
ABSTRACT
The gut microbiota is widely considered to be involved in several diseases, including atherosclerosis, obesity, chronic obstructive pulmonary disease (COPD) and pulmonary arterial hypertension (PAH). This study aimed to determine if changes in the gut microbiome and metabolome play a major role in the early pathogenesis of PAH. Male Wistar rats were injected with monocrotaline (MCT) (55 mg/kg) at day 1 and injected with calcium-sensing receptor (CaSR) antagonist NPS2143 (4.5 mg/kg/d) from days 1 to 21. Fecal samples were obtained. The gut microbiota and metabolome were analyzed by 16S rRNA gene sequencing and mass spectrometry-based analysis to investigate the effect of PAH in this rat model. MCT injection had a marked effect on the composition of the gut microbiota. This finding was further confirmed by metabolomic analysis with identification of several metabolites relevant to the gut microflora. However, NPS2143 partially abrogated this intestinal flora disorder and reversed fecal metabolite abnormalities. In conclusion, our study shows correlations between changes in the gut microbiome and the metabolome in PAH, which are affected by NPS2143.
Graphical abstract
Introduction
Pulmonary arterial hypertension (PAH) has the distinctive features of decreased pulmonary arterial compliance (PAC) together with elevated pulmonary vascular resistance (PVR). Therefore, PAH could become fatal and is thus considered to be a major concern in human health [Citation1]. Various issues with PAH include a lack of rapid and reliable screening and detection assays for early diagnosis of PAH, a lack of specific and reliable biomarkers for PAH, and no novel treatment and therapy are available [Citation2]. Consequently, determination of the mechanism of PAH and development of novel therapeutic treatments are important.
Calcium (Ca2+)-sensing receptor (CaSR) is a member of family C of G-protein coupled receptor amino acids. In combination with G protein, multiple intracellular signaling pathways are activated or inhibited [Citation3]. The ligands or activators of CaSR consist of polyvalent cations (e.g., Ca2+, Mg2+, and Gd3+), polypeptides (e.g., amyloid-β peptide), polyamines (e.g., spermine, spermidine, and putrescine), aminoglycoside antibiotics (e.g., neomycin and kanamycin), and amino acids (e.g., phenylalanine, tyrosine, tryptophan, and glutamate). Moreover, synthetic CaSR agonists (calcimimetics) (e.g., NPS-R-568 and NPS-R-467) and CaSR antagonists (calcilytics) (e.g., NPS 2143) affect CaSR function [Citation4]. The main function of CaSR is to maintain the balance of serum Ca2+, and it is also involved in cellular proliferation and differentiation, and hormone secretion. CaSR is upregulated with a rise in cytosolic-free Ca2+ concentrations triggered by extracellular Ca2+ in pulmonary arterial smooth muscle cells in patients with idiopathic pulmonary arterial hypertension. Blocking the pharmacological effect of CaSR considerably alleviates the development and progression of PAH in rats [Citation5].
For the past decade, the human microbiome has been well established a leading factor in human health and is involved in various diseases. Of these, the gut microbiota is considered to be the most important aspect, and it is associated with respiratory diseases (e.g., PAH) through effects on the gut lung axis [Citation6–8]. Studies have also shown an association between the gut microbiota and produced metabolites, which greatly affects PAH and other diseases [Citation9–11]. Callejo et al. [Citation12] reported that a misbalanced bacterial ecosystem might play a pathophysiological role in PAH by altering immunological, hormonal, and metabolic homeostasis. Another study showed that antibiotic-induced modification of the gut microbiota suppressed the development of PAH [Citation13]. Kim et al. [Citation14] reported that PAH-associated species, such as Blautia, Bifidobacterium, and Collinsella, contributed genes for carnitine metabolism or biosynthesis of arginine, ornithine, and proline. This provided evidence that a change in gut microbiome function in a PAH cohort is closely associated with taxonomic alterations. Patients with PAH have a unique microbiome profile that has a high predictive potential for PAH. Identification and characterization of PAH-specific bacteria and metabolites in the gut could lead to the development of innovative strategies for the control and treatment of PAH.
Multiple omics and bioinformatics analyses are widely used to investigate the molecular mechanism of various diseases [Citation15,Citation16]. However, the combination of determining the gut microbiota and metabolomics analysis in PAH has not been reported. Therefore, in the current study, we investigated the gut microbiota and performed metabolomic analysis to determine the mechanism of how the host and microbiome are involved in PAH in a rat model.
Materials and methods
Animal model for PAH
For the animal model, male Wistar rats that weighed from 250 to 350 g and were aged from 6 to 8 weeks old, were used in this study. The acquisition and care of rats are described in the Acknowledgment section. Twenty-four rats, in three groups (control, PAH, and PAH+NPS2143), were used in this study. All experiments on rats were performed by following a standard procedure as previously published. Induction of PAH was performed by monocrotaline (MCT) injection on the first day at a dose of 55 mg/kg. Subsequently, during days 1 to 21, intraperitoneal injection of NPS2143 was performed on a daily basis at a dose of 4.5 mg/kg/day. Finally (day 21), sodium pentobarbital at a dose of 30 mg/kg was used to sacrifice the rats. The development of the PAH model was evaluated by measuring the ratio of the right ventricle/left ventricle + septum, and right ventricular systolic pressure.
Analysis of the gut microbiota
For analysis of the gut microbiota, we collected and processed fresh fecal samples of rats according to a standard procedure. Template DNA isolation was further performed, followed by sequencing on the Illumina HiSeq 2500 platform. The primer sequences were as follows: 341 F 5′-CCTAYGGGRBGCASCAG-3′ and 806 R 5′-GGACTACNNGGGTATCTAAT-3′. Sequence assembly, quality control, and clustering were then performed.
Metabolomic profiling by liquid chromatography and mass spectrometry (LC-MS)
Metabolomic analysis of all samples was performed using the Thermo Ultimate 3000 system according to previous studies [Citation17]. Additionally, ESI-MSn analysis was conducted as previously described [Citation18,Citation19].
Correlation analysis of gut microbiome and host metabolome
The correlation between the gut microbiota and the host metabolome was analyzed. Analysis was performed as previously described [Citation20].
Statistical analysis
Data are showed as the mean ± standard error. Data were analyzed using one-way analysis of variance. Pearson’s correlation analysis was used to determine the correlation between the gut microbiota and the host metabolome. Relative indices were analyzed using SPSS version 22.0 software. The data were graphically plotted using GraphPad Prism 7. Differences were considered statistically significant at p < 0.05.
Results
The gut microbiome is altered in rats with PAH
Ca2+ can be a stimulation factor depending on different concentrations, and the receptor for Ca2+also plays an important role [Citation5]. Because the effect of NPS 2143 and MCT on PAH still requires further clarification, we analyzed the gut microbiota in rats with PAH to determine the pharmacological effects of these compounds on PAH.
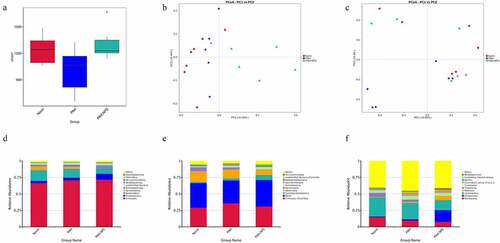
Microbial composition of the gut microbiota showed a lower microbial diversity in the PAH group compared with the control group. However, this change was reversed in the PAH+NPS2143 group, in which NPS2143 led to a significant increase in bacterial diversity ()). Various bacterial species were identified in all three groups () and (c)). Despite sharing similarity in dominating species, changes in the abundance of the gut microbiota were found in all three groups. For the phylum level, the relative abundance of Firmicutes, Proteobacteria, and Actinobacteria was higher in the PAH and PAH+NPS2143 groups than in the control group. However, the relative abundance of Bacteroidota and Spirochaetota was lower in the PAH and PAH+NPS 2143 groups than in the control group ()). For class level, the relative abundance of Bacilli decreased in the PAH but increased in the PAH +NPS2143 group compared with the control group; Firmicutes-Clostridia increased in the PAH group but decreased in the PAH +NPS2143 group, and Gammaproteobacteria was increase in the PAH and PAH+NPS 2143 groups compared with the control group. However, the relative abundance of Bacteroidia and Spirochaetia was lower in the PAH and PAH+NPS2143 groups than in the control group ()). For the genus level, the relative abundance of Allobaculum, Ralstonia, and Bifidobacterium was higher in the PAH and PAH+NPS2143 groups than in the control group. However, the relative abundance of Lactobacillus and Romboutsia was lower in the PAH and PAH+NPS2143 groups than in the control group. Additionally, the relative abundance of Turicibacter, Candidatus_Saccharimonas, and Clostridium_sensu_stricto_1 was increased in the PAH group but decreased in the PAH+NPS 2143 groups ()).
Figure 1. Intestinal microbial diversity and composition among three groups. (a) Variation in diversity within the three groups by Chao index. PCoA plots based on weighted UniFrac metrics (b) and unweighted UniFrac metrics (c). Average relative abundances of dominant bacterial phylum level (d), class level (e), and genus level (f) in the intestine under different treatments
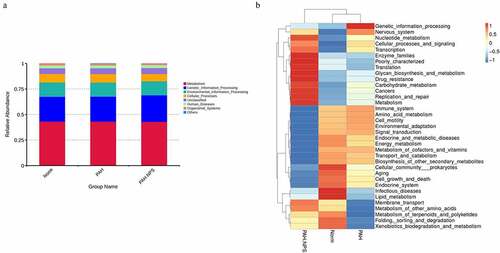
Using the Tax4Fun package, we found that a few pathways were involved in the pathogenicity of the gut bacteria in PAH ()). These pathways were cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and organismal systems. ) shows the significantly different functional genes associated with PAH in the three different groups.
Figure 2. Genes were involved in the pathogenicity of the gut bacteria among three groups. (a) Genes involved in the regulation. (b) All comparisons genes listed in the heatmap
The fecal metabolome is altered in rats with PAH
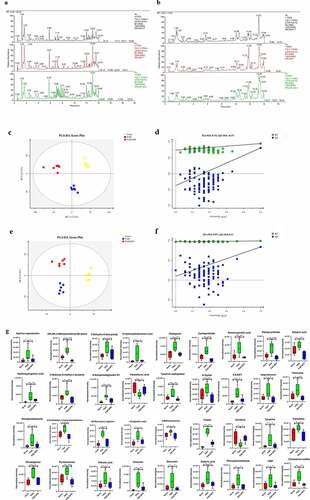
In recent years, alterations in the metabolome due to the microbiome have been reported. Accordingly, we investigated the association between the gut microbiota and the metabolome in this study. Typical LC/MS total ion current (TIC) chromatograms in positive and negative modes of nontarget metabolomics from three groups were shown in ). Visual inspection of these spectra showed obvious difference among the three groups. Using our optimized LC/MS analysis protocol in association with a software-based peak deconvolution procedure, the most peaks were identified. Regardless of positive or negative ionization modes, a significant difference was found for all three groups (–f)). As shown in ) and F, R2, and Q2 are similar, indicating that each of the subjects contributes equally and uniformly to the observed
Figure 3. Effect of disruption on the fecal metabolome. Derived PLS-DA score plots and corresponding permutation testing of PLS-DA from the LC-MS metabolite profiles among three groups. Typical LC/MS total ion current (TIC) chromatograms of non target metabolomics from three groups in positive (a) and negative (b) modes. (c) PLS-DA score plot of positive ions. (d) Permutation testing of positive ions. (e) PLS-DA score plot of negative ions. (f) Permutation testing of negative ions. (g) Fecal metabolites disorder among three groups. The data are presented as the mean ± SD, significant differences between groups are indicated as ***P < 0.001, **P < 0.01 and *P < 0.05
group separation. Hence, the modeling of PLS-DA utilized in this study is stable and reproducible.
This finding indicated that MCT and NPS2143 may be capable of altering metabolomics in rats. A similar finding was also observed ()). We found significantly dysregulated metabolites in the PAH group compared with the control group. Among these metabolites, the levels of 34 were higher, while those of 2 were lower in the PAH group than in the control group. However, these changes were significantly abrogated in the PAH+NPS 2143 group. Therefore, dramatic changes in metabolites of the gut ecosystem appeared to occur in NPS 2143-treated rats with PAH.
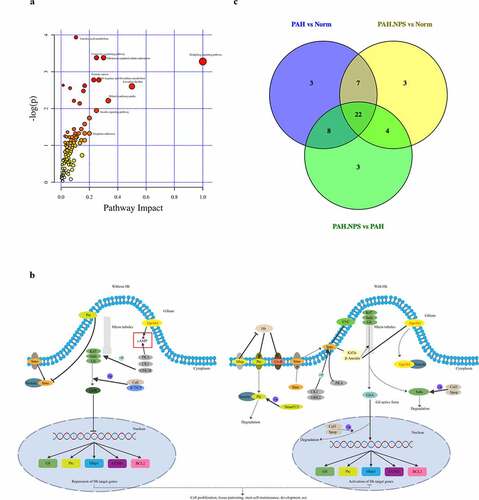
The above-mentioned significantly dysregulated metabolites were enriched in different pathways ()). On the basis of their p value or impact value, 22 metabolic pathways were identified () and KEGG annotation was performed ()). The Venn diagram of common pathways among the three groups compared with each other is shown in ). Therefore, untargeted metabolomics support the global changes in metabolomics predicted from related changes in the composition of the microbiome.
Table 1. Results from Pathway Analysis
Figure 4. The most enriched KEGG pathways analysis. (a) Ingenuity pathway analysis. The size and color are based on the p-value and impact value, small p-value and big pathway impact value indicate that the pathway is greatly influenced. (b) Overall perspective of hedgehog signaling pathway metabolism map. (c) Venn diagram for common pathway among the three groups
Correlation between the gut microbiota and the metabolome
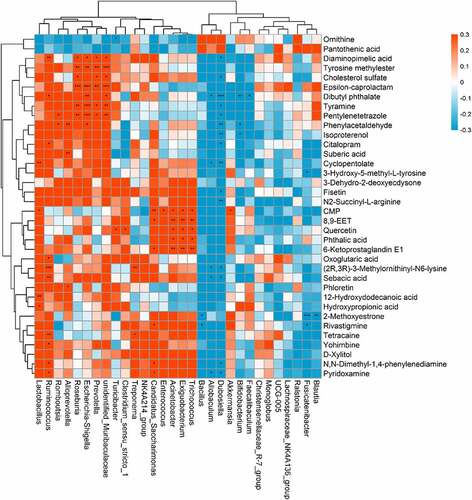
The indirect influence of the gut microbiota composition on the host metabolome has been recently reported [Citation21,Citation22]. We further investigated this correlation specifically after NPS 2143 treatment in rats with PAH (). Allobaculum was negatively correlated with changes in pyridoxamine, sebacic acid, (2 R,3 R)-3-methylornithinyl-N6-lysine, cyclopentolate, and dibutyl phthalate. Bifidobacterium was negatively correlated with changes in isoproterenol and phenylacetaldehyde. Lactobacillus was positively correlated with changes in hydroxypropionic acid, 12-hydroxydodecanoic acid, CMP, and cyclopentolate. Romboutsia was positively correlated with changes in phenylacetaldehyde and pentylenetetrazole. There were also positive correlations between Candidatus_Saccharimonas and changes in pyridoxamine, N,N-dimethyl-1,4-phenylenediamine, rivastigmine, 6-ketoprostaglandin E1, 8,9-EET, and CMP. These findings suggested correlations between changes in the gut microbiota and the host metabolome, which were attenuated through NPS 2143 treatment in rats with PAH.
Figure 5. Relationship between gut microbiome and host metabolome. Heat maps indicated positive (red) and negative (blue) correlations between the levels of host metabolites and the identified gut microbiome at the genus levels of NPS2143-treated PAH rats as compared with PAH rats. The legend shows correlation values from −1 to 1 and assigns the appropriate color to them (Red for positive correlations and blue for negative correlations)
Discussion
There is a considerably large number of species of microorganisms, including bacteria and fungi, in the gastrointestinal tract. Species of microorganisms in the gastrointestinal tract have been widely studied regarding their association with human health [Citation23–25]. The role of gut dysbiosis in the pathogenesis of many diseases is rapidly emerging. In this study, we used multiple omics approaches to study the correlation between the gut microbiota and the host metabolome in rats with PAH and NPS 2143 treatment.
In this study, the gut microbiota was compared between rats with PAH treated with NPS 2143 and control rats. We found a significantly lower α-diversity in the PAH group compared with the control group, which indicated a ‘poor’ health status. However, the α-diversity was restored with NPS 2143 treatment in rats with PAH ()).
In all three studied groups, we examined the composition of the bacterial gut microbiome in the three groups () and (c)). Bacterial composition at the phylum, class, and genus levels was significantly altered by NPS 2143 exposure in rats with PAH. This finding suggests that dysbiosis of the gut microbiota is associated with PAH, which provides new evidence of how the gut microbiome affects the respiratory system. A decrease in potential pathogens and restoration of the normal gut flora by NPS 2143 exposure might alleviate lung injury and improve the host’s metabolism.
In our study, we also showed that changes in the host microbiome reflected alterations in gene expression induced by MCT-treated rats with PAH. MCT exposure also increased the pathogenicity of the gut bacteria in this study () and (b)). Genes involved in the regulation of metabolism, genetic information processing, environmental information processing, cellular processes, human diseases, and organismal systems were significantly altered in MCT-treated rats with PAH. NPS 2143 partly reversed this change.
The intestinal microbiome is considered as an additional metabolic organ for the host, and its metabolites can influence the host’s health [Citation26]. Along with changes in the gut microbiome, a disrupted metabolome was also observed in NPS 2143-treated rats with PAH in our study. In the PAH group, 34 and 2 metabolites were up- and downregulated, respectively, compared with the control group ()). Levels of epsilon-caprolactam, (2 R,3 R)-3-methylornithinyl-N6-lysine, 3-dehydro-2-deoxyecdysone, 12-hydroxydodecanoic acid, citalopram, cyclopentolate, diaminopimelic acid, dibutyl phthalate, suberic acid, hydroxypropionic acid, 3-hydroxy-5-methyl-L-tyrosine, 6-ketoprostaglandin E1, tyrosine methyl ester, d-xylitol, 8,9-EET, isoproterenol, tetracaine, pentylenetetrazole, N,N-dimethyl-1,4-phenylenediamine, N2-succinyl-L-arginine, oxoglutaric acid, 2-methoxyestrone, fisetin, tyramine, yohimbine, rivastigmine, pyridoxamine, phthalic acid, phloretin, quercetin, sebacic acid, phenylacetaldehyde, CMP, and cholesterol sulfate were higher in the PAH group than in the control group. In contrast, pantothenic acid and ornithine levels were significantly lower in the PAH group than in the control group. Changes to these metabolites were significantly abrogated in NPS 2143-treated rats with PAH. A number of previously found important functional metabolites were altered in the fecal samples of MCT-treated rats with PAH, but NPS 2143 abrogated these changes. An increasing number of reports have shown that the gut microbiota is involved in various pathways, including signaling, which is associated with the occurrence and development of PAH. In this study, we provided further insightful analysis on pathways, specifically in consideration of the potential changes and functional roles. We identified 22 pathways ()). These pathways were the hedgehog signaling pathway, vasopressin-regulated water, the longevity-regulating pathway, longevity regulating pathway-multiple species, dilated cardiomyopathy, circadian rhythm, human papillomavirus infection, aldosterone, D-arginine and D-ornithine metabolism, oocyte meiosis, the insulin signaling pathway, progesterone-mediated oocyte maturation, growth hormone synthesis, leukocyte transendothelial migration, secretion, and action, prostate cancer, the MAPK signaling pathway, prion disease, R human T-cell leukemia virus 1 infection, the chemokine signaling pathway, the peroxisome proliferator-activated receptor signaling pathway, the AP1 signaling pathway, and morphine addiction (). The hedgehog signaling pathway was the most affected ()).
In recent years, an increasing number of studies has suggested that the gut microbiota is capable of influencing host metabolism, which eventually leads to changes in the pathogenesis of respiratory diseases [Citation27]. In the current study, the correlation between the microbial composition of the gut microbiota and the metabolome was investigated to determine the mechanisms involved in NPS 2143-treated PAH rats. Importantly, we found the following correlations (). The genus Allobaculum was negatively correlated with changes in pyridoxamine, sebacic acid, (2 R,3 R)-3-methylornithinyl-N6-lysine, cyclopentolate, and dibutyl phthalate. Bifidobacterium was negatively correlated with changes in isoproterenol and phenylacetaldehyde. Lactobacillus was positively correlated with changes in hydroxypropionic acid, 12-hydroxydodecanoic acid, CMP, and cyclopentolate Romboutsia was positively correlated with changes in phenylacetaldehyde and pentylenetetrazole. Turicibacter and Clostridium_sensu_stricto_1 were positively correlated with changes in quercetin. Candidatus_Saccharimonas was positively correlated with changes in pyridoxamine, N,N-dimethyl-1,4-phenylenediamine, rivastigmine, 6-ketoprostaglandin E1, 8,9-EET, and CMP. These results indicate that alterations in the microbial composition from the gut microbiota could eventually lead to changes in certain metabolites by NPS 2143 treatment. Additionally, such influence looks more like in a casual way than specifically.
Conclusion
Our study shows correlations between changes in the gut microbiome and the metabolome in PAH, which are affected by NPS 2143. NPS 2143 treatment attenuates changes in the gut microbiota and subsequent effects on the host metabolome in rats with PAH. The current study may provide important evidence and better understanding of molecular mechanisms, and help in development of novel drug targets and therapeutic treatments for PAH.
Highlights
The gut microbiome is altered in rats with PAH.
The fecal metabolome is altered in rats with PAH.
Correlation in alteration of gut microbiota and the host metabolome through NPS2143 treatment in PAH rats.
Acknowledgements
Rats in this study were obtained from the Experimental Animal Center of Guangdong (Guangdong, China). Approval of animal care and experiments was issued with the Animal Care and Use Committee of Guangzhou Medical University (Approval No. GY2019-064) and in accordance with the principles and guidelines of the National Institutes of Health.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Thenappan T, Khoruts A, Chen Y, et al. Can intestinal microbiota and circulating microbial products contribute to pulmonary arterial hypertension? Am J Physiol Heart Circ Physiol. 2019;317:H1093–H1101.
- Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1):1801887.
- Smith KA, Ayon RJ, Tang H, et al. Calcium-sensing receptor regulates cytosolic [Ca (2+)] and plays a major role in the development of pulmonary hypertension. Front Physiol. 2016;7:517.
- Tang H, Yamamura A, Yamamura H, et al. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;310:L846–859.
- Yamamura A, Guo Q, Yamamura H, et al. Enhanced Ca(2+)-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ Res. 2012;111:469–481.
- Dumas A, Bernard L, Poquet Y, et al. The role of the lung microbiota and the gut-lung axis in respiratory infectious diseases. Cell Microbiol. 2018;20:e12966.
- Vaughan A, Frazer ZA, Hansbro PM, et al. COPD and the gut-lung axis: the therapeutic potential of fibre. J Thorac Dis. 2019;11:S2173–S2180.
- Wedgwood S, Warford C, Agvatisiri SR, et al. The developing gut-lung axis: postnatal growth restriction, intestinal dysbiosis, and pulmonary hypertension in a rodent model. Pediatr Res. 2020;87:472–479.
- Nagpal R, Yadav H. Bacterial translocation from the gut to the distant organs: an overview. Ann Nutr Metab. 2017;71(Suppl 1):11–16.
- Takiishi T, Fenero CIM, Camara NOS. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers. 2017;5:e1373208.
- Pascale A, Marchesi N, Marelli C, et al. Microbiota and metabolic diseases. Endocrine. 2018;61:357–371.
- Callejo M, Mondejar-Parreno G, Barreira B, et al. Pulmonary arterial hypertension affects the rat gut microbiome. Sci Rep. 2018;8:9681.
- Sanada TJ, Hosomi K, Shoji H, et al. Gut microbiota modification suppresses the development of pulmonary arterial hypertension in an SU5416/hypoxia rat model. Pulm Circ. 2020;10:2045894020929147.
- Kim S, Rigatto K, Gazzana MB, et al. Altered gut microbiome profile in patients with pulmonary arterial hypertension. Hypertension. 2020;75:1063–1071.
- Zhao YY, Lin RC. UPLC-MS(E) application in disease biomarker discovery: the discoveries in proteomics to metabolomics. Chem Biol Interact. 2014;215:7–16.
- Su G, Wang H, Bai J, et al. A metabonomics approach to drug toxicology in liver disease and its application in traditional Chinese medicine. Curr Drug Metab. 2019;20:292–300.
- Niu JL, Zhang J, Wei LQ, et al. Effect of fermented cottonseed meal on the lipid-related indices and serum metabolic profiles in broiler chickens. Animals (Basel). 2019;9 (11):930.
- Li H, He Z, Gao D, et al. Characteristics of the intestinal microbiota in very low birth weight infants with extrauterine growth restriction. Front Pediatr. 2019;7:99.
- Niu J, Zhang J, Wei L, et al. Cottonseed meal fermented by candida tropical reduces the fat deposition in white-feather broilers through cecum bacteria-host metabolic cross-talk. Appl Microbiol Biotechnol. 2020;104:4345–4357.
- Duan Y, Xiong D, Wang Y, et al. Toxic effects of ammonia and thermal stress on the intestinal microbiota and transcriptomic and metabolomic responses of litopenaeus vannamei. Sci Total Environ. 2021;754:141867.
- Yang J, Yang Y, Ishii M, et al. Does the gut microbiota modulate host physiology through polymicrobial biofilms? Microbes Environ. 2020;35 (3):ME20037.
- Zhang Y, Wang Y, Ke B, et al. TMAO: how gut microbiota contributes to heart failure. Transl Res. 2021;228:109–125.
- Das B, Nair GB. Homeostasis and dysbiosis of the gut microbiome in health and disease. J Biosci. 2019;44(5):117.
- Durack J, Lynch SV. The gut microbiome: relationships with disease and opportunities for therapy. J Exp Med. 2019;216:20–40.
- Quaranta G, Sanguinetti M, Masucci L. Fecal microbiota transplantation: a potential tool for treatment of human female reproductive tract diseases. Front Immunol. 2019;10:2653.
- Chung HJ, Sim JH, Min TS, et al. Metabolomics and lipidomics approaches in the science of probiotics: a review. J Med Food. 2018;21:1086–1095.
- Budden KF, Gellatly SL, Wood DL, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15:55–63.