated in conjunction with orthogonal encoding. Additional evaluations were carried out using combinations of the most promising feature sets: {Orthogonal, Physicochemical, Niu}, {Orthogonal, Physicochemical, Niu, z-Scales}, and combining the most promising feature set {Orthogonal, Physicochemical, Niu} with the occurrence percentage, as defined by Jaeger.
Twenty training sets were generated from the {746, 1625, Schilling} datasets using the approach described in the Methods section. Each data set was used to train MFF-NEAT classifiers using 10 different alternative amino acid encodings approaches. Summary statistics for each encoding method evaluated on the Impens dataset in terms of ROC-AUC are presented in . Accuracy is defined as ((true positives + true negatives) / (true positives + true negatives + false negatives + false positives)) at a threshold of 0.5. The columns IQR, 1Q and 3Q correspond to the values for the interquartile range, the first quartile and third quartile respectively.
Table 3. Summary statistics for the evaluated amino acid encoding approaches. Each row represents the performance of classifiers trained on same 20 samplings of the {746, 1625, Schilling} dataset and evaluated on the Impens data set. The performance of the LSVM using orthogonal encoding trained on the 20 samplings is included for reference. Results are rounded to 3 decimal places.
MFF-NEAT ensembles
In the literature review, a number of examples have been identified where performance on determining the HIV-1 protease specificity has been improved through the use of ensembles of classifiers.Citation26,29,44,47,49 Therefore, the set of 20 MFF-NEAT classifiers trained using each encoding was also evaluated as a single ensemble using the sum rule. The results are presented in . The performance of the ensembles in terms of ROC-AUC relative to the ensemble members is presented graphically by the solid black dots in . The whiskers show the maximum and minimum performance of individual classifiers within 1.5 times the interquartile range of the upper and lower quartiles respectively. The performance of individual classifiers outside the whiskers, denoted by the hollow circles, can be considered outliers.Citation61
Figure 2. ROC-AUC performance of each amino acid encoding evaluated as an ensemble of size 20 (solid black dot) overlapped with the performance of the individual classifiers used in each ensemble, represented as a box plot.
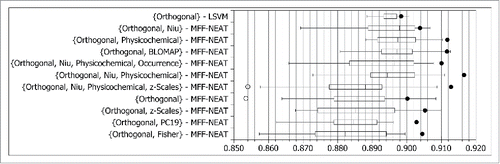
Table 4. The performance of the classifiers used to generate for each amino acid encoding approach, when taken as an ensemble.
Further evaluation of the {Orthogonal, Niu, Physicochemical} encoding
For the most promising encoding set identified, an additional 80 MFF-NEAT classifiers were trained using different samplings of the training data. The distribution of the performance of the full set of 100 classifiers using the {Orthogonal, Niu, Physicochemical} encoding, evaluated individually, is presented in . The performance of the 100 classifiers taken as an ensemble is presented in . To evaluate the potential error of the evaluation of the ensemble for this encoding presented in , the distribution of performances of 100 ensembles of size 20 taken from the pool of 100 is given in .
Figure 3. Distribution of the performances of the 100 classifiers trained using the {Orthogonal, Niu, Physicochemical} encoding in terms of ROC-AUC. The performances of the 20 classifiers used in for the same encoding are included for reference.
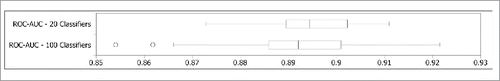
Figure 4. The distribution of ROC-AUC performances of 100 different ensembles each of size 20 taken from a pool of 100 classifiers using the {Orthogonal, Niu, Physicochemical} encoding.
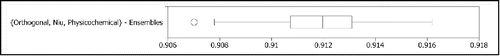
Table 5. Performance of an ensemble of 100 MFF-NEAT classifiers using {Niu, Physicochemical, Orthogonal} encoding, trained on various samplings of the {746, 1625, Schilling} dataset and evaluated on the Impens data set. An ensemble of LSVMs trained on the same samplings is included for reference.
Discussion
From the presented results, it appears that a single LSVM using orthogonal encoding can in general outperform a single MFF-NEAT classifier employing direct encoding of physicochemical properties. Each of the 100 training sets used to generate , was used to train an LSVM with orthogonal encoding, and the results compared against the corresponding MFF-NEAT classifiers using the {Orthogonal, Niu, Physicochemical} encoding. The LSVM achieved superior results to the MFF-NEAT classifier in 63% of the cases in terms of ROC-AUC. The average ROC-AUC over the 100 test sets were 0.896 and 0.892 for LSVM and MFF-NEAT respectively, corresponding to a 0.46% performance decrease for the best MFF-NEAT approach. Although the difference in performance is low, suggesting consistency, a p value of 0.002392 was generated by a paired student's t-Test on the results. It can therefore be stated with confidence that the LSVM approach is able to outperform an MFF-NEAT classifier trained using the most promising physicochemical based encoding evaluated here.
However, it appears that, when taken as an ensemble, the use of physicochemical properties combined with nonlinear classifiers give a consistent improvement in performance over the LSVM benchmark presented in using tuned parameters, the benchmark published by Rögnvaldsson et al.,Citation25 and the ensemble of LSVMs presented in . Although the performance of each LSVM classifier is good, the advantage gained by combining the different perspectives appears limited. Conversely, despite the fact that, in general, the performance of the MFF-NEAT classifiers individually was weaker than that of the SVM, the diversity of the solutions appears to have led to increased generalization on the Impens data set when combined. The under-performance of the individual MFF-NEAT classifiers and the low performance of the LSVM classifiers relative to the MFF-NEAT ensembles suggest that neither the LSVM nor MFF-NEAT classifiers may be best suited to this problem.
Table 6. Performance of an LSVM on the Impens dataset, when trained on the full {746, 1625, Schilling} data set, using a value of 1.284 for C.
If the entire set of cleaved and noncleaved octamers were available, there is evidence to suggest orthogonal encoding and linear classifiers would be sufficient to completely describe the problem. However, the amount of training data currently available is limited, which appears to restrict how well the learning algorithms can infer the mappings from a high level representation such as orthogonal encoding. In the context of limited data being available, a good classifier requires good generalization ability, and directly encoding relevant properties does appear to ameliorate performance. From both and it can be observed that the selection of physicochemical features to include in the input vector does impact performance, and indicates that MFF-NEAT is indeed able to take advantage of this additional information, where relevant, providing an increased performance relative to the use of orthogonal encoding only.
Next we examine the performance of the 100 classifiers trained using the {Niu, Physicochemical, Orthogonal} feature encoding. The distribution of the performance of the classifiers taken individually, as presented in , is roughly consistent with the distribution of the 20 classifier subset used in , meaning that the 20 classifiers used in the earlier experiments is a representative sampling. From , it appears that the performance of the ensemble for the {Niu, Physicochemical, Orthogonal} feature encoding set presented in is at the high end of what we should expect from this encoding (overly optimistic). The interquartile range in , however, still represents a good level of performance relative to the LSVM benchmark. Using the full set of 100 classifiers as a single ensemble actually reduced the performance, as shown in . This was later verified by charting the average performance of 100 different ensembles for each ensemble size ranging from 2 to 40 in steps of 2 (data not shown). Performance was observed to fit a curve which peaked at an ensemble size of 18, followed by a steady decline. In the same experiment, the resubstitution error for the full {746, 1625, Schilling} dataset continued to decrease with the increase in ensemble size, so it appears as though the larger ensembles are overfitting the training data.
HIV-1 protease specificity is a high dimensional problem, with limited exemplars, where even the relevant input features are under debate. The scope of the problem, limited amount of data, biased sampling, potentially misclassified data, and importance of underrepresented patterns make this data set representative of common problems encountered in bioinformatics. These properties make it a very appealing benchmark for putative machine learning bioinformatics tools such as MFF-NEAT.
In this paper, we have improved accuracy in predicting HIV-1 protease specificity through the combination of direct encoding of physicochemical properties and ensembles of nonlinear classifiers. However, the specific classifier used, although offering robust performance across all experiments, is not considered as optimal for this problem. Additionally, although the physicochemical properties worked well, it is likely that improvement can be made through further refining the input vector features. In light of these findings, future work will focus on the definition of a reduced set of the physicochemical properties that are relevant to describing the protease substrate, as well as interrogating previously unused properties, prior to the evaluation of different classifiers. The potential transfer of this research to other related topics will also be investigated.
Methods
Parameter selection
For the MFF-NEAT algorithm, the parameters used were as specified in the previously published evaluations, with 2 exceptions. Firstly, the expert speciation threshold was made more dynamic to account for the wider range of network sizes resultant from the range in cardinality of the input vectors evaluated. The expert network speciation threshold was initially set as ((number inputs * number outputs) / 10), and adjusted such that 1 new expert species was added to the archive approximately every 10 generations, i.e., the expert archive should have roughly 200 elements after 2000 generations. Secondly, the parameters used for controlling the islands are novel as this modification had not previously been published. The island was set to run for 500 additional generations at the 500th generation.
For the LSVMs, the only parameter that requires tuning is the C value. The published state of the art evaluated a range of C values where log(C) = {0, 0.25, 0.5, 0.75…… 4.75, 5}, but the actual selected value is not provided.Citation25 Each C value in this range was reevaluated by training an LSVM on 80% of the data in the {746, 1625, Schilling} datasets, and evaluating it on the remaining 20%. For each C value, this was repeated 10 times using different samplings of the data. Using this approach, the value selected for C for setting a benchmark LSVM performance on generalization to the Impens data set was 1.284, favoring the highest average ROC-AUC, as suggested by Rögnvaldsson et al.Citation25 The performance achieved when training an LSVM on the full {746, 1625, Schilling} dataset, and testing on the Impens data set is presented in . The performance achieved is close to the benchmark published by Rögnvaldsson et al..Citation25
Generating the training data
For out-of-sampling testing on the Impens dataset, the {746, 1625, Schilling} data sets were combined to form a single set of 5643 exemplars. The 9 exemplars of the same octamer, but with different classifications across datasets were removed. Reducing the repeated exemplars to a single instance each further reduced the size of the data set from 5625 to 4955, comprising 852 case and 4103 control exemplars. The experimental design which was decided upon requires 100 different permutations of the dataset. Generating the 100 data sets a priori rather than randomly at runtime allows the same datasets to be reused across evaluations of different encodings, and allows evaluation of the results using paired t-tests.
For each permutation, the first 80% of the case exemplars, and first 80% of the control exemplars were designated the “training set,” and the remainder designated the “generalization set.” The training set is used to set the weights of the classifiers. As neural networks are susceptible to over training,Citation62 the generalization set is used to select the best generalizing classifier. Each training set at this point contained 681 case exemplars and 3282 control exemplars. This corresponds roughly to a ratio of 1:4.82 for case to control exemplars. Neural networks however have poor ability to intelligently handle imbalanced training data.Citation63 To reduce the impact of this imbalance on the training of the MFF-NEAT algorithm, the number of control exemplars in the training set is randomly undersampled such that the ratio of case to control was reduced to 1:3. This ratio was selected as it had previously been used successfully in the construction of the PROSPER web serverCitation31 as well as other similar projects.Citation64,65 The generalization set remained unchanged. Therefore, each training set comprised 681 case and 2043 control exemplars, and each corresponding generalization set comprised 171 case and 821 control exemplars.
Abbreviations
| AA | = | amino acid |
| AUC | = | area under the curve. |
| HIV-1 | = | human immunodeficiency virus type 1 |
| LSVM | = | SVM with a linear kernel |
| MFF-NEAT | = | modular feedforward neuroevolution of augmenting topologies |
| ROC | = | receiver operating characteristic |
| RSVM | = | SVM with a radial basis function (RBF) kernel |
| SVM | = | support vector machine |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by the European Commission, Horizon 2020 Marie Skłodowska-Curie Research and Innovation Staff Exchange, under grant no 644186 to P.W. and a Cork Institute of Technology Rísam Scholarship to T.M.
References
- Barre-Sinoussi F, Chermann J, Rey F, Nugeyre M, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vezinet-Brun F, Rouzioux C, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science (80- ) 1983; 220:868-71; PMID:6189183; http://dx.doi.org/10.1126/science.6189183
- Gallo R, Sarin P, Gelmann E, Robert-Guroff M, Richardson E, Kalyanaraman V, Mann D, Sidhu G, Stahl R, Zolla-Pazner S, et al. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science (80- ) 1983; 220:865-7; PMID:6601823
- Sousa SF, Tamames B, Fernandes PA, Ramos MJ. Detailed Atomistic Analysis of the HIV-1 Protease Interface. J Phys Chem B 2011; 115:7045-57; PMID:21545127; http://dx.doi.org/10.1021/jp200075s
- Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci 1988; 85:4686-90; PMID:3290901; http://dx.doi.org/10.1073/pnas.85.13.4686
- WHO. Number of deaths due to HIV/AIDS. [cited 2015 Oct 22]; Available from: http://www.who.int/gho/hiv/epidemic_status/deaths_text/en/
- CDC. HIV in the United States, Statistics Overview [Internet]. [cited 2015 Oct 22]; Available from: http://www.cdc.gov/hiv/statistics/basics/ataglance.html
- Coffin J. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science (80- ) 1995; 267:483-9; PMID:7824947
- Kontijevskis A, Wikberg JES, Komorowski J. Computational proteomics analysis of HIV-1 protease interactome. Proteins 2007; 68:305-12; PMID:17427231; http://dx.doi.org/10.1002/prot.21415
- Nalam MNL, Schiffer CA. New approaches to HIV protease inhibitor drug design II: testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr Opin HIV AIDS 2008; 3:642-6; PMID:19373036; http://dx.doi.org/10.1097/COH.0b013e3283136cee
- Ohtaka H, Freire E. Adaptive inhibitors of the HIV-1 protease. Prog Biophys Mol Biol 2005; 88:193-208; PMID:15572155; http://dx.doi.org/10.1016/j.pbiomolbio.2004.07.005
- Abramowitz N, Schechter I, Berger A. On the size of the active site in proteases II. Carboxypeptidase-A. Biochem Biophys Res Commun 1967; 29:862-7; PMID:5624785; http://dx.doi.org/10.1016/0006-291X(67)90299-9
- duVerle DA, Mamitsuka H. A review of statistical methods for prediction of proteolytic cleavage. Brief Bioinform 2012; 13:337-49; PMID:22138323; http://dx.doi.org/10.1093/bib/bbr059
- Salzberg S. On comparing classifiers: Pitfalls to avoid and a recommended approach. Data Min Knowl Discov 1997; 1:317–28. http://dx.doi.org/ 10.1023/A:1009752403260
- You L, Garwicz D, Rognvaldsson T. Comprehensive Bioinformatic Analysis of the Specificity of Human Immunodeficiency Virus Type 1 Protease. J Virol 2005; 79:12477-86; PMID:16160175; http://dx.doi.org/10.1128/JVI.79.19.12477-12486.2005
- Schilling O, Overall CM. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol 2008; 26:685-94; PMID:18500335; http://dx.doi.org/10.1038/nbt1408
- Impens F, Timmerman E, Staes A, Moens K, Ariën KK, Verhasselt B, Vandekerckhove J, Gevaert K. A catalogue of putative HIV-1 protease host cell substrates. Biol Chem 2012; 393:915-31; PMID:22944692; http://dx.doi.org/10.1515/hsz-2012-0168
- Alvarez E, Castelló A, Menéndez-Arias L, Carrasco L. HIV protease cleaves poly(A)-binding protein. Biochem J 2006; 396:219-26; PMID:16594896; http://dx.doi.org/10.1042/BJ20060108
- Nie Z, Bren GD, Vlahakis SR, Schimnich AA, Brenchley JM, Trushin SA, Warren S, Schnepple DJ, Kovacs CM, Loutfy MR, et al. Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J Virol 2007; 81:6947-56; PMID:17442709; http://dx.doi.org/10.1128/JVI.02798-06
- Gerencer M, Burek V. Identification of HIV-1 protease cleavage site in human C1-inhibitor. Virus Res 2004; 105:97-100; PMID:15325085; http://dx.doi.org/10.1016/j.virusres.2004.04.010
- Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. Substrate Shape Determines Specificity of Recognition for HIV-1 Protease. Structure 2002; 10:369-81; PMID:12005435; http://dx.doi.org/10.1016/S0969-2126(02)00720-7
- Kim H, Zhang Y, Heo Y-S, Oh H-B, Chen S-S. Specificity rule discovery in HIV-1 protease cleavage site analysis. Comput Biol Chem [Internet] 2008 [cited 2016 Jan 6]; 32:72-9; PMID:18006382. Available from: http://www.sciencedirect.com/science/article/pii/S147692710700120X
- Xing E, Jordan M, Karp R. Feature Selection for High-Dimensional Genomic Microarray Data. In: Brodley CE, Danyluk AP, editors. Proc. 18th International Conf. on Machine Learning Morgan Kaufmann; 2001. page 601-8.
- Rögnvaldsson T, You L, Garwicz D. Bioinformatic approaches for modeling the substrate specificity of HIV-1 protease: an overview. Expert Rev Mol Diagn 2007; 7:435-51; PMID:17620050; http://dx.doi.org/10.1586/14737159.7.4.435
- Rögnvaldsson T, You L. Why neural networks should not be used for HIV-1 protease cleavage site prediction. Bioinformatics 2004; 20:1702-9; PMID:14988129; http://dx.doi.org/10.1093/bioinformatics/bth144
- Rognvaldsson T, You L, Garwicz D. State of the art prediction of HIV-1 protease cleavage sites. Bioinformatics 2015; 31:1204-10; PMID:25504647; http://dx.doi.org/10.1093/bioinformatics/btu810
- Niu B, Yuan X-C, Roeper P, Su Q, Peng C-R, Yin J-Y, Ding J, Li H, Lu W-C. HIV-1 Protease Cleavage Site Prediction Based on Two-Stage Feature Selection Method. Protein Pept Lett 2013; 20:290–8; PMID: 22591479
- Kawashima S. AAindex: Amino Acid index database. Nucleic Acids Res 2000; 28:374-374; PMID:10592278; http://dx.doi.org/10.1093/nar/28.1.374
- Freund Y, Schapire RE. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. J Comput Syst Sci 1997; 55:119-39; http://dx.doi.org/10.1006/jcss.1997.1504
- Nanni L, Lumini A. MppS: An ensemble of support vector machine based on multiple physicochemical properties of amino acids. Neurocomputing 2006; 69:1688-90; http://dx.doi.org/10.1016/j.neucom.2006.04.001
- Oztürk O, Aksaç A, Elsheikh A, Ozyer T, Alhajj R. A consistency-based feature selection method allied with linear SVMs for HIV-1 protease cleavage site prediction. PLoS One 2013; 8:e63145; PMID:24058397; http://dx.doi.org/10.1371/journal.pone.0063145
- Song J, Tan H, Perry AJ, Akutsu T, Webb GI, Whisstock JC, Pike RN. PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One 2012; 7:e50300; PMID:23209700; http://dx.doi.org/10.1371/annotation/920bd689-3af7-418f-8149-43e683e18852
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics 2000; 16:404-5; PMID:10869041; http://dx.doi.org/10.1093/bioinformatics/16.4.404
- Cheng J, Randall AZ, Sweredoski MJ, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res 2005; 33:W72-6; PMID:15980571; http://dx.doi.org/10.1093/nar/gki396
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004; 337:635-45; PMID:15019783; http://dx.doi.org/10.1016/j.jmb.2004.02.002
- Gök M, Özcerit AT. A new feature encoding scheme for HIV-1 protease cleavage site prediction. Neural Comput Appl 2012; 22:1757-61; http://dx.doi.org/10.1007/s00521-012-0967-5
- Taylor WR. The classification of amino acid conservation. J Theor Biol 1986; 119:205-18; PMID:3461222; http://dx.doi.org/10.1016/S0022-5193(86)80075-3
- Gök M, Özcerit AT, Istanbullu A. A New Feature Extraction Technique for HIV-1 Protease Cleavage Site Analysis. Glob. J. Technol 3; PMID:NOT_FOUND
- Gök M, Özcerit AT, Istanbullu A. A New Feature Extraction Technique for HIV-1 Protease Cleavage Site Analysis. Proc. 3rd World Conf. Inf. Technol. 2013; 3:1-6.
- Newell NE. Cascade detection for the extraction of localized sequence features; specificity results for HIV-1 protease and structure-function results for the Schellman loop. Bioinformatics 2011; 27:3415-22; PMID:22039211; http://dx.doi.org/10.1093/bioinformatics/btr594
- Li X, Hu H, Shu L. Predicting human immunodeficiency virus protease cleavage sites in nonlinear projection space. Mol Cell Biochem 2010; 339:127-33; PMID:20054614; http://dx.doi.org/10.1007/s11010-009-0376-y
- Kim G, Kim Y, Lim H, Kim H. An MLP-based feature subset selection for HIV-1 protease cleavage site analysis. Artif Intell Med 2010; 48:83-9; PMID:19945261; http://dx.doi.org/10.1016/j.artmed.2009.07.010
- Lasko TA, Bhagwat JG, Zou KH, Ohno-Machado L. The use of receiver operating characteristic curves in biomedical informatics. J Biomed Inform 2005; 38:404-15; PMID:16198999; http://dx.doi.org/10.1016/j.jbi.2005.02.008
- Jaeger S, Chen S. Information fusion for biological prediction. J Data Sci 2010; 8:269–88.
- Nanni L, Lumini A. Using ensemble of classifiers for predicting HIV protease cleavage sites in proteins. Amino Acids 2008; 36:409-16; PMID:18401541; http://dx.doi.org/10.1007/s00726-008-0076-z
- Nanni L, Lumini A. A new encoding technique for peptide classification. Expert Syst Appl 2011; 38:3185-91; http://dx.doi.org/10.1016/j.eswa.2010.09.005
- Yuan Y, Liu H, Qiu G. A new approach for HIV-1 protease cleavage site prediction combined with feature selection. J Biomed Sci Eng 2013; 06:1155-60; http://dx.doi.org/10.4236/jbise.2013.612144
- Nanni L, Brahnam S, Lumini A. Artificial intelligence systems based on texture descriptors for vaccine development. Amino Acids 2010; 40:443-51; PMID:20552381; http://dx.doi.org/10.1007/s00726-010-0654-8
- Nanni L, Lumini A. Coding of amino acids by texture descriptors. Artif Intell Med 2010; 48:43-50; PMID:19892537; http://dx.doi.org/10.1016/j.artmed.2009.10.001
- Nanni L, Brahnam S, Lumini A. Matrix representation in pattern classification. Expert Syst Appl 2012; 39:3031-6; http://dx.doi.org/10.1016/j.eswa.2011.08.165
- Oğul H. Variable context Markov chains for HIV protease cleavage site prediction. Biosystems 2009; 96:246-50; PMID:19758550; http://dx.doi.org/10.1016/j.biosystems.2009.03.001
- Lipman DJ, Pastor RW, Lee B. Local sequence patterns of hydrophobicity and solvent accessibility in soluble globular proteins. Biopolymers 1987; 26:17-26; PMID:3801594; http://dx.doi.org/10.1002/bip.360260106
- Manning T, Walsh P. Automatic task decomposition for the neuroevolution of augmenting topologies (NEAT) algorithm Berlin, Heidelberg: Springer Berlin Heidelberg; 2012.
- Jacobs RA, Jordan MI, Nowlan SJ, Hinton GE. Adaptive Mixtures of Local Experts. Neural Comput 1991; 3:79-87; http://dx.doi.org/10.1162/neco.1991.3.1.79
- Stanley KO, Miikkulainen R. Evolving neural networks through augmenting topologies. Evol Comput 2002; 10:99-127; PMID:12180173; http://dx.doi.org/10.1162/106365602320169811
- Radcliffe NJ. Genetic set recombination and its application to neural network topology optimisation. Neural Comput Appl 1993; 1:67-90; http://dx.doi.org/10.1007/BF01411376
- Whitley D, Rana S, Heckendorn R. The island Model Genetic algorithm: On separability, population size and convergence. CIT J Comput Inf Technol 1999; 7:33–47.
- Hellberg S, Sjoestroem M, Skagerberg B, Wold S. Peptide quantitative structure-activity relationships, a multivariate approach. J Med Chem 1987; 30:1126-35; PMID:3599020; http://dx.doi.org/10.1021/jm00390a003
- Sandberg M, Eriksson L, Jonsson J, Sjöström M, Wold S. New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J Med Chem 1998; 41:2481-91; PMID:9651153; http://dx.doi.org/10.1021/jm9700575
- Wu C, Whitson G, Mclarty J, Ermongkonchai A, Chang T-C. Protein classification artificial neural system. Protein Sci 1992; 1:667-77; PMID:1304365; http://dx.doi.org/10.1002/pro.5560010512
- Maetschke S, Towsey M, Boden M. BLOMAP: An encoding of amino acids which improves signal peptide cleavage site prediction. In: Chen Y-PP, Wong L, editors. Proceedings Third Asia Pacific Bioinformatics Conference. 2005. page 273-89;
- Patsopoulos NA. Relative Citation Impact of Various Study Designs in the Health Sciences. JAMA 2005; 293:2362; PMID:15900006; http://dx.doi.org/10.1001/jama.293.19.2362
- Manning T, Sleator RD, Walsh P. Biologically inspired intelligent decision making: a commentary on the use of artificial neural networks in bioinformatics. Bioengineered 2013; 5:80-95; PMID:24335433; http://dx.doi.org/10.4161/bioe.26997
- Mazurowski MA, Habas PA, Zurada JM, Lo JY, Baker JA, Tourassi GD. Training neural network classifiers for medical decision making: the effects of imbalanced datasets on classification performance. Neural Netw 2008; 21:427-36; PMID:18272329; http://dx.doi.org/10.1016/j.neunet.2007.12.031
- Song J, Tan H, Shen H, Mahmood K, Boyd SE, Webb GI, Akutsu T, Whisstock JC. Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 2010; 26:752-60; PMID:20130033; http://dx.doi.org/10.1093/bioinformatics/btq043
- Shao J, Xu D, Tsai S-N, Wang Y, Ngai S-M. Computational Identification of Protein Methylation Sites through Bi-Profile Bayes Feature Extraction. PLoS One 2009; 4:e4920; PMID:19290060; http://dx.doi.org/10.1371/journal.pone.0004920