
ABSTRACT
Clostridium difficile (C. difficile) is a kind of obligate anaerobic gram-positive Bacillus related with intestinal diseases and antibiotic treatment. In present study, the C. difficile genome was studied employing met genomic technology. Genome sequencing identified C. difficile LCL126 has total size of 4,301,949 bp with a 27.97% of GC content. Specifically, 4119 predicted coding genes, 188 repeat sequences, 13 prophages and 8 gene islands were detected. Additionally, gene function analysis aspect of the function annotation, effector, and virulence were concluded that total of 3367 cluster of orthologous groups of proteins genes and classified into 24 categories, while the most outstanding class was metabolic process (1533) and catalytic activity (1498). The carbohydrate-active enzymes have been detected 127 genes, pathogenicity analysis revealed that 133 reduced and 22 increased virulence (hypervirulence) genes, while 54 unaffected and 10 loss of pathogenicity genes were found. Furthermore, perform the visualization and methylation expression were revealed that the dominant types comprised m4C, m5C, and m6C with the number of 6989, 36,666, and 3534, respectively. Overall, whole genome sequence information of C. difficile LCL126 was obtained and functional prediction was revealed its possible toxicological potential genes existence.
GRAPHICAL ABSTRACT
1. Introduction
Clostridium difficile (C. difficile) is a kind of obligate anaerobic gram-positive Bacillus, and considered as main causative agents of intestinal diseases related with antibiotic treatment, which can cause series of diseases such as clinical manifestations with acquired diarrhea and pseudomembranous enteritis [Citation1,Citation2]. The incidence of C. difficile infection within hospital and communities have risen prominently during the past 20 years, mainly derived from the virulence of toxins [Citation3,Citation4]. Alcala et al. reported that C. difficile becoming the quite general phenomenon of diarrhea in developed countries hospital, accounting for probably 25% of antibiotic-associated diarrhea [Citation5], and the U.S. Disease Control and Prevention Centers has been designated the strain C. difficile act as an urgent threat to human health [Citation6].
Interestingly, the feature worthy noting that self of Clostridium is not a pathogen while Clostridium infection is formed attributed by producing various toxins [Citation7]. The various pathologies noticed from Clostridium infections are usually related to the toxin produced types. Clinically isolated from Clostridium has been classified as five toxin types from A to E in line with the major secreted lethal toxins. Each toxin can cause specific disease in human and animals from mild poisoning to potentially severe life-threatening, and the severity of symptoms varied might be partly attributable to the toxin production level [Citation6,Citation7]. Especially, the pathogenesis of C. difficile is reported in view of the action of major toxins by toxic A of C. difficile (TcdA) and toxic B of C. difficile (TcdB) encoded which disturb the intestinal epithelium powerful monoglycosy-transferase [Citation3]. TcdA is an enterotoxin that can cause intestinal mucosal tissue damage and led to hemorrhagic fluid secretion, although TcdB lacks obvious enteric toxicity, and it is a powerful cytotoxin, most of entero-toxigenic strains producing TcdA and TcdB (A+ B+), and TcdA express missing A− B+ strains can also cause clinical disease [Citation8]. The typical toxin producer strains of tcdA and tcdB are found in a 19.6 kb DNA element on a similar chromosome and are called pathogenic locus [Citation9,Citation10], has so far been found only in pathogenic strains which inactivate the guanosine triphosphate (GTP) binding protein, caused a series of chain reactions that eventually lead to diarrhea as well as colitis from mild to life threatening [Citation11].
The production of toxins varies greatly between distinct strains and seems to be highly affected by environmental conditions, comprising the nutrients availability and temperature changes [Citation4]. The methods to explore the molecular perspective of C. difficile constantly developed, for instant pulsed-field gel electrophoresis (PFGE), multilocus variable-tandem repeat, and sequence typing, single region PCR karyotype analysis and amplified fragment length polymorphism (AFLP) [Citation12,Citation13]. However, these approaches application limited due to long turnaround duration, high cost while poor reproducibility and portability, as well as difficulty in data analysis. Subsequently, advanced technology developing with binary typing [Citation14,Citation15] and single-molecule real-time (SMRT) sequencing [Citation16]. Recently, high-throughput sequencing genomics has been applied owing to advantages aspect of shorten turnaround duration, simplicity and distinguishability as well as portability [Citation15].
The objective of the present study was to identify the whole genome sequencing of C. difficile LCL126 with attention to phylogenetic using genome analysis. In addition, to identify the information about the co-evolution and diversity of phages and bacteria, as well as attempt to elucidate the mechanism of toxicity from the perspective of genomics and metabolomics.
2. Materials and methods
2.1. Bacterial strains and DNA extraction
The C. difficile LCL126 strain was provided by the Clostridium Laboratory of Lanzhou Institute of Biological Products (Gansu, China). Genomic DNA extracted using SDS method [Citation17]. Agarose gel electrophoresis was performed to check the purity and stability, and then further constructed database based on Pacbio and Illumina platforms. After qualified the library, carry out the selection by PacBio Sequel and Illumina NovaSeq PE150 according to the effective concentration and target data at the Beijing Novogene Bioinformatics Technology Co., Ltd., China.
2.2. Genome composition analysis
Genome component analysis was performed based on the valid data after quality control of each sample, and using SMRT Link v5.0.1 software to assemble the reads [Citation17]. Align the reads to the assembled genome sequence and count the distribution were conducted to identify the sequencing depth mapping and longest sequence. Finally, by comparing the original data to the preliminary assembly sequence and optimize the assembly results by arrow software. Subsequently, perform coding and repeated sequence prediction on the newly sequenced genome by using software Gene-MarkS (Version 4.17) [Citation18] and Repeat-Masker (Version open-4.0.5) [Citation19]. Based on the sequence composition, IslandPath-DIOMB (Version 0.2) [Citation20] and phiSpy (Version 2.3) [Citation19] were used to predict gene islands and prophages that were related to a variety of biological functions including pathogenicity and environmental adaptability.
2.3. Genome function analysis and visualization display
Diamond comparison of the protein sequence of the predicted gene with each functional database and the result filtering and annotation. Functional analysis is mainly carried out from the following: gene ontology (GO) annotation based on cell components, molecular functions, and biological processes [Citation21]. Kyoto encyclopedia of genes and genomes (KEGG) [Citation22] annotations were including data on genomes, chemical molecules, and biochemical systems, aspect of metabolic pathways (KEGG PATHWAY), drugs (KEGG DRUG), diseases (KEGG DISEASE), functional models (KEGG MODULE), gene sequences (KEGG GENES), genomes (KEGG GENOME), and etc. Annotation of cluster of orthologous groups of proteins (COG) [Citation23] was constructed according to the systematic evolutionary relationship classification of encoded proteins of the complete genome. Carbohydrate-active enZYmes database (CAZy) [Citation24] annotation was evaluated based on the family of related enzymes that can catalyze carbohydrate degradation, modification, and biosynthesis, mainly contains five categories of glycoside hydrolases (GHs), glycosyl transferases (GTs), polysaccharide lyases (PLs) and carbohydrate esterases (CEs), and oxidation reductase (Auxiliary Activities, AAs). In addition, to comprehensively predict whether the protein sequence was a secreted protein and the effector protein using SignalP (Version 4.1), TMHMM (Version 2.0 c) [Citation25] and EffectiveT3 (Version 1.0.1) [Citation26]. Furthermore, pathogen-host interactions database (PHI) annotations were performed to identify virulence or pathogenicity [Citation27].
Finally, the SMRT Link v5.0.1 [Citation17,Citation18] was used to perform methylation site detection and possible methylation transferase-recognized nucleotide motifs on the final genome assembly results (motif) prediction. For the assembled genome sequence of the sequenced sample and combined with the prediction result of the coding gene were performed by the using of Circos software [Citation28] and then display the whole genome map of the sample genome.
3. Results and discussion
Clostridium difficile has attracted more and more attention in recent years due to its high prevalence, plasticity and virulence potential. The genomic diversity of C. difficile has been continuously explored with the advancement of biotechnology, and there is a close relation between metabolism and virulence. Based on this background, this study focuses on the LCL126 strain through genome sequencing and trying to reveal the possible potential toxic mechanism from genome component analysis and functional annotation.
3.1. The Genome overview component characteristics of Clostridium difficile LCL126
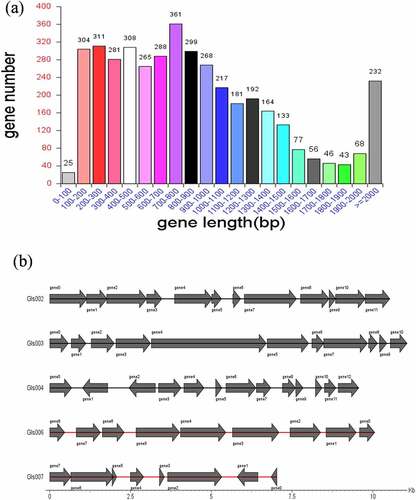
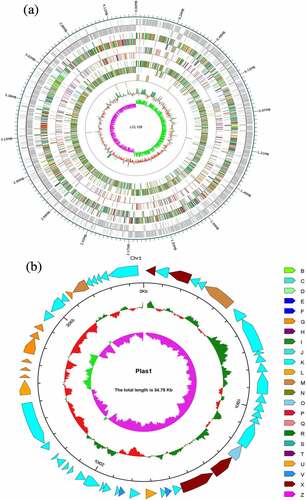
The identified total size of the genome Clostridium difficile LCL126 was 4,301,949 bp with a 27.97% of GC content using high-throughput sequencing, and including a 4,267,198 bp (28.93% GC) circular chromosome and one circular plasmids [plas1 (34,751 bp)] (). The number of total 4119 predicted coding genes were found with average gene length of 875 bp (account total length 83.76%). One hundred twenty-four RNAs (89 tRNAs, 11 rRNAs (5s denovo), 12 rRNAs (5s and 23s denovo) were obtained. A total 188 repeat sequence were detected comprising 105 long terminal repeat array, 31 DNA transposable, 16 short and 34 long interspersed nuclear elements as well as 2 rolling circle. The number of total 13 prophages was predicted with total length 638,171 and average 49,090.1, a total of 17 clustered regularly interspaced short palindromic repeat sequences (CRISPR) array was predicted with average length 543.412. The gene island (GI) prediction sequence was obtained number of 8 GI with 187,976 bp total and 23,497 bp average length, the statistical map of gene distribution in gene islands is presented in . Whole genome of C. difficile identified 622–3000 genes and 4.1–4.3 molecular base pairs (Mbp) length attributed to higher plasticity and degree of recombination, the difference was varied due to techniques and strains [Citation29,Citation30]. Some bacteriophages might be involved in regulate bacterial toxin genes, thereby altering virulence-related phenotypes while the specific role of prophages is unclear but they were involved in the virulence and pathogens evolution [Citation31–33].
Figure 1. The Clostridium difficile LCL126 gene length statistics, the abscissa is the gene length and the ordinate is the number of the corresponding genes (a); and the statistical map of gene distribution in gene islands (b)
3.2. The genome function study of Clostridium difficile LCL126
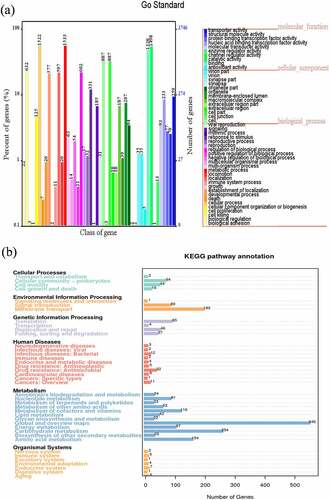
Gene function analysis of Clostridium difficile LCL126 was demonstrated in view of three aspects: function annotation, effector and virulence or pathogenicity analysis. First of all, perform functional annotations through different functional databases. In GO annotation, total number of 12,427 was detected and most outstanding class was cell (887), catalytic activity (1498) and metabolic process (1533) during classified ontology of cellular component, molecular function and biological process (). The KEGG annotation were shown 3175 genes and classified six mainly pathways of cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and organismal systems (). The predominant pathway was global and overview maps and carbohydrate metabolism with 546 and 254 unigenes, followed by membrane transport and amino acid metabolism with 193 and 154 unigenes, 116 unigenes of metabolism of cofactors and vitamins, while translation and cellular community-prokaryotes account 85 and 64 unigenes. As to COG annotation was identified total of 3367 genes and classified into 24 categories, which most abundant term was Transcription with 392 array, followed main richness term was General function prediction only (286) and Carbohydrate transport and metabolism (283) (Fig. S1a). Additionally, CAZy database obtained total 127 genes including 23 Carbohydrate-related modules (CBM), 12 CEs, 63 GHz and 29 GTs (Fig. S1b).
Figure 2. Gene function analysis of Clostridium difficile LCL126 based on Gene Ontology (GO) annotation: the abscissa represents the GO function classification on the sample annotation, the right ordinate represents the number of genes on the annotation, and the left ordinate represents the percentage of the number of genes on the annotation to all coding genes (a); and Kyoto Encyclopedia of Genes and Genomes metabolic pathway classification: and the number on the bar graph represents the number of genes in the annotation, and the other axis is the code of each function class of level1 in the database (b)
Meanwhile, the number of signal peptide, transmembrane structure, and secreted proteins were 84, 993, and 54. Among all the 4119 genes encode, the predicted numbers of T3SS effector and non-T3SS effector proteins were 79 and 4040, respectively. The virulence or pathogenicity analysis was revealed that 133 identified genes belong to reduced virulence, 22 increased virulence (hypervirulence) was found, while 54 unaffected pathogenicity and 10 loss of pathogenicity genes was detected (Fig. S1c).
3.3. Genome methylation and visual analysis of Clostridium difficile LCL126
In biological systems, methylation is enzymatically catalyzed and involves heavy metal modification, gene expression regulation, protein function regulation, and ribonucleic acid (RNA) processing. The presence of methylation modification plays an important role in the substitution of DNA transcription, and abnormal methylation will cause many of diseases usually, thus the systematic mapping of methylation groups has attracted increasing interest. In the epigenetic modification, the final genome assembly results were detected by methylation sites and three types of modified sites were obtained, the number of modification sites type m4C, m5C, m6A, and modified base was 6989, 36,666, 3534, and 351,658, which account for 1.75%, 9.1930%, 0.8861%, and 88.1686%, respectively. The distribution statistics of methylation motifs on genetic regions (GRs)/intergenic region (IRGs) shows that the number of m4C, m5C, and m6C methylation types on the genome was 6989, 36,666, and 3534. The number of methylated types in a gene region account for 83.3, 16.69, and 78.04%. Additionally, the number of nucleotide motif sequence CAAAAA and TV recognized by unmethylated methyltransferase on the genome is 2927 and 1,782,530, and the number of unmethylated motif sequences on gene regions account 78.1 and 83.34%. Visualization of apparent modification distribution methylation circle diagram shown in . Whole-genome visualization map is demonstrated in and the detailed information includes analysis of non-coding RNA, and gene function annotations were stated in the above first part of the genome overview component characteristics. Some strains were relevance to serious illness such as ribo-type 176, 244, 27, 17, 78 by encoding or destroying genes for different enzymes [Citation14]. While as the separation of more detailed strains with the technology advances, it seems to be more diversification than previously record [Citation34,Citation35]. But no reports related to serious diseases found till now. Previous study described the number and type of the pro-inflammatory strain surface protein SlpA that may relate to toxic behavior in C. difficile [Citation14,Citation36,Citation37]. The genomic information was provided in present study confirmed that C. difficile LCL126 has some relevant toxin genes, while in-depth study of its metabolome and functional genes needs to be further studied to explore the toxicological mechanism of C. difficile LCL126, aimed to improve the comprehensive understanding of microorganisms and promote the C. difficile related infectious diseases.
Figure 3. Visualization of apparent modification distribution methylation circle of Clostridium difficile LCL126, each circle from outside to inside represents the position of the genome
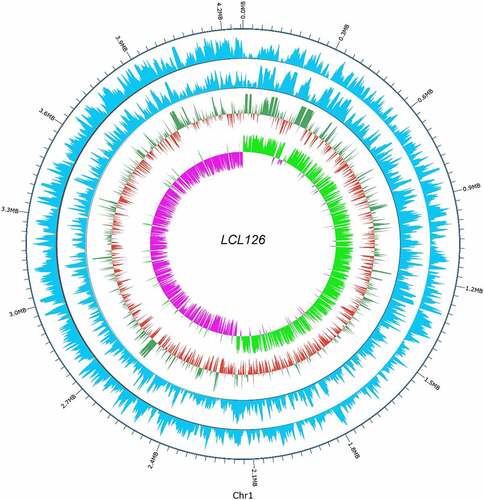
Figure 4. Whole-genome visualization map of Clostridium difficile LCL126. LCL126-Chr1 genome-wide map: the outermost circle is the position coordinates of the genome sequence, from the outside to the inside indicating the coding gene and the result of gene function annotation (a); LCL126-Plas1 genome-wide map: From the outside to the inside is the Cluster of Orthologous Groups (COG) functional annotation classification genes (the arrow indicates the positive chain code in a clockwise direction), the genome sequence position coordinates, and the genome GC content (b)
4. Conclusion
Genome sequencing could be revealing the importance and comprehensive information aspect of biology. Present study has profound insights into genomics and diversity of C. difficile bacteriophages, while this field was still at infancy. The Clostridium difficile LCL126 exist virulence-related gene including 22 increased virulence genes, it might be having virulence potential. In view of the increasing number of C. difficile cases and the severity of the disease, it was essential to expand current limited scientific knowledge about C. difficile bacteriophages. Finally, the ultimate purpose was to develop a non-antibiotic approach via the encode of antibacterial genes.
Highlights:
1. The whole genome information of C. difficile LCL126 was identified.
2. C. difficile LCL126 has total size of the genome 4,301,949 bp with a 27.97% of GC content.
3. 133 reduced virulence and 22 increased virulence (hypervirulence) was detected.
4. Methylation is expressed mainly distributed in m4C, m5C, and m6C with 6989, 36,666, and 3534 genes.
5. The strain of C. difficile LCL126 was predicted to have toxic potential.
Supplemental Material
Download ()Acknowledgements
The authors are thankful to all laboratory colleagues for their advice and help.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- Rineh A, Kelso MJ, Vatansever F, et al. Clostridium difficile infection: molecular pathogenesis and novel therapeutics. Expert Review of Anti-infective Therapy. 2014;12(1):131–150.
- Cao H, Wong S, Yam W, et al. Genomic investigation of a sequence type 67 Clostridium difficile causing community-acquired fulminant colitis in Hong Kong. Int J Med Microbiol. 2019;309(5):270–273.
- Monot M, Eckert C, Lemire A, et al. Clostridium difficile: new insights into the evolution of the pathogenicity locus. Sci Rep. 2015;5(1):15023.
- Marvaud JC, Quevedo-Torres S, Eckert C, et al. Virulence of new variant strains of Clostridium difficile producing only toxin A or binary toxin in the hamster model. Microbes Infect. 2019;32:100590.
- Alcala L, Reigadas E, Marín M, et al. Impact of clinical awareness and diagnostic tests on the underdiagnosis of Clostridium difficile infection. Eur J Clin Microbiol Infect Dis. 2015;34:1515–1525.
- Solomon SL, Oliver KB. Antibiotic resistance threats in the United States: stepping back from the brink. Am Fam Physician. 2014;89:938.
- Dupuy B, Matamouros S. Regulation of toxin and bacteriocin synthesis in Clostridium species by a new subgroup of RNA polymerase σ-factors. Res Microbiol. 2006;157:201–205.
- Knight DR, Elliott B, Chang BJ, et al. Diversity and evolution in the genome of Clostridium difficile. Clin Microbiol Rev. 2015;28:721–741.
- Mani N, Dupuy B. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. P Nat Acad Sci. 2001;98:5844–5849.
- Wang S, Hong W, Dong S, et al. Genome engineering of Clostridium difficile using the CRISPR-Cas9 system. Clin Microbiol Infect. 2018;24:1095–1099.
- Bouillaut L, Dubois T, Sonenshein A, et al. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol. 2015;166:375–383.
- Sim JHC, Truong C, Minot SS, et al. Determining the cause of recurrent Clostridium difficile infection using whole genome sequencing. Diagn Micr Infec Dis. 2017;87(1):11–16.
- Zeng Z, Zhao H, Dorr MB, et al. Bezlotoxumab for prevention of Clostridium difficile infection recurrence: distinguishing relapse from reinfection with whole genome sequencing. Anaerobe. 2019;61:102137.
- Quesada-Gómez C, Murillo T, Arce G, et al. Proteogenomic analysis of the Clostridium difficile exoproteome reveals a correlation between phylogenetic distribution and virulence potential. Anaerobe. 2020;62:102151.
- Li Z, Liu X, Zhao J, et al. Comparison of a newly developed binary typing with ribotyping and multilocus sequence typing methods for Clostridium difficile. J Microbiol Methods. 2018;147:50–55.
- Riedel T, Wittmann J, Bunk B, et al. A Clostridioides difficile bacteriophage genome encodes functional binary toxin-associated genes. J Biotechnol. 2017;250:23–28.
- Lim HJ, Lee EH, Yoon Y, et al. Portable lysis apparatus for rapid single‐step DNA extraction of Bacillus. Portable lysis apparatus for rapid single‐step dna extraction of bacillus subtilis. J Appl Microbiol. 2016;120:379–387.
- Reiner J, Pisani L, Qiao W, et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of aBardet–Biedl Syndrome 9(BBS9) deletion. Npj Genom Med. 2018;3.
- Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Nucleic Acids Res. 2001;29:2607–2618.
- Hsiao W, Wan I, Jones SJ, et al. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics. 2003;19:418–420.
- Minoru M, Susumu G, Shuichi K, et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:277–280.
- Minoru M, Susumu G, Masahiro H, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:354–357.
- Galperin MY, Makarova KS, Wolf YI, et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:261–269.
- Cantarel BL, Coutinho PM, Corinne R, et al. EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:233–238.
- Petersen TN, Brunak S, Heijne G, et al. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786.
- Valerie E, Thomas N, Alexander P, et al. DB—updates and novel features for a better annotation of bacterial secreted proteins and type III, IV, VI secretion systems. Nucleic Acids Res. 2016;44:669–674.
- Martin U, Rashmi P, Arathi R, et al. Interactions database (PHI-base): additions and future developments. Nucleic Acids Res. 2015;43:645–655.
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones S, Marra M. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;1:1639–1645.
- Holt K, Seth-Smith H, Quail M, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci. 2010;107:7527–7532.
- Forgetta V, Oughton MT, Marquis P, et al. Fourteen-genome comparison identifies DNA markers for severedisease- associated strains of Clostridium difficile. J Clin Microbiol. 2011;49:2230–2238.
- Xu P, Zhang X, Su H, et al. Genome-wide analysis of PYL-PP2C-SnRK2s family in Camellia sinensis. Bioengineered. 2020;11:103–115.
- Kuehne S, Minton N. ClosTron-mediated engineering of Clostridium. Bioengineered. 2012;3:247–254.
- Andres-Lasheras S, Bolea R, Mainar-Jaime R, et al. Presence of Clostridium difficile in pig faecal samples and wild animal species associated with pig farms. J Appl Microbiol. 2017;122:462–472.
- Kang Z, Zhang J, Jin P, et al. Directed evolution combined with synthetic biology strategies expedite semi-rational engineering of genes and genomes. Bioengineered. 2015;6:136–140.
- Sheehan D, O’Sullivan S. Online homology modelling as a means of bridging the sequence-structure gap. Bioengineered. 2011;2:299–305.
- Janoir C. Virulence factors of Clostridium difficile and their role during infection. Anaerobe. 2016;37:13–24.
- Tan Y, Yang X, Pei M, et al. A genome-wide survey of interaction between rice and Magnaporthe oryzae via microarray analysis. Bioengineered. 2021;12:108–116.