
ABSTRACT
Myocardial hypertrophy is a pathological thickening of the myocardium, leading to various ailments, such as myocardial infarction and heart failure. RBM38 is critical in modulating mRNA translation for multiple protective activities such as p53 tumor repressor and p21 kinase cell cycle inhibitors. Liver X receptors (LXR-α) agonists reduce cellular hypertrophy initiated by various hypertrophic stimuli as lipopolysaccharides and Ang II. This research investigates the possible cooperation between RBM38 and LXR-α and mechanisms in modulating myocardial hypertrophy. H9C2 cells were treated with PE, TNF-α, and AngII to induce myocardial hypertrophy. RBM38 and LXR- α were overexpressed or silenced in H9C2 cells, and hypertrophy markers (ANF and Myh7) were determined with Western blot and RT-qPCR. Binding assays were done through RNA immunoprecipitation. H&E and Rhodamine-labeled phalloidin staining assays were used to assess the relative cell surface change. The results demonstrated RBM38 downregulation in in vitro models of myocardial hypertrophy. Modulation of RBM38 expression also exerted inverse effects on myocardial hypertrophy markers. Further observations also showed that LXR-α expression regulates the myocardial hypertrophy markers in H9C2 cells and RBM38 binds with LXR-α mRNA, consequently inhibiting LXR-α expression. Finally, overexpression of RBM38 rescues Angiotensin II–induced myocardial hypertrophy by regulating LXR-α dependent lipogenesis pathway. In conclusion, RBM38 Overexpression rescues Angiotensin II–induced myocardial hypertrophy by regulating LXR-α dependent lipogenesis pathway.
1. Introduction
Myocardial hypertrophy is a pathological thickening of the myocardium, leading to a reduction of the heart chamber. The disease is often caused by stenosis of the heart valve or high blood pressure, and it is independently a risk factor for various cardiovascular ailments, such as myocardial infarction, heart failure, arrhythmia, and abrupt death[Citation1]. Heart failure and arrhythmia often lead to fibrosis, which is an advanced pathological demonstration of various diseases [Citation2].
At the cellular level, myocardial hypertrophy is characterized by cytoskeletal reorganization and overgrown cardiomyocytes. The disease further demonstrates elevated fetal-type gene expression at the molecular level [Citation3,Citation4]. Mechanically, myocardial hypertrophy is initially an adaptive heart remodeling for rising cardiac load, encompassing intense hemodynamic afterload due to hypertension5[Citation5]. Working under an increased load to counteract the effect, myocardial cell volume, heart weight, left ventricle, and ventricular septum thickness increase in size [Citation6]. However, following intense stress and prolonged work-load, hypertrophy can minimize ventricle function, heart failure, and eventual death.
RNA-binding proteins (RBPs) are critical modulators of several cellular pathways. RBPs precisely regulate numerous RNA functions such as stability, activity, and biogenesis [Citation7]. During the regulation of post-transcriptional gene expression, RBPs control various metabolic steps and non-coding RNA translation [Citation8]. MicroRNAs (miRNAs) are small, non-coding RNAs that cooperate with RBPs in the regulation process. Examples of critical molecules in the RBP families include muscleblind-like 1 (MBNL1 and MBNL2), CUGBP, ELAV-like family 1 (CEFL1), RBM15, RBM20, RBM24, and RBM38.
Of critical interest are RBM38 (RNPC1), which has been confirmed to modulate the mRNA translation of tumor repressor p53, p21 kinase inhibitor of the cell cycle, HuR RNA binding protein, p53 family member genes p73, and p63, pro-apoptotic transcripts MIC-1 and MDM2 regulator of p53 [Citation9]. Besides, RBM38 has been critical in regulating endothelial cells’ response to vascular injuries [Citation10,Citation11]. Studies have also elucidated RBM38 role in cardiac remodeling [Citation12]. However, the possible pathway involved during cardiac remodeling is yet to be studied. Lack of knowledge of the involved mechanisms thus creates various challenges in the target and treatment of myocardial hypertrophy. Consequently, recognizing precise pleiotropic targets integrated through various organ systems may shed light on efficacious therapeutic modalities in reversing or curbing heart failure.
Liver X receptors (LXR) α and β are proteins of nuclear receptor ligand-activated transcription factors superfamily. LXR induces transcription of target genes via retinoid X receptor (RXR) heterodimerization and interaction with LXR response element (LXRE). LXRs, at the molecular level, acts as ligand-activated transcription factors essential for the regulation and coordination of cholesterol homeostasis and inflammatory signaling. In myocardial hypertrophic remodeling, LXR agonists have proven to reduce cellular hypertrophy initiated by various hypertrophic stimuli as lipopolysaccharides and Angiotensin II (Ang II) [Citation13]. The possibility of targeting and integrating RBM38 and LXR-α as a therapeutic approach against cardiac remodeling has never been investigated. Herein, we provide evidence to support that RBM38 is a potential modulator of myocardial hypertrophy via the LXR-α-Dependent lipogenesis pathway.
The study hypothesized that RNA Binding Motif Protein-38 could regulate myocardial hypertrophy in the LXR-α-Dependent lipogenesis pathway. The objective of this investigation was to understand the role of RNA Binding Motif Protein-38 in myocardial hypertrophy. The specific goals were to determine whether RBM38 is downregulated in myocardial hypertrophy in vitro, to understand the effects of Modulating RBM38 expression on myocardial hypertrophy markers, and to determine the effects of LXR-α expression on myocardial hypertrophy markers in H9C2 cells. Therefore, the overall final goal was to investigate the LXRα/SREBP/ChREBP pathway used by RBM38 in myocardial hypertrophy.
2. Materials and methods
2.1. Cell culture
The study was carried out using adherent H9C2 cell lines of rat embryonic cardiomyocytes. The cells were grown in culture plates in Dulbecco’s modified Eagle’s medium DMEM, supplemented with 10% fetal bovine serum,100 U/ml penicillin, and 100 µg/ml streptomycin. The growth and maintenance condition was at 37°C, and 5% CO2. Cell splitting was done at the exponential phase when 70–80% confluence had been achieved. The cells were rinsed in PBS, digested with 0.25% trypsin and EDTA solution, and centrifuged at 1000 rpm for 5 minutes. Cardiomyocytes were subsequently inoculated into new culture plates.
2.2. PE, TNF-α, and Ang II stimulation
H9C2 cells at a confluence of 70% were rinsed with PBS Later, 2 ml DMEM supplemented with 10% FBS was added and then treated with Phenylephrine (PE) (100 µM), Ang II (1 µM), or TNF-α (50 U/ml) for 48 hrs under the tissue culture conditions. The control groups were only incubated in the complete DMEM. Cells were then harvested by trypsinization and washed using cold PBS.
2.3. Plasmid and siRNA transfection
H9C2 cells were seeded in 6-well plates for 24 hours before transfection. The cells were then transfected using RBM-OE, si-RBM38, LXR-α, or si-LXR-α. For the RBM38- Overexpression (OE), the RBM38-OE plasmid was constructed by cloning the full-length coding sequence of RBM38 into the pcDNA3.1 plasmid (Invitrogen). The oligonucleotide sequences used were purchased from Genepharma (Shanghai). Transfection was done with Lipofectamine 2000TM (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s guidelines [Citation14].
2.4. Knockdown and over-expression
For the RBM38 and LXR-a gene knockdown assays with siRNA transfections, Lipofectamine RNAiMAX (Invitrogen) was used following the manufacturer’s guidelines [Citation15]. Transfection with RBM38 siRNA, LXR-a siRNA, or nonspecific siRNA (Stealth RNAi; Invitrogen) was done after H9C2 cells had reached a confluence of 100%. The medium was then changed to allow for cell differentiation. For the over-expression assays, transfection was done a day before the H9C2 cells had a 100% confluence to increase transfection efficiency. Lipofectamine 2000 (Invitrogen) was used for the transfection as per the manufacturer’s guidelines. Growing H9C2 cells (70%–80% confluent) were transfected with pcDNA3.1-RBM38 or the control plasmid and grown for one more day to obtain 100% confluence. The cells were later differentiated by the addition of new medium. For the rescue assays, plasmid transfection was carried out a day before siRNA transfection and induction of differentiation.
2.5. RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total cellular RNA was isolated from cultured H9C2 cells using Trizol (Takara, Beijing). Extracted RNA was then reverse-transcribed to synthesize cDNA using a PrimeScript II 1st Strand cDNA Synthesis kit (TakaRa, Japan) according to the manufacturers’ guidelines. SYBR green was used as Master Mix. RT-qPCR was then carried out using the SYBR Premix Ex TaqTM II Kit (TakaRa) in a LightCycler480 SW 1.51 System from LightCycler480 II. β-actin was used as the internal control for mRNA detection. The relative expression levels of mRNA were calculated using the 2−DDCT method [Citation16]. The primer sequences are shown in .
Table 1. List of primers
2.6. Protein extraction and Western blot
Total cellular protein was isolated from H9C2 cells using a RIPA lysis buffer (50mM Tris-HCl pH8, 150 mM NaCl, 1% NP-40, 0.2% sodium deoxycholate, 0.1% SDS, 1mMNa3VO4, 1mMPMSF) supplemented with protease inhibitor cocktail (Roche). Sonication of the cells was then done on a Bioruptor (TMUCD-200 (Diagenode) at a low speed for 15 minutes. Cells were then centrifuged at 4°C, 15,000 g for 20 minutes, and supernatants were finally harvested. A Bradford assay was subsequently used for protein concentrations assessment. For electrophoresis, the protein sample lysates (40 µg) were next loaded on 10% (w/v) Tris-HCl sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels, ran on a 100 V power, and transferred to polyvinylidene fluoride (PVDF) membrane (Millipore). To carry out Western blot assays, the PVDF membrane was blocked in 5% skimmed milk powder for 1 hr and then incubated with RBM38 (sc-365,898; Santa Cruz), Myh7(sc-53,090; Santa Cruz), ANF (sc-515,701; Santa Cruz), LXR-α (sc-377,260; Santa Cruz), β-actin rabbit monoclonal antibodies (Cell signaling) at 4°C overnight. The usage of a housekeeper gene, β-actin, was ensured as an internal control. The following day, the membranes were then washed three times with phosphate-buffered saline-Tween 20 (PBS-T) and then further incubated with anti-mouse or anti-rabbit peroxidase-conjugated secondary antibody (Cell Signaling Technology). The bands, indicating Immunological complexes, were finally visualized using ImmobilonTM Western Chemiluminescent HRP substrate (Millipore). Three independent assays were performed and data analyzed using ImageJ software and graph pad prism. Protein expression was normalized to levels of β-actin expression.
2.7. RNA immunoprecipitation (RIP)
RNA immunoprecipitation (RIP) assay RIP was done [Citation17]. Briefly, approximately 2 × 107 H9C2 cells were lysed with RNA immunoprecipitation lysis buffer (Millipore, USA) and then incubated with 5 μg of rabbit polyclonal anti-RBM38 or non-immunized rabbit IgG at 4°C overnight. The RNA-protein immune-complexes were brought down by protein A/G magnetic beads and finally followed by RNA purification. The purified RNA was subsequently subjected to RT-qPCR to measure the level of LXR-α.
2.8. Hematoxylin and Eosin (H&E) Staining
H&E staining was performed as previously described4. Briefly, adhered H9C2 cells on microscope slides were fixed on alcohol and hydrated by immersion in water for 30 seconds. The slides were then dipped into a jar containing Mayer’s hematoxylin and agitated for 30 sec, and rinsed in water for 1 min. The slides were subsequently stained with 1% eosin solution for 30 sec with agitation, dehydrated with two changes of 95% alcohol, and two changes of 100% alcohol for 30 sec each. The alcohol was later extracted with two changes of xylene. Finally, two drops of glycerol mounting medium were added and covered using a coverslip and observed under a microscope.
2.9. Rhodamine-labeled phalloidin staining
Rhodamine-labeled phalloidin staining was done as previously described [Citation18]. Briefly, H9C2 cells fixation and permeabilization were done using 4% paraformaldehyde and 0.5% Triton X-100, respectively. Permebilized cells were then blocked for 30 min using 5% BSA at room temperature. Cells were later incubated overnight with a Myh7 (sc-53,090; Santa Cruz), ANF (sc-515,701; Santa Cruz), LXR-α (sc-377,260; Santa Cruz), and Ang II (MA1-82,996; ThermoFisher) antibodies (1:100) at 4°C. The following day, cells’ washing was done thrice and incubated in the dark at 37°C using fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibody for 1 h. The cells were subsequently washed in PBS, then incubated for 30 minutes with 0.5 μM rhodamine-labeled phalloidin and 10 minutes with 0.5 μM DAPI. Cells were finally examined and photographed using Leica DMI3000B fluorescence microscope (Wetzlar, Germany).
2.10. Statistical analysis
Data were represented as mean ± SEM. All data were statistically analyzed by a paired t-test and one-way ANOVA, which was used to obtain statistical differences. P < 0.01 was regarded as statistically significant results.
3. Results
3.1. RBM38 is downregulated in in-vitro models of myocardial hypertrophy
Myocardial hypertrophy is a pathological thickening of the myocardium, leading to various ailments, such as myocardial infarction and heart failure. PE, TNF-α, and AngII are proteins that may contribute to myocardial hypertrophy development and maintenance in various ways, for instance, directly via its trophic effects on the cardiac and indirectly through the hemodynamic effects. This work hypothesized that RBM38 is a potential modulator of myocardial hypertrophy via the LXR-α-Dependent lipogenesis pathway.
To assess the RMB38 expression in myocardial hypertrophy in vitro, we treated H9C2 cells with PE, TNF-α, and Ang II, respectively. The ANF and myh7 proteins expressions were then determined using western blot. Our observations indicated significantly increased expressions of these proteins in the PE, TNF-α, and AngII treated cells compared to the control group (). We then assessed the mRNA expression of ANF and Myh7 through RT-qPCR assays. As shown in , treatment of cells with PE, TNFα, and Ang II significantly increased Myh7 mRNA expression compared to the control group.
Figure 1. RBM38 is downregulated in in-vitro models of myocardial hypertrophy
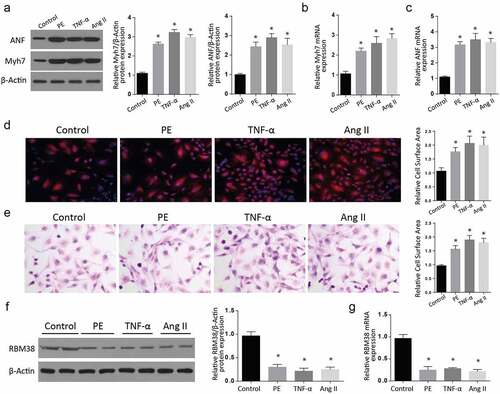
Similarly, assessment of ANF mRNA showed significantly increased mRNA expressions in the treatment of cells with PE, TNFα, and Ang II, compared to the control group (). Further, H&E staining and Rhodamine-labeled phalloidin staining assays showed a significant relative cell surface area increase in PE, TNF-α, and Ang II-treated cells compared to the control cells (). Next, we analyzed the RBM38 protein expression in control, PE-, TNF-α, and the Ang II-treated group of cells through western blot. As shown in , the expression of RBM38 was significantly downregulated following the PE-, TNF-α and Ang II treatment as compared to the control group. We finally determined the RBM38 mRNA expression in the same treatment groups, and in agreement with the western blot results, RBM38 mRNA levels were significantly reduced in both the PE, TNF-α and Ang II treatment, compared to the untreated cells (). Taken together, these observations confirm that RBM38 is downregulated in myocardial hypertrophy in vitro.
3.2. Modulating RBM38 expression exerts inverse effects on myocardial hypertrophy markers
We then aimed at studying the impact of RMB38 modulation on myocardial hypertrophy markers. We transfected H9C2 cells with RBM38 in the pcDNA-3.1 plasmid. An empty plasmid without RBM38 was used as a negative control. Following a successful transfection, we analyzed for RBM38 expression in the study and control group using western blot assay. As shown in , RMB38 expression was significantly increased in the RBM38-OE group compared to the pcDNA3.1 group. RT-qPCR studies also showed significant elevation of RBM38 mRNA (), ANF mRNA (), and Myh7 mRNA () in the RBM38-OE compared to the control groups.
Figure 2. Modulating RBM38 expression exerts inverse effects on myocardial hypertrophy markers
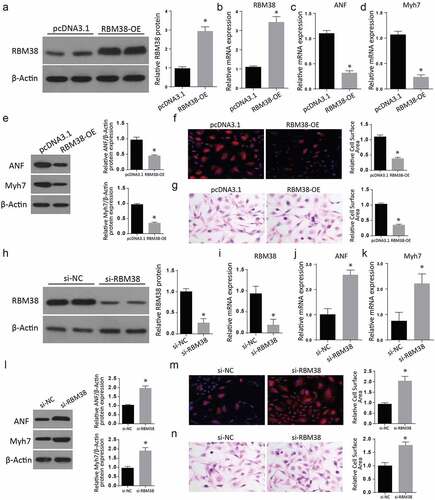
We further analyzed the expression of myocardial hypertrophy markers ANF and Myh7 in H9C2 cells transfected with RBM38-OE plasmid through western blot. Our findings demonstrated a significant decrease of ANF and Myh7 proteins in the RBM-OE than in the control cells (). Further, H&E staining and Rhodamine-labeled phalloidin staining assays showed a significantly reduced relative cell surface area in RBM38-OE than in the pcDNA3.1 cells ().
Later, the RBM38 protein in RBM38-OE cells was silenced through small interfering RNA (siRNA). The western blot assays result demonstrated a significantly reduced RBM38 expression in the si-RBM38 than in the si-NC group (2 H and 2I). Later, we carried out RT-qPCR assays that showed a significantly reduced RBM38 mRNA expression (2 J), but significantly increased ANF and Myh7 mRNA expression expressions () in the si-RBM38 compared to the si-NC groups. Western blot assays demonstrated significantly increased ANF and Myh7 proteins expressions in si-RBM38 cells compared to the si-NC group (). Further, H&E staining and Rhodamine-labeled phalloidin staining assays showed a significant increase in the relative cell surface area in si-RBM38 than in the si-NC cells (). These observations all confirm that regulation of RBM38 has inverse effects on myocardial hypertrophy markers.
3.3. LXR-α expression regulates the myocardial hypertrophy markers in H9C2 cells
To understand the role played by Liver X receptor-α (LXR-α) in myocardial hypertrophy, we treated H9C2 cells with PE, TNF-α, and Ang II. We then used western blot assays to study the expression of LXR-α. Compared to the control group, the western blot results showed a significantly increased LXR-α protein in the various treatments (). The quantification of LXR-α confirmed a significant increase in all the cells after the treatments with PE, TNF-α, or Ang II compared to the control group. Further RT-qPCR investigation also confirmed a significantly elevated LXR-α mRNA expression in the treated cells compared to the control group. (). Next, we transfected H9C2 cells with si-LXR-α and then lysed proteins for western blot assays. Our findings demonstrated a significant reduction of LXR-α protein expression in the si-LXR-α cells compared to the si-NC group (). The quantification of the LXR-α protein confirmed a significantly increased expression in the si-LXR-α group. Further, the LXR-α mRNA expression assessment through RT-qPCR also indicated a significantly down-regulated LXR-α mRNA expression () but significantly increased ANF mRNA expression () and Myh7 mRNA expression () in Si-LXR-α cells as compared to the si-NC groups.
Figure 3. LXRα expression regulates the myocardial hypertrophy markers in H9C2 cells
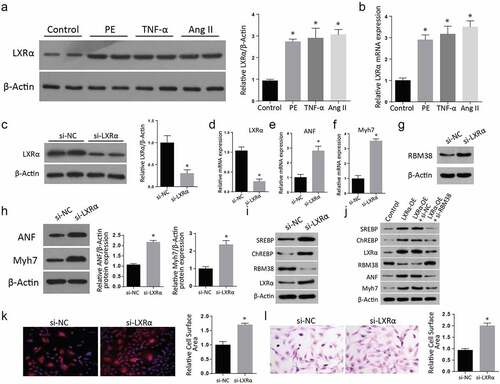
We later determined the RBM38 protein level in si-LXR- α cells through western blot, where a significantly increased expression was reported compared to the si-NC (3 G). Similarly, the expressions of myocardial hypertrophy markers ANF and Myh7 protein expression were significantly increased in the Si-LXR-α as compared to the Si-NC cells (). Further western blot analysis of proteins demonstrated a remarkable increment of SREBP, ChREBP, and LXR- α, but a drop in the RBM38 protein level in the si-LXR- α cells compared to the si-NC (). Later, the cells were transfected with LXRα-OE, LXRα-OE+si-NC, LXRα-OE+ si-RBM38, and proteins expression was analyzed. According to the outcomes, SREBP, ChREBP, LXR- α, RBM38, ANF, and Myh7 levels were significantly elevated in the LXR-α -OE, LXR-α-OE+si-NC and LXRα-OE+ si-RBM38 cells compared to the control group (). Further, H&E staining () and Rhodamine-labeled phalloidin staining () assays showed a significant relative cell surface area increase in si-LXR-α as compared to the si-NC. Summarily, these observations demonstrated that myocardial hypertrophy markers are regulated by the expression of LXR-α in H9C2 cells
3.4. RBM38 binds with LXR-α mRNA to inhibit its expression
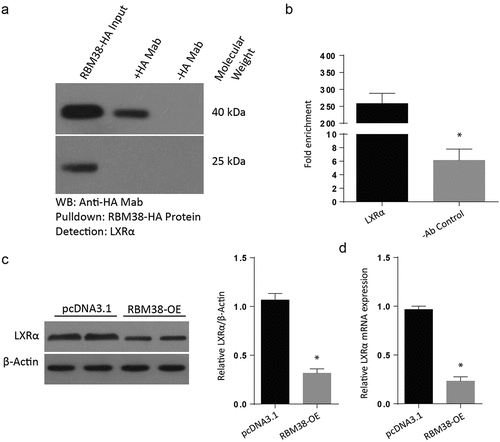
Next, we examined the mechanism through which LXR-α regulates myocardial hypertrophy. RNA immunoprecipitation experiment followed by RT-PCR and RT-qPCR were undertaken to determine whether RBM38 could bind to LXR-α mRNA directly in H9C2 cells. It was noted that LXR-α mRNA transcript was present in RBM38 but not in the control Immunoglobulin G immune-complexes (). From the H9C2 cells transfected with RBM38OE plasmid and its negative counterpart, we assessed for the relative LXR-α protein expression through western blot and observed a lack of band in the RBM38OE cells compared to the pcDNA3.1, which demonstrated a visible protein band (). Finally, we determined the relative mRNA expression of LXR-α in the cells transfected with RBM38OE plasmid. The western blot results indicated a significantly reduced LXR-α protein expression in the RBM-OE compared to the pcDNA3.1 group, as shown in . The RT-qPCR assessment also confirmed significantly reduced mRNA expressions in the RBM38-OE group compared to the pcDNA3.1 group (). These observations demonstrate that RBM38 binds with LXR-α mRNA to suppress its expression.
Figure 4. RBM38 binds with LXRα mRNA to inhibit its expression
3.5 Overexpression of RBM38 rescues Angiotensin II-induced myocardial hypertrophy by regulating LXR-α dependent lipogenesis pathway
Finally, we intended to understand the pathway utilized by RBM38 in regulating myocardial hypertrophy induced by Angiotensin II. An assessment of lipogenic transcription factor carbohydrate response element-binding protein (ChREBP) and sterol regulatory element-binding proteins (SREBPs) was done through western blot. Results indicated significantly reduced levels of SREBPs, ChREBP, RBM38, and LXR-α proteins in RBM38-OE compared to the pcDNA3.1 group (). Further, H&E staining and Rhodamine-labeled phalloidin staining assays revealed a significant size reduction in RBM38-OE+ Ang II compared to the Ang II group, as demonstrated in , respectively).
Figure 5. Overexpression of RBM38 rescues Angiotensin II–induced myocardial hypertrophy by regulating LXRα dependent lipogenesis pathway
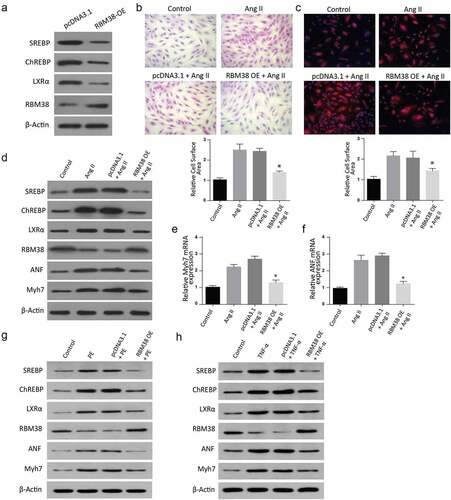
Further, cells were transfected with AngII, pcDNA3.1+ AngII, or RBM38-OE + Ang II. Assessment of SREBPs, ChREBP, LXR-a, RBM38, ANF, and Myh7 expressions was then done through western blot. The findings indicated a significantly increased LXR-α, SREBP, ChREBP, ANF, and Myh7 expression but reduced RBM38 in the AngII and pcDNA3.1+ AngII group compared to the control cells. However, LXR-α, SREBP, and ChREBP expressions were suppressed in RBM38-OE+ Ang II compared to the controls, as indicated in . Further analysis of myocardial hypertrophy ANF and Myh7 mRNA markers through RT-qPCR showed a significantly increased Myh7 mRNA expression () and ANF mRNA expression () in the AngII, pcDNA3.1+ AngII, and RBM38-OE+Ang II compared to the control group. Similarly, the cells were co-trasfected with PE, pcDNA3.1+ PE, or RBM38-OE+PE, and proteins expression was determined through western blot. The findings indicated a significant increase in SREBP, ChREBP, LXR-α, ANF, and myh7 expressions in the PE and pcDNA3.1+ PE transfected cells, which were all significantly down-regulated in the RBM38-OE+PE group. Finally, the cells were transfected with TNF- α, pcDNA3.1+ TNF- α, or RBM38-OE+TNF- α, and proteins expression was analyzed through western blot assays. According to the observations, SREBP, ChREBP, LXR-a, ANF, and myh7 expressions were significantly increased in the TNF- α and pcDNA3.1+ TNF- α transfected cells as compared to the control group. However, in the RBM38OE+TNF- α group, SREBP was significantly reduced while the other proteins were increased compared to the control. In summary, these observations demonstrate that RBM38 Overexpression rescues Angiotensin II–induced myocardial hypertrophy via regulating LXR-α dependent lipogenesis pathway
4. Discussion
In the present study, the previously unrecognized biological role of RBM38 in myocardial hypertrophy is revealed. The main findings of this study include the following; RBM38 is suppressed in myocardial hypertrophy conditions in vitro, and RBM38 expression modulation exerts inverse effects on myocardial hypertrophy markers. Further, LXR-α expression regulates the myocardial hypertrophy markers in cardiomyocytes, RBM38 binds with LXR-α mRNA to inhibit its expression, and overexpression of RBM38 rescues Angiotensin II–induced myocardial hypertrophy by regulating LXR-α dependent lipogenesis pathway.
Tumor necrosis factor- α (TNF- α), PE, and Ang II have roles in inducing myocardial hypertrophy. PE induces myocardial hypertrophy by downregulating Cain/Cabin or AKAP79 molecules associated with calcineurin catalytic subunit [Citation19]. Ang II initiates myocardial hypertrophy by regulating pathways such as mitogen-activated protein kinases (MAPK) and Reactive oxygen species (ROS). TNF-α also induces myocardial hypertrophy through the initiation of ROS production [Citation20].
RBM38 is a critical RBM24 homolog sharing multiple targets with RBM24, such as regulating p63 and p21 mRNA stability [Citation9,Citation21–23]. RBM38 occurs in various cells and tissues, even though its exact expression pattern has never been elucidated since it is enriched in the erythrocytes or the skeletal and heart muscles [Citation24]. Much research has shown the various essential roles of RBM38. For instance, it has been mentioned as a regulator of Fgfr2 splicing in epithelial cells [Citation25]; it regulates RNA processes, including splicing, translation, stability, and the accessibility of microRNA to the specific mRNA target [Citation26].
RBM38 has also been identified as an essential cell maturation regulator in the erythrocytes, where it arbitrates alternative splicing of regulators of blood cell maturation as Mef2d, EPB41, and CYB5A [Citation10]. Indeed, RBM38 deficiency in mice has been associated with susceptibility to hematopoietic malfunctions, spontaneous tumors, and accelerated aging [Citation27].
Further, RBM38 has been linked with the p53 pathway and shown as a necessary protein for proper p53 functioning [Citation28]. Consequently, its knock-out in mice has shown an elevation of p53 in various cells and tissues such as embryonic fibroblasts, thymus, and spleen, which contributed to their enhanced penetrance to tumors [Citation27]. RBM38 binds to various genes such as p53, p21, GDF15, mdm2, HuR, and c-myc through their 3ʹUTR AU-rich elements [Citation29]. RBM38 has also been reported as a critical splice factor of the cardiac, which plays a role in governing heart sarcomerogenesis by regulating the alternative protein isorforms expressions [Citation12].
The role of LXR-α in myocardial hypertrophy regulation has been widely identified. In vitro, LXR agonists have been linked with reduced cellular hypertrophy initiated by various hypertrophic stimuli such as phenylephrine and endothelin-130-[Citation30]. On the other hand, LXR-α knockdown in cardiomyocytes has been associated with increased cellular growth [Citation31]. A significant increase of LXR-α protein has also been observed in the pressure overloaded myocardium [Citation13]. Indeed the findings of this study further emphasize the importance of LXR-α in the modulation of myocardial hypertrophy. In the present study, increased LXR-α levels following induction of cells with LXR-α agonists such as PE, Ang II, and TNF-α was to regulate the development of myocardial hypertrophy.
The liver plays a core function in glucose homeostasis. Glucose stimulates insulin release by the pancreas and regulates ChREBP activity, a transcription factor critical for the modulation of lipogenesis [Citation32]. Glucose additionally binds and activates the liver X receptor (LXR) transcriptional activity, induces LXR at the expected physiological concentrations in the liver, and hastens LXR target gene expression. LXR-α, thus, controls ChREBP and sREBP1 genes, all specific for lipogenesis. LXR-α is essential in the regulation of hypertrophic cardiac remodeling. LXR-α agonists suppress cardiac hypertrophy induced by various stimuli, for instance, lipopolysaccharides, PE, and Ang II [Citation33].
In contrast, LXR-α knockdown leads to a raised cardiomyocytes growth. A significant LXR-α elevation is also reported in pressure overloaded myocardium [Citation34]. During myocardial hypertrophy, there is a shift of metabolism from fatty acid oxidation (FAO) to the utilization of glucose [Citation35]. The change is linked to increased glycolysis in a hypertrophied heart. Mechanistically, the free adenosine monophosphate (AMP) in cardiomyocyte get heightened during a heart pressure overload [Citation36]. Consequently, signaling is transduced through AMP-activated protein kinase, whose outcome is fibrosis and cardiac dysfunctions [Citation37].
5. Conclusion
In connection with our results, RBM38 is downregulated in in vitro models of myocardial hypertrophy, and modulation of its expression exerts inverse effects on myocardial hypertrophy markers. Further, LXR-α expression regulates the myocardial hypertrophy markers in H9C2 cells, and RBM38 binds with LXR-α mRNA, inhibiting LXR-α expression. Finally, the overexpression of RBM38 rescues Angiotensin II–induced myocardial hypertrophy by regulating LXR-α dependent lipogenesis pathway. Collectively, results from the present investigation shed light on an essential role of RNA Binding Motif Protein-38 in regulating myocardial hypertrophy via an LXR-α dependent lipogenesis pathway.
Disclosure statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Funding
References
- Han D, Gao Q, Cao F. Long non-coding RNAs (LncRNAs) - The dawning of a new treatment for cardiac hypertrophy and heart failure. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):2078–2084.
- Chothani S, Schäfer S, Adami E, et al. Widespread translational control of fibrosis in the human heart by RNA-Binding proteins. Circulation. 2019;140(11):937–951.
- Chen Y, Chang Y, Zhang N, et al. Atorvastatin attenuates myocardial hypertrophy in spontaneously hypertensive rats via the C/EBPβ/PGC-1α/UCP3 pathway. Cell Physiol Biochem. 2018;46(3):1009–1018.
- Tang L, Xie J, Yu X, et al. MiR-26a-5p inhibits GSK3β expression and promotes cardiac hypertrophy in vitro. PeerJ. 2020;8:e10371.
- Heinzel FR, Hohendanner F, Jin G, et al. Myocardial hypertrophy and its role in heart failure with preserved ejection fraction. Journal of applied physiology (Bethesda, Md.: 1985). 2015;119(10):1233–1242.
- Bernardo BC, Weeks KL, Pretorius L, et al. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227.
- Ladd AN. New insights into the role of RNA-Binding proteins in the regulation of heart development. Int Rev Cell Mol Biol. 2016;324:125–185.
- Felicetta A, Condorelli G. RNA binding protein and microRNA control of endothelial cell function. Cardiovasc Res. 2019;115(12):1690–1691.
- Jiang Y, Zhang M, Qian Y, et al. Rbm24, an RNA-binding protein and a target of p53, regulates p21 expression via mRNA stability. J Biol Chem. 2014;289(6):3164–3175.
- Heinicke LA, Nabet B, Shen S, et al. The RNA binding protein RBM38 (RNPC1) regulates splicing during late erythroid differentiation. PLoS One. 2013;8(10):e78031.
- Sonnenschein K, Fiedler J, Pfanne A, et al. Therapeutic modulation of RNA-binding protein Rbm38 facilitates re-endothelialization after arterial injury. Cardiovasc Res. 2019;115(12):1804–1810.
- van den Hoogenhof MMG, van der Made I, Beqqali A, et al. The RNA-binding protein Rbm38 is dispensable during pressure overload-induced cardiac remodeling in mice. PloS One. 2017;12(8):e0184093.
- Wu S, Yin R, Ernest R, et al. Liver X receptors are negative regulators of cardiac hypertrophy via suppressing NF-kappaB signalling. Cardiovasc Res. 2009;84(1):119–126.
- Shi X, Zhang B, Chu Z, et al. Wogonin inhibits cardiac hypertrophy by activating Nrf-2-Mediated antioxidant responses. Cardiovasc Ther. 2021;2021:9995342.
- Nguyen TTT, Ishida CT, Shang E, et al. Activation of LXR receptors and inhibition of TRAP1 causes synthetic lethality in solid tumors. Cancers (Basel). 2019;11(6):788.
- Boureima Oumarou D, Ji H, Xu J, et al. Involvement of microRNA-23b-5p in the promotion of cardiac hypertrophy and dysfunction via the HMGB2 signaling pathway. Biomed Pharmacothe. 2019;116:108977.
- Zhang Y, Fan X, Yang H. Long non-coding RNA FTX ameliorates hydrogen peroxide-induced cardiomyocyte injury by regulating the miR-150/KLF13 axis. Open life sciences. 2020;15(1):1000–1012.
- Li K, Lin Y, Li C. MiR-338-5p ameliorates pathological cardiac hypertrophy by targeting CAMKIIδ. Arch Pharm Res. 2019;42(12):1071–1080.
- Frey N, Katus HA, Olson EN, et al. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589.
- Sriramula S, Francis J, Sovari AA. Tumor necrosis factor - Alpha is essential for Angiotensin II-Induced ventricular remodeling: role for oxidative stress. PLoS One. 2015;10(9):e0138372.
- Xu E, Zhang J, Zhang M, et al. RNA-binding protein RBM24 regulates p63 expression via mRNA stability. Mol Cancer Res. 2014;12(3):359–369.
- Zhang J, Jun Cho S, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci U S A. 2010;107(21):9614–9619.
- Cho SJ, Zhang J, Chen X. RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Res. 2010;38(7):2256–2267.
- Alvarez-Dominguez JR, Zhang X, Hu W. Widespread and dynamic translational control of red blood cell development. Blood. 2017;129:619–629.
- Warzecha CC, Sato TK, Nabet B, et al. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33(5):591–601.
- Miyamoto S, Hidaka K, Jin D, et al. RNA-binding proteins Rbm38 and Rbm24 regulate myogenic differentiation via p21-dependent and -independent regulatory pathways. Genes Cells. 2009;14(11):1241–1252.
- Zhang J, Xu E, Ren C, et al. Mice deficient in Rbm38, a target of the p53 family, are susceptible to accelerated aging and spontaneous tumors. Proc Natl Acad Sci U S A. 2014;111(52):18637–18642.
- Léveillé N, Elkon R, Davalos V, et al. Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat Commun. 2011;2(1):513.
- She X, Lin Y, Liang R, et al. RNA-Binding Motif protein 38 as a potential biomarker and therapeutic target in cancer. Onco Targets Ther. 2020;13:13225–13236.
- Cannon MV, Silljé HHW, Sijbesma JWA, et al. LXRα improves myocardial glucose tolerance and reduces cardiac hypertrophy in a mouse model of obesity-induced type 2 diabetes. Diabetologia. 2016;59(3):634–643.
- Kuipers I, Li J, Vreeswijk-Baudoin I, et al. Activation of liver X receptors with T0901317 attenuates cardiac hypertrophy in vivo. Eur J Heart Fail. 2010;12(10):1042–1050.
- Mitro N, Mak PA, Vargas L, et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445(7124):219–223.
- Cannon MV, Yu H, Candido WM, et al. The liver X receptor agonist AZ876 protects against pathological cardiac hypertrophy and fibrosis without lipogenic side effects. Eur J Heart Fail. 2015;17(3):273–282.
- Cannon MV, van Gilst WH, de Boer RA. Emerging role of liver X receptors in cardiac pathophysiology and heart failure. Basic Res Cardiol. 2016;111(1):3.
- Cannon MV, Silljé HH, Sijbesma JW, et al. Cardiac LXR α protects against pathological cardiac hypertrophy and dysfunction by enhancing glucose uptake and utilization. EMBO Mol Med. 2015;7(9):1229–1243.
- Tran DH, Wang ZV. Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc. 2019;8(12):e012673.
- Giudice J, Cooper TA. RNA-binding proteins in heart development. Adv Exp Med Biol. 2014;825:389–429.