
ABSTRACT
Yeast spores can be used as a carrier to produce enzyme capsules. In the present study, this technique was applied to a diagnostic enzyme named creatinase. We found that a secretory form of Pseudomonas putida creatinase could be entrapped in the spore wall, and such spores were used as creatinase capsules. The activity of the encapsulated creatinase was largely improved by mild spore wall defective mutations, such as DIT1 or OSW2 deletions. The advantages of this method include the following: encapsulated and freeze-dried creatinase is produced without preparing the purified enzyme, and it exhibits resistance to environmental stresses, such as high temperature and SDS treatments. Thus, yeast spores could be applied to establish quick and easy clinical diagnostic methods.
Introduction
Creatine amidohydrolase or creatinase can mediate the hydrolysis of creatine to produce sarcosine and urea. This enzyme, together with 2 other enzymes, namely, creatininase and sarcosine oxidase, is used as a clinical enzyme to measure creatinine levels in specimens, such as blood samples.Citation1,2 Studies have been conducted to improve their properties. For example, several chemical immobilization procedures of these enzymes have been studied to make them stable and reusable. These experiments have been performed mainly to develop and improve amperometric creatinine biosensors.Citation3-6
Previously, we reported that spores of the budding yeast Saccharomyces cerevisiae are used to produce enzyme capsules.Citation7 This method is based on unique properties of the yeast spore wall. In the spore wall, the outermost layer or the dityrosine layer is mainly composed of bisformyl-dityrosine, and the second outermost layer or the chitosan layer is composed of chitosan.Citation8-11 Physiologically, these structures are required to protect spores so that yeast spores are resistant to various environmental stresses.Citation12 The presence of these structures provides spores with a distinct property; when the secretory forms of soluble enzymes are expressed in sporulating cells, they are entrapped in the periplasmic space of the spore wall.Citation7 The dityrosine layer is critical for the entrapment of secretory proteins in the spore wall probably because its pore size is sufficiently small to prevent protein diffusion.Citation13 Although the dityrosine layer functions as a diffusion barrier for proteins, relatively small molecules, such as sugars, can pass through it.Citation7,14 Thus, spores can be applied to biological enzyme capsules.Citation7,15 The encapsulated enzyme is protected by the spore wall so that it exhibits resistance to environmental stresses, such as high temperature and digestive enzymes.
The dityrosine and chitosan layers can be eliminated through specific mutations; DIT1 is required for the synthesis of bisformyl dityrosine, and dit1Δ mutation causes a loss of the dityrosine layer.Citation9,16,17 CHS3 encodes a chitin synthase, and its deletion results in a loss of both the dityrosine and chitosan layers.Citation18,19 Although the deletion of the dityrosine layer causes a leak of soluble proteins held in the spore wall, the chitosan layer can also retain the secretory proteins to some extent. Previous studies have shown that dit1Δ spores, not chs3Δ spores, can hold some soluble enzymes in the spore wall, although the capacity of dit1Δ spores to hold enzymes is much lower than that of wild-type spores.Citation7 Interestingly, enzymes held in dit1Δ spores generally exhibit higher activity than that in wild-type spores,Citation7,15 which is probably because the dityrosine layer is a barrier for substrates to access the enzymes. Previous reports have shown that osw2Δ mutation can also improve the activity of enzymes held in the spore wall.Citation7,15 Although the exact function of Osw2 is not yet known, osw2Δ mutation causes a minor spore wall defect probably in the dityrosine layer.Citation13,19 Therefore, the integrity of the spore wall can affect the activity of encapsulated enzymes. A remarkable difference between dit1Δ and osw2Δ mutants is that the latter has the dityrosine layer so it can stably entrap proteins in the spore wall.Citation13
Creatine is a small molecule, so we expect it to pass through the spore wall. Thus, in the present study, we attempted to apply yeast spore-based encapsulation technique to creatinase. Such an enzyme may be used for clinical examinations.
Results and discussion
Yeast spores can hold Pseudomonas putida creatinase
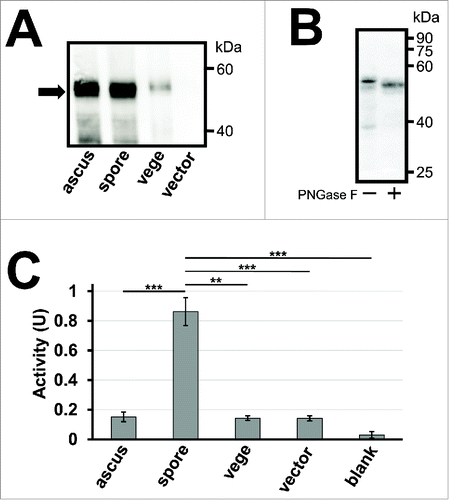
To produce yeast spores encapsulating creatinase, the P. putida creatinase gene was cloned into a yeast expression vector named pRS426TEF.Citation20 A yeast secretory signal sequence from SPR1 was fused at the 5′ end to localize the enzyme to the spore wall, and 3 tandem repeats of the hemagglutinin (HA) epitope gene was added at the 3′ end for its detection. The creatinase expression plasmid was then transformed into a wild-type S. cerevisiae strain AN120, and western blot analysis was performed to verify that the HA fusion (creatinase–HA) was expressed in spores (). Yeast spores are covered with the ascal membrane and ascal wall. In this study, the term “ascus” was used for samples of intact ascospores, and “spore” was used for samples in which spores were released from the asci by breaking down the ascal cell wall and membrane. Given that TEF2 promoter is a constitutive promoter, creatinase–HA was detected in lysates of ascus, spore, and vegetative cell samples (). Molecular weight of creatinase–HA estimated from SDS-PAGE was 52 kDa (data not shown). The molecular weight was slightly larger than the predicted one (49 kDa) because creatinase–HA expressed in yeast was modified by asparagine-linked glycosylation; after PNGas-F treatment, it was decreased to 50 kDa (). The amount of creatinase–HA detected in ascus and spore lysates was much more than that in vegetative cell lysate, probably because creatinase–HA fusion was retained in the spore wall, whereas it was secreted out of the cell wall in the vegetative cells. We found that creatinase activity in the spores expressing creatinase–HA was much higher than that in vegetative cells (). Such a high creatinase activity was not detected in spores harboring the empty vector (). Although a comparable amount of creatinase–HA was detected in ascus and spore lysates, the activity detected in the asci was similar to that in vegetative cells (). This result was consistent with the notion that, in intact asci, its substrate (creatine) cannot access the enzyme because of the existing ascal membrane. These results demonstrated that yeast spores could be used as microcapsules to hold creatinase. Notably, in this experiment, the activity was measured in wet cell samples.
Figure 1. Activity assay of creatinase expressed in yeast spores. (A) 50 µg of lysates from intact asci (ascus), spores (spore) and vegetative cells (vege) expressing creatinase–HA were subjected to western blot analysis using an anti–HA antibody to detect the HA fusion (arrow). Spores harboring an empty vector, pRS426TEF, were used as a control. The spore sample was prepared by treating asci with β-glucanase and sonication to lyse the ascal cell wall and membrane. Vegetative cells were cultured in YPAcetate media. (B) Creatinase–HA expressed in wild-type spores was treated with or without PNGase F, and subjected to western blot analysis using an anti–HA antibody. (C) Creatinase activity was detected from intact asci (ascus), spores (spore) or vegetative cells (vege) expressing creatinase–HA, and spores harboring the empty vector (vector). The activity detected in the reaction mixture without yeast cells is also shown (blank). 5×108 of cells or asci were used for the assay. Cell amounts were adjusted by turbidity and, for the spore sample, it was measured before asci were broken. One unit of activity was defined as the amount (mg) of enzyme which produces 1 µmol of urea per minute at 37°C. Data presented are the mean ± SE of 3 independent samples. **P< 0.01; ***P< 0.001.
Activity of encapsulated creatinase is improved by dit1Δ and osw2Δ mutations
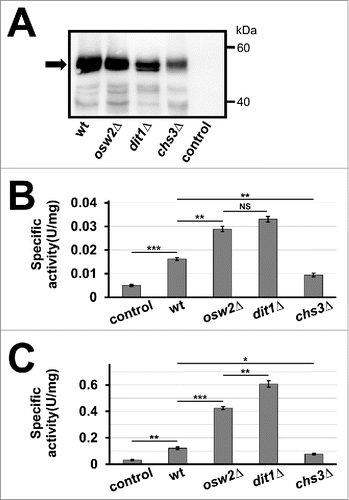
We examined whether the creatinase activity is improved in spore wall deficient mutants, namely, osw2Δ and dit1Δ. Western blot analysis showed that both mutant spores could retain creatinase–HA (). Although the amount of creatinase-HA held in dit1Δ spores was much less than that in wild-type spores, the activity of the dit1Δ spores was almost 2-fold higher than that of wild-type spores (). osw2Δ mutation could also improve the activity of creatinase in spores; the amount of creatinase–HA detected in the mutant spores was slightly lower than that in wild-type spores (). Under this condition, a statistically significant difference was not observed between the activities of dit1Δ and osw2Δ spores (). The levels of creatinase–HA protein and its activity detected in chs3Δ spores were significantly lower than those in the other spores (), thereby suggesting that the chitosan layer could retain creatinase even in the absence of the dityrosine layer. These experiments were performed with purified wet spores, and quantity was adjusted by weight. Subsequently, we tested whether the encapsulated creatinase can survive through freeze-drying. As shown in , creatinase activity was detected in the freeze-dried spores, and dit1Δ spores exhibited the highest activity among spores.
Figure 2. Activities of creatinase expressed in spore wall deficient mutants, osw2Δ, dit1Δ, and chs3Δ. (A) 50 µg of lysates from wild-type (wt), osw2Δ, dit1Δ, or chs3Δ spores expressing creatinase–HA were subjected to western blot analysis using an anti–HA antibody to detect the HA fusion (arrow). Wild-type spores harboring the empty vector was used as a control. (B, C) Creatinase activity was measured for 28 mg of indicated wet spores (B) or 5 mg of freeze-dried spores (C) expressing creatinase–HA. Spore samples were purified by percoll gradient centrifugation before measuring weight or freeze-drying. Wild-type spores harboring the empty vector were used as a control. Data presented are the mean ± SE of 3 independent samples. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant.
osw2Δ mutant spores can retain creatinase as wild-type spores
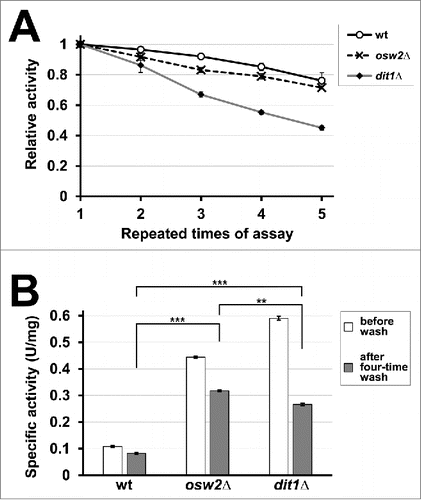
dit1Δ spores carrying creatinase–HA exhibited higher activity than the other spores, probably because its substrate (creatine) could easily access the enzyme without being obstructed by the dityrosine layer. However, the lack of the dityrosine layer makes the spore wall leaky, so dit1Δ spores are less capable of holding creatinase–HA (). In accordance with this notion, creatinase in dit1Δ spores was susceptible to high salt and detergent wash: osw2Δ spores carrying creatinase–HA exhibited higher activity than dit1Δ after 4 washes with high salt and detergent solution (). Therefore, under some conditions, osw2Δ spores were the superior option to produce the encapsulated creatinase. Note that, dit1Δ spores also exhibited higher activity than wild-type spores even after 4 high salt and detergent washes ().
Figure 3. Repetitive use of creatinase encapsulated in spores. (A) For 5 mg of purified wild-type (wt), osw2Δ, and dit1Δ spores expressing creatinase–HA, first creatinase activity was assayed (first assay). After washing with 0.6 M NaCl solution containing 0.5% Triton X-100, the activities were assayed again (second assay). This cycle was repeated 4 times. For each sample, the activity obtained at the first assay was determined as 1.0 and relative activities are shown. Data presented are the mean ± SE of 3 independent samples. (B) Specific activities of the above spores before wash and after 4-time wash are shown. Data presented are the mean ± SE of 3 independent samples. **P < 0.01; ***P < 0.001.
Creatinase is stabilized through encapsulation in yeast spores
Km of the encapsulated creatinase measured at 50°C are shown in . Free creatinase–HA, which was expressed and secreted from vegetative yeast cells, was used as a control. Creatinase–HA in osw2Δ and dit1Δ spores exhibited lower Km compared with those in wild-type spores.
Table 1. Km values of creatinase–HA in wild-type, osw2Δ, dit1Δ, or chs3Δ spores, and the free creatinase–HA at 50°C are shown.
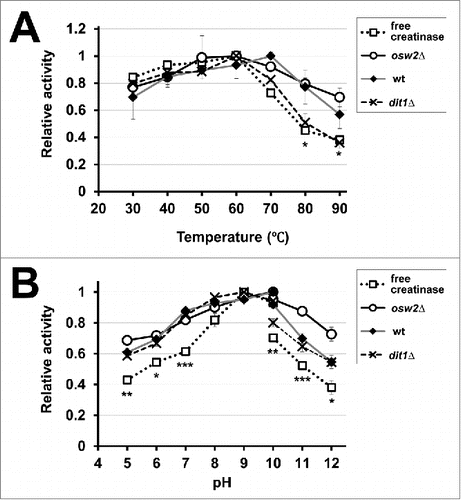
Subsequently, we measured the activities of creatinase expressed in spores in various temperatures and pH. As shown in , the activities of creatinase in wild-type and osw2Δ spores were more stable than those of free creatinase at higher temperatures. Such thermostability was not observed for creatinase in dit1Δ spores. For pH sensitivity, the activities of creatinase expressed in spores were generally stable than those of free creatinase (). Notably, creatinase in dit1Δ spores and the free enzyme exhibited high activity at pH 9, whereas the high activity of creatinase in wild-type and osw2Δ spores was detected at pH 10 (). Additionally, creatinase activity in osw2Δ spores was more stable than that in other spores at pH 11 and 12 (statistical differences were observed between osw2Δ and wild-type or dit1Δ spores at these pH values).
Figure 4. Assessment of the activities of the encapsulated creatinases at various temperatures (A) and pH (B). To assess temperature and pH sensitivities for indicated spores harboring creatinase–HA or free creatinase–HA, their activities were assayed at various temperatures (30°C to 90°C) and pH (5 to 12). As the free enzyme, creatinase–HA secreted from vegetative yeast cells was used. The maximum activity obtained for each assay was determined as 1.0 and relative activities are shown. For the pH sensitivity, potassium phosphate buffer (pH 5 to 10) and sodium carbonate buffer (pH 10 to 12) were used. Data presented are the mean ± SE of 3 independent samples. Statistic analysis was performed between free creatinase and osw2Δ spores, and asterisks were deposited when statistically significant differences were found. *P < 0.05; **P < 0.01; ***P < 0.001.
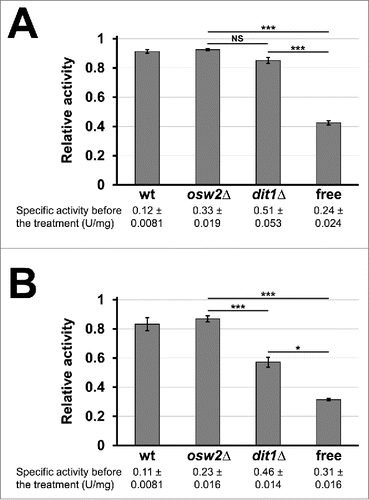
To further verify that creatinase encapsulated in spores is protected from environmental stresses, we examined whether this encapsulated creatinase is resistant to proteinase K. As shown in , creatinases in wild-type and osw2Δ, as well as dit1Δ spores, were tolerant to proteinase K treatment. Moreover, the encapsulated creatinase was resistant to SDS treatment. As shown in , activity of the creatinases in wild-type and osw2Δ spores was retained by more than 80% after 5% SDS treatment, whereas that of the free enzyme was decreased to about 30%. Creatinase in dit1Δ spores was less tolerant to SDS compared with that in wild-type and osw2Δ spores, even though dit1Δ spores were more resistant to SDS than the free enzyme (). Thus, the encapsulated creatinase, particularly in osw2Δ spores, is useful under adverse conditions because it exhibits resistance to various environmental attacks.
Figure 5. Assessment of sensitivities of the encapsulated creatinase toward environmental stresses. 4 mg of indicated purified freeze-dried spores containing creatinase–HA were treated with proteinase K for 12 h (A) or 5% SDS for 10 min (B) at 30°C, and then creatinase activities were assayed. For each sample, the activities obtained before the treatments were determined as 1.0 and relative activities are shown. Culture media containing the soluble creatinse–HA was used as a control (free). Specific activities before the treatments were indicated under the sample names. Data presented are the mean ± SE of 3 independent samples. Results of the t-test among osw2Δ, dit1Δ spores and the free enzyme were shown. *P < 0.05; ***P < 0.001; NS, not significant.
Overall, we demonstrated that encapsulated creatinase could be produced using yeast spores. Our results showed that dit1Δ spores could produce creatinase capsules with the highest activity. However, the disadvantages of using dit1Δ spores are that encapsulated creatinase is leaky and sensitive to environmental stresses. Thus, under certain conditions, osw2Δ mutant may be the most desirable option to produce the enzyme capsule. Compared to chemical procedures for enzyme immobilization, the advantage of this method is that immobilized and stress-resistant creatinase is produced without preparing the purified enzyme. Although serum creatinine levels are generally measured in clinical diagnosis, salivary creatinine levels are also elevated in kidney diseases.Citation21 Considering these distinct properties, the spore-based encapsulated creatinase may be used to produce a quick and easy method for diagnosing creatinine levels in salivary samples. Immobilized creatinase, creatininase and sarcosine oxidase have been applied to develop amperometric creatinine biosensors.Citation3-6 Since previous studies have shown that microorganisms, including S. cerevisiae are available to develop amperometric biosensors,Citation22 it would be intriguing to investigate if creatinase encapsulating spores are applied to this purpose.
Materials and methods
Yeast strains and growth media
Yeast strains and oligonucleotide primers used in this study are listed in and , respectively. All strains used in this study are in the fast-sporulating SK-1 strain background. YPAD media (10 g/L yeast extract, 20 g/L peptone, 30 mg/L adenine, 20 g/L glucose) was used to grow yeast cells without plasmids. Yeast cells harboring plasmids were grown in synthetic defined (SD) media (6.7 g/L yeast nitrogen base, 2 g/L dropout mix without appropriate selectable supplements, 20 g/L glucose or galactose). Twenty g/l agar was added for making plates. The drop out mix was prepared by mixing the following supplements: 0.5 g adenine, 2 g l-alanine, 2 g l-arginine, 2 g l-asparagine, 2 g l-aspartic acid, 2 g l-cysteine, 2 g l-glutamine, 2 g l-glutamic acid, 2 g glycine, 2 g l-histidine, 2 g myo-inositol, 2 g l-isoleucine, 10 g l-leucine, 2 g l-lysine, 2 g l-methionine, 0.2 g para-aminobenzoic acid, 2 g l-phenylalanine, 2 g l-proline, 2 g l-serine, 2 g l-threonine, 2 g l-tryptophan, 2 g l-tyrosine, 2 g uracil, 2 g l-valine.
Table 2. Yeast strains used in this study.
Table 3. Oligo nucleotide primers used in this study.
Plasmids
pRS426TEF-ss-CI-3HA was used to express a yeast signal peptide and 3 tandem repeat of the hemagglutinin (3 × HA) epitope fusion to creatinase in yeast cells. This plasmid was constructed as follows. The creatinase without the stop codon was amplified by PCR using HXO588 and HXO590 as primers and P. putida KT2440 genomic DNA as a template. The resulting PCR fragment was digested with HindIII and XhoI, and cloned into pRS426TEF.Citation20 The resulting plasmid was digested with SpeI and HindIII, and a DNA fragment containing the SPR1 signal sequence (first 24 amino acids) digested out of pRS424TEF-spRFPCitation7 with the same enzymes was ligated. Finally, using pFA6a-3HAHis3MX6Citation23 as a template and HXO609 and HXO610 as primers, a DNA fragment containing the 3 × HA gene and the ADH1 terminator was amplified. The DNA fragment was cloned into the creatinase containing plasmid using XhoI and KpnI sites. All DNA sequences of genes amplified by PCR were verified by sequencing.
Creatinase activity assay
P. putida creatinase secreted into culture media was used as a control of the free enzyme. To prepare this, wild-type yeast cells harboring pRS426TEF-ss-CI-3HA were cultured in SD media and the culture was centrifuged at 6,200 × g for 1 minute to remove cells. Then the supernatant was filtrated through Amicon-Ultra (molecular weight cut off 10 kDa, Millipore, Shanghai, China) to concentrate and exchange solvent to enzyme dilution buffer (10 mM potassium phosphate pH 8.0, 0.16 mL/L 2-mercaptoethanol): the enzyme was concentrated about 5 times and the media was diluted with the buffer about 30 times. Yeast cells harboring creatinase or the free creatinase were suspended in the enzyme dilution buffer to measure their activities. Creatinase activity was assayed by following the protocol accompanied with Kikkoman creatinase (Kikkoman, Noda Japan). 0.1 mL of 0.3 M potassium phosphate buffer (pH 7.7) and 0.8 mL of 0.1 M creatine solution (creatine was dissolved in water) were mixed and incubated at 37°C for 5 min. Then, 0.1 mL of creatinase containing solution was added and incubated for 10 min at 37°C. To detect urea calorimetrically, the reaction mixture was mixed with 2 mL of p-dimethylaminobenzaldehyde solution (2 g of p-dimethylaminobenzaldehyde, 100 mL of ethanol, 15 mL of concentrted HCl, 115 mL of water), allowed to stand for 30 min at 25°C, and OD435 was measured by spectrophotometer (Ultrospec 2100 pro, Amersham biosciences, USA). The Michaelis-Menten constant (Km) was determined using 5–40 mM of creatine. The reactions were stopped after 10 min and the urea was determined as described above. Km was obtained using the Lineweaver-Burk equation. To assess pH stability, 10 mM potassium phosphate buffer (pH 5 to 10) or 10 mM sodium carbonate buffer (pH 10 to 12) were used instead of the potassium phosphate buffer (pH 7.7). One unit of activity was defined as the amount of urea (µmol) released per minute at 37°C.
Yeast culture, sporulation and spore purification
Yeast spores and vegetative cells were prepared as follows. First, yeast cells derived from a single transformant colony were grown overnight in 5 mL of SD liquid media with appropriate supplemental amino acids. For sporulation, 1 mL of the culture was then transferred into 30 mL of YPAcetate (1% w/v yeast extract, 2% w/v peptone, 2% w/v potassium acetate) and grown for 24 h. The cells were harvested by centrifugation at 3000 × g for 1 minute, washed with 30 mL of H2O, resuspended in 30 mL of 2% w/v potassium acetate medium, and cultured for 24 h. Sporulation efficiency was determined by counting spores under the light microscope. Under our experimental condition, sporulation efficiency was greater than 90%. Vegetative cells were similarly cultured in YPAcetate media: 1 mL of SD culture was shifted to 30 mL of YPAcetate and grown for 24 h and harvested.
To release spores from asci, the ascal wall was first digested by β-glucanase (lyticase, Sigma-Aldrich, Shanghai, China). For this, spores prepared as above were resuspended in 1 mL of spheroplast buffer (50 mM potassium phosphate buffer pH7.5, 1.4 M sorbitol, 40 mM β-mercaptoethanol) and mixed with 50 µL of β-glucanase stock solution (200 U of β-glucanase was dissolved in 500 µL of 50% v/v glycerol). After 3 h of incubation at 37°C, spores were washed twice with spheroplast buffer. Then, they were resuspended in spheroplast buffer and sonicated to disrupt the ascal membrane.
All spores as prepared above were purified by percoll gradient centrifugation based on a previously described method.Citation24 Spores were washed 3 times with 5 ml of 0.5% v/v Triton-X. After the washes, the resulting pellet was resuspended in 1 mL of 0.5% v/v Triton-X and layered on top of Percoll (Sigma-Aldrich, Shanghai, China) gradients (50-80% v/v Percoll, 10% v/v 2.5 M sucrose and 0.5% v/v Triton-X). After centrifugation at 15,000 × g at 4°C for 1 h, the top of 3 layers which consist of vegetative cells and debris were removed. The remaining layer containing spores was washed with 10 mL of 0.6 M NaCl once and then 10 mL of 0.5% v/v Triton-X twice. Purified spores were freeze-dried as follows. First, spores were frozen in a -20°C freezer for more than 2 h. They were then freeze-dried by EYELA FD-1000 freeze-dryer (Tokyo Rikakikai, Tokyo, Japan) at -50°C for 72 h under the pressure of 25 Pa.
Spore protection assay
For proteinase K treatment, in 100 µL of proteinase buffer (50 mM Tris-HCl pH 7.5, 10 mM CaCl2), proteinase K (Sigma-Aldrich, Shanghai, China) was mixed in the solution at final concentration of 30 U/mL. Then, 4 mg of freeze-dried spores or 20 µL of free creatinase were suspended in the solution, and incubated at 37°C for 12 h. Spores were then washed with 0.6 M NaCl solution and 0.5% v/v TritonX-100 and creatinase activity was assayed as described above.
SDS treatment was performed as follows. 4 mg of freeze-dried spores or 20 µL of the free enzyme was mixed with 100 µL of 5% w/v SDS and incubated at 30°C for 10 minutes. Spores were then washed with 0.6 M NaCl solution and 0.5% v/v TritonX-100 and creatinase activity was assayed.
Western blotting
Western blotting was performed as described beforeCitation7 with some modifications. Spores and vegetative cells were suspended in 500 µL of 8 M urea and lysed by sonication for 1 hour on ice. The cell lysates were then centrifuged at 4,000 × g at 4°C for 5 min and 50 µg of the supernatants were subjected to SDS-PAGE (5% stacking gel and 10% separating gel). Protein concentration was determined by BCA protein assay kit (Beyotime, Jiangsu, China). A mouse anti-HA antibody (Transgen Biotech, Beijing, China) was used as a primary antibody at 1:6000 dilutions. Goat anti mouse IgG-HRP (Transgen Biotech, Beijing, China) were used as secondary antibodies at 1:6000 dilution. TBST (10 mM Tris-HCl pH 8.0, 160 mM NaCl, 0.05% v/v Tween 20) supplemented with 5% w/v non-fat dry milk was used for blocking and antibody incubations. Signals were visualized by Clarity Western ECL Substrate (Biorad, Shanghai, China) and images were obtained by using ImageQuant LAS4000 (GE Healthcare Bio-Science, Uppsala, Sweden).
For PNGase F treatment, 10 µg of spore lysate was incubated with 0.5 U of PNGase F (New England Biolabs, Beijing, China) following the protocol accompanied with the enzyme at 37°C for 12 h. Total volume of the reaction mixture was 20 µl and 8 µl was subjected to protein gel blot analysis.
Statistics
Data presented are the mean ± SE of 3 independent samples obtained from different cultures. Statistical significance was determined with Student's t-test (2-tail, heteroscedastic) using Microsoft Excel software. Differences between the analyzed samples were considered significant at P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| HA | = | hemagglutinin |
| P. putida | = | Pseudomonas putida |
| S. cerevisiae | = | Saccharomyces cerevisiae |
| SD | = | synthetic defined |
Acknowledgments
The authors are grateful to G. Du (Jiangnan University, Wuxi, China) for providing P. putida KT2440 genomic DNA.
Funding
This work was supported by National Natural Science Foundation of China (21576118) and Fundamental Research Funds for the Central Universities (JUSRP51629B) to H. Nakanishi, Fundamental Research Funds for the Central Universities (JUSRP51319B) to X.-D. Gao, and National Key Laboratory of Biochemical Engineering, China (2014KF-02) to Z. Li.
References
- Kinoshita T, Hiraga Y. A fluorophotometric determination of serum creatinine and creatine using a creatinineamidohydrolase-creatineamidinohydrolase-sarcosine oxidase-peroxidase system and diacetyldichlorofluorescin. Chem Pharm Bull 1980; 28:3501-6; PMID:7226292; https://doi.org/10.1248/cpb.28.3501
- Miller BF, Dubos R. Determination by a specific, enzymatic method of the creatinine content of blood and urine from normal and nephritic individuals. J Biol Chem 1937; 121:457-64
- Berberich JA, Yang LW, Madura J, Bahar I, Russell AJ. A stable three-enzyme creatinine biosensor. 1. Impact of structure, function and environment on PEGylated and immobilized sarcosine oxidase. Acta Biomaterialia 2005; 1:173-81; PMID:16701794; https://doi.org/10.1016/j.actbio.2004.11.006
- Sakslund H, Hammerich O. Effects of Ph, Temperature and Reaction-Products on the Performance of an Immobilized Creatininase-Creatinase-Sarcosine Oxidase Enzyme-System for Creatinine Determination. Analytica Chimica Acta 1992; 268:331-45; https://doi.org/10.1016/0003-2670(92)85229-Y
- Berberich JA, Chan A, Boden M, Russell AJ. A stable three-enzyme creatinine biosensor. 3. Immobilization of creatinine amidohydrolase and sensor development. Acta Biomaterialia 2005; 1:193-9; PMID:16701796; https://doi.org/10.1016/j.actbio.2004.11.008
- Berberich JA, Yang LW, Bahar I, Russell AJ. A stable three enzyme creatinine biosensor. 2. Analysis of the impact of silver ions on creatine amidinohydrolase. Acta Biomaterialia 2005; 1:183-91; PMID:16701795; https://doi.org/10.1016/j.actbio.2004.11.007
- Shi LB, Li ZJ, Tachikawa H, Gao XD, Nakanishi H. Use of yeast spores for microencapsulation of enzymes. Appl Environ Microbiol 2014; 80:4502-10; PMID:24837390; https://doi.org/10.1128/AEM.00153-14
- Briza P, Ellinger A, Winkler G, Breitenbach M. Chemical composition of the yeast ascospore wall. The second outer layer consists of chitosan. J Biol Chem 1988; 263:11569-74; PMID:3042773
- Briza P, Kalchhauser H, Pittenauer E, Allmaier G, Breitenbach M. N,N'-Bisformyl dityrosine is an in vivo precursor of the yeast ascospore wall. Eur J Biochem 1996; 239:124-31; PMID:8706696; https://doi.org/10.1111/j.1432-1033.1996.0124u.x
- Briza P, Winkler G, Kalchhauser H, Breitenbach M. Dityrosine is a prominent component of the yeast ascospore wall. A proof of its structure. J Biol Chem 1986; 261:4288-94; PMID:3512567
- Neiman AM. Ascospore formation in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev 2005; 69:565-84; PMID:16339736; https://doi.org/10.1128/MMBR.69.4.565-584.2005
- Coluccio AE, Rodriguez RK, Kernan MJ, Neiman AM. The yeast spore wall enables spores to survive passage through the digestive tract of drosophila. PLoS One 2008; e2873.
- Suda Y, Rodriguez RK, Coluccio AE, Neiman AM. A screen for spore wall permeability mutants identifies a secreted protease required for proper spore wall assembly. PLoS One 2009; 4:e7184
- Herman PK, Rine J. Yeast spore germination: a requirement for Ras protein activity during re-entry into the cell cycle. EMBO J 1997; 16:6171-81; PMID:9321396; https://doi.org/10.1093/emboj/16.20.6171
- Li ZJ, Li Y, Duan SL, Liu J, Yuan P, Nakanishi H, Gao XD. Bioconversion of d-glucose to d-psicose with immobilized d-xylose isomerase and d-psicose 3-epimerase on Saccharomyces cerevisiae spores. J Ind Microbiol Biotechnol 2015; 42:1117-28; PMID:26065389; https://doi.org/10.1007/s10295-015-1631-8
- Briza P, Breitenbach M, Ellinger A, Segall J. Isolation of two developmentally regulated genes involved in spore wall maturation in Saccharomyces cerevisiae. Genes Dev 1990; 4:1775-89; PMID:2249774; https://doi.org/10.1101/gad.4.10.1775
- Briza P, Eckerstorfer M, Breitenbach M. The sporulation-specific enzymes encoded by the DIT1 and DIT2 genes catalyze a two-step reaction leading to a soluble ll-dityrosine-containing precursor of the yeast spore wall. Proc Natl Acad Sci U S A 1994; 91:4524-8; PMID:8183942; https://doi.org/10.1073/pnas.91.10.4524
- Pammer M, Briza P, Ellinger A, Schuster T, Stucka R, Feldmann H, Breitenbach M. DIT101 (CSD2, CAL1), a cell cycle-regulated yeast gene required for synthesis of chitin in cell walls and chitosan in spore walls. Yeast 1992; 8:1089-99; PMID:1293886; https://doi.org/10.1002/yea.320081211
- Coluccio A, Bogengruber E, Conrad MN, Dresser ME, Briza P, Neiman AM. Morphogenetic pathway of spore wall assembly in Saccharomyces cerevisiae. Eukaryot Cell 2004; 3:1464-75; PMID:15590821; https://doi.org/10.1128/EC.3.6.1464-1475.2004
- Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995; 156:119-22; PMID:7737504; https://doi.org/10.1016/0378-1119(95)00037-7
- Celec P, Tothova L, Sebekova K, Podracka L, Boor P. Salivary markers of kidney function - Potentials and limitations. Clin Chim Acta 2016; 453:28-37; PMID:26633856; https://doi.org/10.1016/j.cca.2015.11.028
- Nakamura H, Shimomura-Shimizu M, Karube I. Development of microbial sensors and their application. Adv Biochem Eng Biotechnol 2008; 109:351-94; PMID:18004516
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998; 14:953-61; PMID:9717241; https://doi.org/10.1002/(SICI)1097-0061(199807)14:10%3c953::AID-YEA293%3e3.0.CO;2-U
- Kloimwieder A, Winston F. A Screen for Germination Mutants in Saccharomyces cerevisiae. G3 (Bethesda) 2011; 1:143-9; PMID:22384326; https://doi.org/full_text
- Neiman AM, Katz L, Brennwald PJ. Identification of domains required for developmentally regulated SNARE function in Saccharomyces cerevisiae. Genetics 2000; 155:1643-55; PMID:10924463
- Zhang H, Tachikawa H, Gao XD, Nakanishi H. Applied usage of yeast spores as chitosan beads. Appl Environ Microbiol 2014; 80:5098-105; PMID:24907339; https://doi.org/10.1128/AEM.00677-14