
ABSTRACT
Massive production of reactive oxygen species (ROS) in human periodontal ligament fibroblasts (HPdLFs) by nitrogen-containing bisphosphonates (BPs) is the main factor causing BP-related osteonecrosis of the jaw. Further, oxidative stress and apoptosis of fibroblasts induced by ROS are closely associated with the activation of MAPK. Parathyroid hormone-related protein (PTHrP) can block the activity of MAPK by regulating the levels of MAPK phosphatase 1 (MKP1). Therefore, it is speculated that PTHrP can inhibit the apoptosis of HPdLFs caused by nitrogen-containing BP via regulating the expression levels of MKP1. Herein, alendronate sodium salt trihydrate (nitrogen-containing BP, FOS) and HPdLFs were co-cultured for 24 h, 48 h, and 72 h, and the levels of ROS and apoptosis were determined, respectively. After 48 h co-culture, FOS significantly increased the levels of ROS and apoptosis, and high phosphorylation levels of p38, ERK1/2 and p66Shc were found in this study. However, the inhibitors of p38 and ERK1/2 significantly reduced the apoptosis of HPdLFs. Interestingly, PTHrP pre-treatment significantly reduced the phosphorylation levels of p38, ERK1/2, and p66Shc. More importantly, MKP1 inhibitor sanguinarine inhibited the dephosphorylation levels of p38, ERK1/2, and p66Shc caused by PTHrP. Altogether, PTHrP can inhibit nitrogen-containing BP-induced apoptosis of HPdLFs by activating MKP1 phosphatase.
Graphical abstract
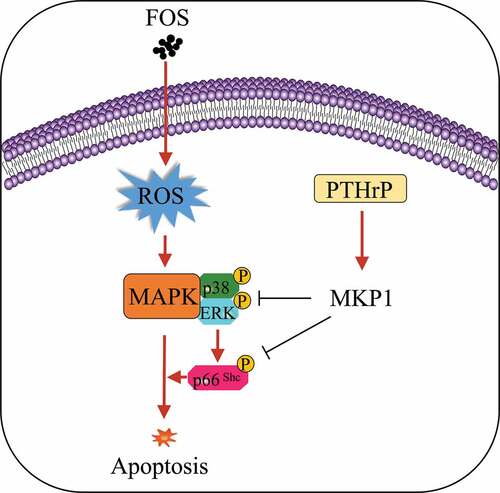
1. Introduction
Bisphosphonates (BPs), which functionally serve as bone resorption inhibitors, have been used in medicine for nearly 30 years, and play an important role in the treatment of malignant bone tumors (multiple myeloma and bone metastasis) or metabolic bone diseases (Paget’s disease and severe osteoporosis) [Citation1]. BPs are divided into first, second, and third generations, among which the second and third generations of BPs containing nitrogen exhibit a stronger inhibitory effect on bone resorption [Citation2,Citation3]. Nitrogen-carrying BPs such as alendronate sodium salt trihydrate (FOS) and zoledronate acid (ZOL), play an important role in the treatment of osteoporosis and bone metastasis [Citation3-6]. However, in recent years, with an increasing use of nitrogen-carrying BPs, the number of cases of nitrogen-carrying BP-related osteonecrosis of the jaw (BRONJ) is gradually increasing, which is mainly related with the fact that nitrogen-carrying BPs can cause strong oxidative stress and cellular apoptosis through the overproduction of reactive oxygen species (ROS) [Citation7,Citation8].
ROS are produced during oxidative phosphorylation of the mitochondria or may be produced by exogenous substances, such as xenobiotic compounds [Citation9]. When the ROS content exceeds the antioxidant defense capacity of cells, it can damage the nucleic acids, proteins, and lipids in cells, causing some clinical symptoms [Citation10,Citation11]. Pharmacological and genetic studies on mice have confirmed the harmful effects of oxidative stress on bones [Citation10]. The harmful effects of oxidative stress involve various molecular mechanisms of cellular processes, including apoptosis and aging [Citation11,Citation12]. ROS can cause the reversible oxidation of cysteine residues, resulting in auto-phosphorylation of proteins, such as apoptosis signal-regulated kinases (ASKs), and activation of downstream mitogen-activated protein kinases (MAPKs) p38, ERK1/2, jun N-terminal kinases (JNKs), and phosphatidylinositol 3-kinase (PI3K)/Akt [Citation13-15]. Cellular stress stimulation, as observed during hypoxia and mechanical and oxidative challenges, can regulate the apoptosis of many cell types, such as osteoblasts [Citation12,Citation16], cardiomyocytes [Citation17], fibroblasts [Citation18], and periodic ligament stem cells [Citation19], by activating the above mentioned kinases. In many cell types, such as osteoblasts [Citation20] and marginal cells [Citation21], p66Shc, an amplifier of ROS, is an important mediator for ROS-induced apoptosis.
Hormones such as thyroid hormones and melatonin, which are extracellular regulators of oxidative stress, can target various signaling pathways in a variety of tissues [Citation10,Citation20,Citation22,Citation23]. Parathyroid hormone (PTH) and its bone counterpart PTH-related protein (PTHrP) combine with the common PTH type 1 receptor (PTH1R), which can increase the survival rate of cells by regulating intracellular signaling pathways such as ERK1/2 and p38MAPKs, thus promoting partial bone formation [Citation24-27]. MAPK phosphatase-1 (MKP1) belongs to the threonine-tyrosine phosphatase family, which can inhibit the activity of ERK, p38MAPKs, and JNK through dephosphorylation [Citation28,Citation29]. Previous studies have indicated that PTHrP can inhibit the activity of MAPKs by regulating the expression of MKP1, and thus inhibit the apoptosis of osteoblasts [Citation30].
In view of the fact that the oxidative stress and apoptosis of fibroblasts induced by ROS are closely associated with the activation of MAPK and that PTHrP can regulate the activity of MAPK by regulating MKP1, it can be speculated that PTHrP can inhibit ROS-induced apoptosis of fibroblasts by regulating the expression of MKP1. To verify this hypothesis, the levels of ROS and apoptosis were determined after FOS and HPdLFs were co-cultured, and the relationship between PTHrP and the expression level of MKP1 was comprehensively analyzed to disentangle whether and how PTHrP pre-treatment inhibits FOS-induced apoptosis of HPdLFs at the molecular level.
2. Materials and methods
2.1 Reagents and antibodies
Nitrogen-containing BP, alendronate sodium salt trihydrate (FOS) was purchased from Wako in Japan. SCGMTM BulletKitTM, N-acetyl-cysteine (NAC), fluorescein isothiocyanate (FITC)-conjugated annexin V, and ECL chemiluminescence were acquired from Sigma in China. PTHrP, cell-permeable 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), U0126 (inhibitor of ERK1/2), SB203580 (inhibitor of p38), sanguinarine chloride (inhibitor of MKP1), lipofectamine LTX transfection reagent, Tris HCl, NaCl, EDTA, 1% Triton X-100, 1% sodium hydroxide, 0.1% sodium dodecyl sulfate (SDS), protease inhibitor cocktail, cocktail Set II, bovine serum albumin (BSA), Considering bicinchoninic acid (BCA), nitrocellulose membrane were obtained from ThermoFisher in China. In addition, α-Tubulin, peroxidase-conjugated goat anti-rabbit IgG and the antibodies of p-ERK1/2, ERK1/2, p-Akt, Akt, p-p38, p38, p-JNK, JNK, MKP-1, and p-p66Shc (Ser36) were acquired from ThermoFisher in China. Propidium iodide was purchased from Solarbio in China.
2.2. Cell culture condition and experimental design
The normal adult HPdLFs used in this study were purchased from ScienCell (USA). The HPdLFs were cultured with SCGMTM BulletKitTM (Sigma, China) at 37°C with 5% CO2.
To investigate the effect of FOS on HPdLFs, after the HPdLFs were treated with 30 µM FOS for 24 h, 48 h, and 72 h, the levels of ROS and apoptosis were determined. Based on the result, HPdLFs treated with 30 µM FOS for 48 h were selected as experimental subject, and then the phosphorylation levels of p38, ERK1/2, Akt, JNK and p66Shc were analyzed. In addition, the inhibitors of p38 (SB203580 [5 µM]) and ERK1/2 (U0126 [10 µM]) were used to analyze the roles of p38 and ERK1/2 in apoptosis of HPdLFs.
To disentangle whether and how PTHrP inhibits FOS-induced apoptosis of HPdLFs, following the treatment with 30 µM FOS, the HPdLFs were treated with 100 nM PTHrP for 30 min, and then with sanguinarine chloride [0.5 µM] for 1 h to analyze the relationship between PTHrP and the expression level of MKP1.
2.3 Determination of ROS
The HPdLFs (1 × 103) were incubated in a 96-well plate for 24 h, and then treated with 30 µM FOS for 24 h, 48 h, and 72 h, respectively. The cells untreated with FOS in the corresponding time points were considered as controls. Approximately 12 h before the cells were treated with FOS, 90 mM NAC was added to the cell suspension [Citation7,Citation31]. To determine the ROS content, CM-H2DCFDA was added to the culture medium until the final concentration was 10 µM. The fluorescence was determined using a fluorescence spectrophotometer (Infinite M200 Pro) at 488 nm excitation and 525 nm emission.
2.5. Detection of apoptosis by flow cytometry
To measure apoptosis, the cells were double-stained with fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide for 5 min at room temperature. The number of annexinV(-)/propidium iodide(-) living cells and annexinV(+) apoptotic cells was examined by flow cytometry (ThermoFisher, USA). The data were collected with FACSCaliburTM (Becton Dickinson) and analyzed with CellQuestTM (Becton Dickinson).
2.6. Western blot analysis
Total cellular protein was extracted at pH 7.4 with 50 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium hydroxide, 0.1% sodium dodecyl sulfate (SDS), protease inhibitor cocktail, and cocktail Set II. Considering bovine serum albumin (BSA) as the standard, bicinchoninic acid (BCA) was used to determine the content of protein in the cellular extract. The proteins were isolated using 5%-10% polyacrylamide-SDS gels (30–60 μg/Lane). After electrophoresis, the sample was transferred to a nitrocellulose membrane, and blocked with 5% skim milk containing 50 mmol/L Tris-HCl (pH 7.5) and 150 mmol/L NaCl containing 0.05% Tween 20. The corresponding antibodies p-ERK 1/2 (1:1000), ERK 1/2 (1:2000), p-Akt (1:1000), Akt (1:2000), p-p38 (1:1000), p38 (1:2000), p-JNK (1:1000), JNK (1:2000), MKP-1 (1:500), and p-p66Shc (Ser36) (1:500) were added, followed by incubation overnight at 4°C. α-Tubulin was used as the loading control. After washing, peroxidase-conjugated goat anti-rabbit IgG was incubated with the membrane, and ECL chemiluminescence was added. The optical density of the protein band was normalized to the corresponding α-tubulin band.
2.7. Statistical analysis
The data are expressed as mean±standard error of mean (SEM). Nonparametric variance analysis (Kruskal–Wallis) was used to evaluate changes in kinase phosphorylation and apoptosis, followed by post-hoc Dunn’s test. P < 0.05 was considered significant.
3. Results
3.1. FOS induces the apoptosis of HPdLFs by regulating the generation of ROS
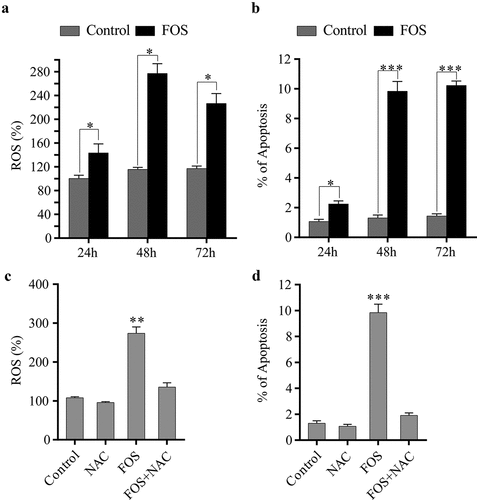
Previous studies have shown that BPs can induce the generation of ROS in HPdLFs. To verify this conclusion, HPdLFs were treated with 30 μM of FOS for different time points (24 h, 48 h, and 72 h). Subsequently, the ROS content was determined and it was found that the ROS content was significantly increased after FOS and HPdLFs were co-cultured for 48 h (). Previous studies have shown that ROS are the main factor causing cellular apoptosis. Therefore, the degree of apoptosis of HPdLFs was examined, and we found that the degree of apoptosis was significantly increased after FOS and HPdLFs were co-cultured for 48 h (). To further study whether the apoptosis of HPdLFs induced by FOS was associated with ROS, after HPdLFs were co-cultured with FOS and the ROS scavenger NAC for 48 h, and the level of ROS and the degree of apoptosis were subsequently determined. It was found that NAC can significantly reduce the level of ROS and the degree of apoptosis (, d), indicating that FOS induced the apoptosis of HPdLFs by promoting the generation of ROS.
Figure 1. ROS generation by FOS treatment induced cell apoptosis in HPdLFs. (a) FOS increased ROS production in HPdLFs after 24, 48, and 72 h. (b) FOS increased apoptosis in HPdLFs after 24, 48, and 72 h. (c) NAC suppressed FOS-induced ROS generation in HPdLF after 48 h. (d) NAC suppressed FOS-induced apoptosis in HPdLF after 48 h. Data represented mean±standard error of mean (SEM) of six independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control
3.2. The role of different protein kinases in FOS-induced apoptosis of HPdLFs
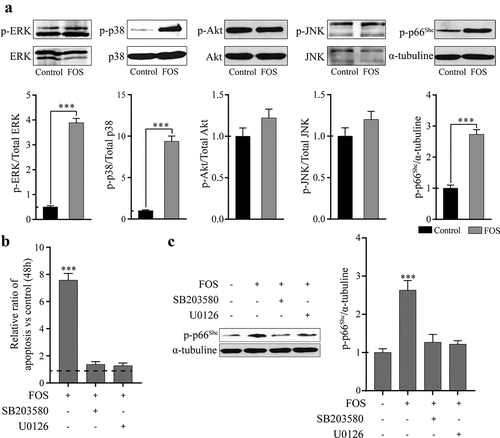
It has been reported that ROS can regulate intracellular kinases and activate the related cell signaling pathways [Citation9,Citation12]. Therefore, to study whether ROS produced by FOS can activate the kinases related to cell activity, the effect of FOS on the phosphorylation of cell survival-related kinases was examined, and it was found that the phosphorylation degrees of ERK1/2, p38MAPK, and the pro-apoptotic and oxidative stress marker p66Shc were significantly increased (), whereas no significant changes were observed in the phosphorylation levels of Akt and JNK. To study whether FOS affects the survival of HPdLFs by regulating the phosphorylation of ERK1/2 and p38, FOS and HPdLFs were co-cultured in a media containing ERK1/2 and p38 inhibitors, respectively, and the degree of cell apoptosis was then determined. We found that ERK1/2 and p38 inhibitors can significantly reduce the apoptosis induced by FOS (). Phosphorylation levels of ERK1/2 and p38 have been reported to affect the activity of p66Shc. Thus, we analyzed the effect of ERK1/2 and p38 inhibitors on the phosphorylation of p66Shc and found that ERK1/2 and p38 inhibitors can inhibit p66Shc phosphorylation induced by FOS (). This indicated that FOS could promote the phosphorylation of p66Shc by regulating ERK1/2 and p38, and subsequently induce apoptosis.
Figure 2. FOS induced apoptosis by triggering phosphorylation of p38, ERK1/2, and p66Shc in HPdLFs. (a) FOS (30 μM) significantly increased ERK1/2 (44/42 KDa), p38 (43 KDa), and p66Shc (66 KDa) (pro-apoptotic and oxidative marker) phosphorylation, at 48 h in HPdLFs. (b) FOS-induced apoptosis was inhibited by pretreatment with p38 (5 mM SB203580) or ERK1/2 (10 mM U0126) inhibitors for 30 min in HPdLFs. (c) FOS-induced phosphorylation of the oxidative stress and apoptosis marker p66Shc was suppressed by pretreatment with p38 (5 mM SB203580) or ERK1/2 (10 mM U0126) inhibitors for 30 min in HPdLFs. Data represented mean±standard error of mean (SEM) of six independent experiments. ***P < 0.001 versus control
3.3. PTHrP inhibits the phosphorylation of kinases in an MKP1-dependent manner
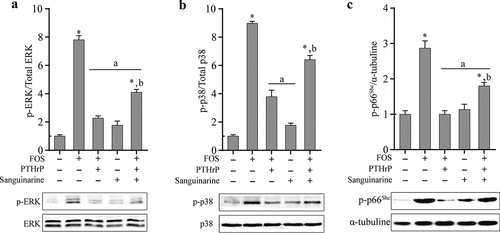
It has been reported that PTHrP can inhibit the phosphorylation of MAPKs induced by H2O2-induced ROS in osteoblasts [Citation30]. To study whether PTHrP can inhibit the phosphorylation of ROS-induced kinases induced by FOS in HPdLFs, HPdLFs pre-treated with PTHrP were co-cultured with FOS for 48 h, followed by the examination of phosphorylation. It was found that PTHrP can significantly reduce the FOS-induced phosphorylation levels of ERK1/2, p38, and p66Shc (). Previous studies have shown that PTHrP can regulate the activities of p38, ERK1/2, and p66Shc through MKP1 [Citation30]. Therefore, we evaluated whether PTHrP inhibits the phosphorylation levels of p38, ERK1/2, and p66Shc induced by FOS through MKP1. To verify this hypothesis, as described previously [Citation32-34], we used MKP1 inhibitor sanguinarine to pre-treat HPdLFs and found that the phosphorylation levels of ERK1/2 and p66Shc in HPdLFs treated with PTHrP and FOS were significantly higher (), indicating that MKP1 inhibitor sanguinarine can significantly inhibit the dephosphorylation ability of PTHrP for ERK1/2 and p66Shc.
Figure 3. PTHrP inhibited FOS-dependent kinase phosphorylation by mediating MKP1 in HPdLFs. PTHrP reduced FOS-induced ERK1/2 activation in HPdLFs, and the effect was suppressed by MKP1 inhibitor, sanguinarine (0.5 mM) (a). PTHrP reduced FOS-induced p38 activation in HPdLFs, and the effect was suppressed by MKP1 inhibitor, sanguinarine (0.5 mM) (b). PTHrP reduced FOS-induced p66Shc activation in HPdLFs, and the effect were suppressed by MKP1 inhibitor, sanguinarine (0.5 mM) (c). Data represented mean±standard error of mean (SEM) of six independent experiments. *P < 0.05 versus control; aP<0.05 versus FOS stimulation; bP<0.05 versus corresponding condition without inhibitor
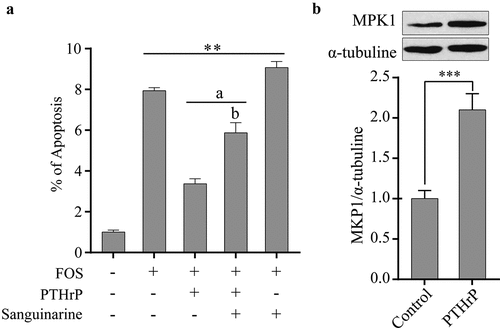
3.4. PTHrP inhibits FOS-induced apoptosis by regulating the expression of MKP1
To determine whether PTHrP can inhibit the apoptosis of HPdLFs induced by FOS, HPdLFs cells were co-cultured with PTHrP and FOS for 48 h and the degree of apoptosis was examined. It was found that PTHrP could significantly inhibit FOS-induced apoptosis of HPdLFs (). However, sanguinarine can completely abolish this inhibitory effect of PTHrP on the apoptosis of HPdLFs (). To investigate whether PTHrP can promote the expression of MKP1 in HPdLFs, HPdLFs were pre-treated with PTHrP for 30 min, and the protein level of MKP1was then determined. We found that PTHrP can promote the expression of MKP1 (). The above mentioned results indicated that PTHrP can inhibit the phosphorylation levels of p38 and ERK1/2 by promoting the expression of MKP1, thus inhibiting the apoptosis of HPdLFs induced by FOS.
Figure 4. PTHrP inhibited FOS-induced apoptosis by MKP1 in HPdLFs. (a) PTHrP inhibited FOS-induced apoptosis in HPdLFs, and the effect was suppressed by MKP1 inhibitor, sanguinarine, (0.5 mM). (b) PTHrP increased protein level of MKP1 in HPdLFs. Data represented mean±standard error of mean (SEM) of six independent experiments. **P < 0.01 and ***P < 0.001 versus control; aP<0.05 versus FOS stimulation; bP<0.05 versus the corresponding FOS + PTHrP (without inhibitor)
4. Discussion
The massive production of ROS can damage HPdLFs, and subsequently cause BRONJ [Citation7]. During the process of cellular response to oxidative damage, MAPKs, such as ERK1/2, p38, and JNK, can be activated, and can eventually affect the survival, proliferation, and differentiation ability of various types of cells, such as cardiomyocytes [Citation17], hepatocytes [Citation35], T lymphocytes [Citation36], osteoblasts [Citation12,Citation16], and fibroblasts [Citation18]. Previous studies have shown that PTHrP can inhibit the activation of MAPKs induced by ROS [Citation37]. Therefore, we hypothesized that PTHrP can inhibit the pro-apoptotic effect of ROS on HPdLFs by regulating the MAPK-signaling pathway. Our results indicated that PTHrP can inhibit the FOS-induced apoptosis of HPdLFs by activating MKP1.
It has been reported that BPs can induce the generation of ROS in many types of cells, such as human gingival fibroblasts [Citation38], osteoclast precursor cells [Citation39], and mature osteoclast-like cells [Citation39]. In this study, after FOS and HPdLFs were co-cultured for 48 h, we found that the level of ROS in HPdLFs and the degree of HPdLF apoptosis both significantly increased. Previous studies have shown that ROS can induce the apoptosis of many types of cells, such as osteoblasts [Citation10] and human embryonic fibroblasts [Citation40], by activating MAPKs (such as ERK1/2, JNK, p38, and Akt). This study indicated that ERK1/2 and p38 were activated by FOS in HPdLFs. In some cell lines, the activated ERK can induce the release of mitochondrial cytochrome c, the activation of caspase-8, permanent cell cycle arrest, or the formation of autophagic vacuoles [Citation41]. Under normal condition, the redox regulatory protein thioredoxin (Trx) binds to and inhibits the activity of the protein apoptosis signal-regulated kinase 1 (ASK1), which is associated with p38 and JNK activation. However, ROS can cause the dissociation of the Trx-ASK1 complex, which in turn activates p38 and JNK and triggers apoptosis [Citation12,Citation42]. Although the activation of JNK and Akt induced by ROS plays an important role in the regulation of apoptosis, our results show that FOS cannot activate JNK and Akt [Citation43]. Therefore, JNK and Akt may not be involved in the FOS-mediated apoptosis pathway of HPdLFs.
PTHrP regulates various biological processes of various mammalian cells, such as cell differentiation, proliferation, and apoptosis [Citation44,Citation45]. Previous studies have shown that PTHrP can inhibit H2O2-induced apoptosis of osteoblasts by regulating the phosphorylation of p38 MAPKs and ERK [Citation30]. This study showed that exogenous PTHrP can inhibit the phosphorylation of p38 MAPK and ERK and FOS-induced apoptosis of HPdLFs, indicating that PTHrP can inhibit FOS-induced apoptosis of HPdLFs by regulating p38 MAPK and ERK signaling pathway. In addition, PTHrP can inhibit the phosphorylation of ERK and p38 by promoting the expression of MKP1, thereby inhibiting the apoptosis of osteoblasts induced by H2O2 [Citation30]. This study indicated that the protein level of MKP1 in HPdLFs pre-treated with PTHrP was significantly increased, and that sanguinarine an inhibitor of MKP1 can significantly reduce the dephosphorylation of p38 MAPK and ERK1/2 induced by PTHrP. Further, we found that PTHrP can inhibit the apoptosis of HPdLFs by promoting the expression of MKP1 and inhibiting the phosphorylation of p38 and ERK1/2 induced by FOS. It is worth noting that sanguinarine completely inhibited the dephosphorylation of PTHrP to p38, and partially inhibited the dephosphorylation of PTHrP to ERK. MKP1 has been reported to dephosphorylate MAPK family proteins according to affinity (p38 ≥ JNK > > ERK1/2) [Citation46]. Therefore, the expression of MKP1 induced by PTHrP may completely inhibit the activity of p38 in HPdLFs, but it cannot completely inhibit the activity of ERK.
It has been reported that the phosphorylation of p66Shc at Ser36 can regulate oxidative stress-related apoptotic response and cell senescence [Citation47]. The phosphorylation of p66Shc can open the mitochondrial permeability transition pore, leading to the swelling of organelles [Citation48]. Subsequently, the disruption of mitochondrial integrity can lead to the release of a variety of pro-apoptotic mitochondrial factors into the cytosol, thus activating the apoptotic cascade pathway and causing cell death [Citation49]. Phosphorylated p66Shc can not only regulate apoptotic signals but also continuously enhance the production of ROS through different mechanisms. Therefore, regulation of p66Shc phosphorylation may be the key target for inhibiting apoptosis induced by ROS. Our results show that a part of dephosphorylation of PTHrP to p66Shc is regulated by MKP1 in HPdLFs. In response to the process of oxidative stress, the activation of ERK1/2 and p38 may promote the phosphorylation of p66Shc at Ser36, Ser54, and Thr386 [Citation50,Citation51], indicating that phosphorylated p66Shc may be a downstream effector in the ERK1/2 and p38 pro-apoptosis process. We found that the inhibitors of ERK1/2 and p38 can inhibit the FOS-induced phosphorylation of p66Shc, suggesting that p66Shc is a downstream regulator of ERK1/2 and p38 during the process of FOS-induced HPdLF apoptosis.
5. Conclusion
In summary, our results suggest that PTHrP inhibits nitrogen-containing BP-induced apoptosis of HPdLFs by activating MKP1 phosphatase.
Highlights
FOS-induced ROS cause oxidative stress and cellular apoptosis in HPdLFs
Cellular apoptosis of HPdLFs is closely associated with the activation of MAPK
PTHrP can regulate the activity of MAPK by regulating the levels of MKP1
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bassett CA, Donath A, Macagno F, et al. Diphosphonates in the treatment of myositis ossificans. Lancet. 1969;2(7625):845.
- Rogers MJ, Crockett JC, Coxon FP, et al. Biochemical and molecular mechanisms of action of bisphosphonates. Bone. 2011;49(1):34–41.
- Lindsay R. Modeling the benefits of pamidronate in children with osteogenesis imperfecta. J Clin Invest. 2002;110(9):1239–1241.
- Marx RE. Pamidronate (Aredia) and zoledronate (Zometa) induced avascular necrosis of the jaws: a growing epidemic. J Oral Maxillofac Surg. 2003;61(9):1115–1117.
- Ruggiero SL, Dodson TB, Fantasia J, et al. American association of oral and maxillofacial surgeons. American association of oral and maxillofacial surgeons position paper on medication-related osteonecrosis of the jaw–2014 update. J Oral Maxillofac Surg. 2014;72(10)::1938–1956.
- Ruggiero SL, Mehrotra B, Rosenberg TJ, et al. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 2004;62(5):527–534.
- Taniguchi N, Osaki M, Onuma K, et al. Bisphosphonate-induced reactive oxygen species inhibit proliferation and migration of oral fibroblasts: a pathogenesis of bisphosphonate-related osteonecrosis of the jaw. J Periodontol. 2020;91(7):947–955.
- Lang M, Zhou Z, Shi L, et al. Influence of zoledronic acid on proliferation, migration, and apoptosis of vascular endothelial cells. Br J Oral Maxillofac Surg. 2016;54(8):889–893.
- Ray P, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–990.
- Almeida M, O’Brien CA. Basic biology of skeletal aging: role of stress response pathways. J Gerontol A Biol Sci Med Sci. 2013;68(10):1197–1208.
- Haigis MC, Yankner BA. The Aging Stress Response. Mol Cell. 2010;40(2):333–344.
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15.
- Janssen-Heininger YM, Mossman BT, Heintz NH, et al. van der Vliet A: redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45(1):1–17.
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–990.
- Checa J, Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. J Inflamm Res. 2020;13:1057–1073.
- Haigis MC, Yankner BA. The aging stress response. Mol Cell. 2010;40(2):333–344.
- Xiao L, Pimentel DR, Wang J, et al. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol. 2002;282(4):C926–934.
- Kim BY, Han MJ, Chung AS. Effects of reactive oxygen species on proliferation of Chinese hamster lung fibroblast (V79) cells. Free Radic Biol Med. 2001;30(6):686–698.
- Wu Y, Yang Y, Yang P, et al. The osteogenic differentiation of PDLSCs is mediated through MEK/ERK and p38 MAPK signalling under hypoxia. Arch Oral Biol. 2013;58(10):1357–1368.
- Almeida M, Han L, Ambrogini E, et al. Oxidative stress stimulates apoptosis and activates NF-kappaB in osteoblastic cells via a PKCbeta/p66shc signaling cascade: counter regulation by estrogens or androgens. Mol Endocrinol. 2010;24(10):2030–2037.
- Hao C, Wu X, Zhou R, et al. Downregulation of p66Shc can reduce oxidative stress and apoptosis in oxidative stress model of marginal cells of stria vascularis in Sprague Dawley rats. Drug Des Devel Ther. 2019;13:3199–3206.
- Duan W, Yang Y, Yi W, et al. New role of JAK2/STAT3 signaling in endothelial cell oxidative stress injury and protective effect of melatonin. PLoS One. 2013;8(3):e57941.
- Villanueva I, Alva-Sánchez C, Pacheco-Rosado J. The role of thyroid hormones as inductors of oxidative stress and neurodegeneration. Oxid Med Cell Longev. 2013;2013:218145.
- Miao D, He B, Jiang Y, et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J Clin Invest. 2005;115(9):2402–2411.
- McCauley LK, Martin TJ. Twenty-five years of PTHrP progress: from cancer hormone to multifunctional cytokine. J Bone Miner Res. 2012;27(6):1231–1239.
- Esbrit P, Alcaraz MJ. Current perspectives on parathyroid hormone (PTH) and PTH-related protein (PTHrP) as bone anabolic therapies. Biochem Pharmacol. 2013;85(10):1417–1423.
- Mahalingam CD, Sampathi BR, Sharma S, et al. MKP1-dependent PTH modulation of bone matrix mineralization in female mice is osteoblast maturation stage specific and involves P-ERK and P-p38 MAPKs. J Endocrinol. 2013;216(3):315–329.
- Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60(3):261–310.
- Kirk SG, Samavati L, Liu Y. MAP kinase phosphatase-1, a gatekeeper of the acute innate immune response. Life Sci. 2020;241:11715.
- Ardura JA, Portal-Núñez S, Castelbón-Calvo I, et al. Parathyroid hormone-related protein protects osteoblastic cells from oxidative stress by activation of MKP1 phosphatase. J Cell Physiol. 2017;232(4):785–796.
- Tai TW, Chen CY, Su FC, et al. Reactive oxygen species are required for zoledronic acid-induced apoptosis in osteoclast precursors and mature osteoclast-like cells. Sci Rep. 2017;7(1):44245.
- Trappanese DM, Sivilich S, Ets HK, et al. Regulation of mitogen-activated protein kinase by protein kinase C and mitogen-activated protein kinase phosphatase-1 in vascular smooth muscle. Am J Physiol Cell Physiol. 2016;310(11):C921–C930.
- Chen Y, Wang H, Zhang R, et al. Microinjection of sanguinarine into the ventrolateral orbital cortex inhibits Mkp-1 and exerts an antidepressant-like effect in rats. Neurosci Lett. 2012;506(2):327–331.
- Vogt A, Tamewitz A, Skoko J, et al. The benzo[c]phenanthridine alkaloid, sanguinaraine, is a selective, cell active inhibitor of mitogen-activated protein kinase phosphatase-1. J Biol Chem. 2005;280(19):19078–19086.
- Conde De La Rosa L, Mh S, Te V, et al. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44(5):918–929.
- Griffith CE, Zhang W, Wange RL. ZAP-70-dependent and -independent activation of Erk in Jurkat T cells. Differences in signaling induced by H2o2 and Cd3 cross-linking. J Biol Chem. 1998;273(17):10771–10776.
- Lezcano V, Gentili C, De Boland AR. Role of PTHrP in human intestinal Caco-2 cell response to oxidative stress. Biochim Biophys Acta. 2013;1833(12):2834–2843.
- De Colli M, Zara S, Di Giacomo V, et al. Nitric oxide-mediated cytotoxic effect induced by zoledronic acid treatment on human gingival fibroblasts. Clin Oral Investig. 2015;19:1269–1277.
- Tai TW, Su FC, Chen CY, et al. Activation of p38 MAPK-regulated Bcl-xL signaling increases survival against zoledronic acid-induced apoptosis in osteoclast precursors. Bone. 2014;67:166–174.
- Kang W, Jia Z, Tang D, et al. Fusobacterium nucleatum facilitates apoptosis, ROS generation, and inflammatory cytokine production by activating AKT/MAPK and NF-κB signaling pathways in human gingival fibroblasts. Oxid Med Cell Longev. 2019;2019:1681972.
- Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 2010;277(1):2–21.
- Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94.
- Kim AH, Khursigara G, Sun X, et al. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21(3):893–901.
- Meyer R, Schreckenberg R, Kretschmer F, et al. Parathyroid hormone-related protein (PTHrP) signal cascade modulates myocardial dysfunction in the pressure overloaded heart. Eur J Heart Fail. 2007;9(12):1156–1162.
- Röthig A, Schreckenberg R, Weber K, et al. Effects of nicotine on PTHrP and PTHrP receptor expression in rat coronary endothelial cells. Cell Physiol Biochem. 2012;29(3–4):485–492.
- Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. 1997;272(27):16917–16923.
- Migliaccio E, Giorgio M, Mele S, et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402(6759):309–313.
- Giorgio M, Migliaccio E, Orsini F, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122(2):221–233.
- Orsini F, Migliaccio E, Moroni M, et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem. 2004;279(24):25689–25695.
- Hu Y, Wang X, Zeng L, et al. ERK phosphorylates p66shcA on Ser36 and subsequently regulates p27kip1 expression via the Akt-FOXO3a pathway: implication of p27kip1 in cell response to oxidative stress. Mol Biol Cell. 2005;16(8):3705–3718.
- Fa K, Yamamori T, Mattagajasingh I, et al. Rac1 leads to phosphorylation-dependent increase in stability of the p66shc adaptor protein: role in Rac1-induced oxidative stress. Mol Biol Cell. 2006;17(1):122–129.