
ABSTRACT
Morphine tolerance poses a great challenge for clinicians, whose pathogenesis has a close connection with microglial activation and neuroinflammation. Dihydroartemisinin (DHA) that derives from artemisinin, may serve as a potential anti-inflammatory drug. In this study, the effects as well as the underlying mechanism of DHA on suppressing microglial activation and neuroinflammation were explored. The microglial cell line BV-2 cells were induced by morphine and treated with DHA or minocycline. With the application of CCK-8, the cell viability was detected. Western blot was employed to assess the expressions of Ki67, IBa-1, and TLR4 and quantitative real-time PCR (qRT-PCR) was adopted to evaluate miRNA-16 (miR-16) expression. With the adoption of ELISA kits and qRT-PCR, the release of inflammatory cytokines was evaluated. Besides, luciferase reporter assay was applied to testify the binding relationship between miR-16 and TLR4. NF-κB expression was measured by immunofluorescence. DHA reduced cell viability and decreased protein expression of Ki67 and IBa-1 in morphine-induced BV-2 cells. Additionally, DHA contributed to the declined release of pro-inflammatory cytokines. miR-16 was down-regulated by morphine but was up-regulated by DHA concentration-dependently in BV-2 cells. The inhibition of miR-16 partly abolished the inhibitory effects of DHA on morphine-induced microglial activation and neuroinflammation. Moreover, TLR4 was found to be bound to miR-16, and the inhibitory effect of DHA on TLR4/NF-κB was partly reversed by miR-16 inhibition. In conclusion, DHA remarkably suppressed microglial activation and neuroinflammation through regulating miR-16-mediated TLR4/NF-κB signaling. This study may provide a new solution to improve clinical analgesic efficacy of morphine.
Introduction
Morphine, a classic opioid analgesic drug, has been widely applied in clinic and has become the first-line drug for treating pain in cancer or non-cancer patients [Citation1]. Nevertheless, long-term application of morphine results in analgesic tolerance and numerous adverse effects, greatly limiting the clinical use of morphine. Therefore, it is of great importance to understand the mechanisms of morphine tolerance and delineate specific protocols to handle this problem.
Among the previous studies, a plenty of scholars paid more attention to morphine tolerance on neurons, including the changes in neuronal plasticity caused by the continuous stimulation of G protein uncoupling, and the increase in the synthesis of anti-opioid substances [Citation2,Citation3]. However, an increasing number of convincing evidence revealed that microglia activation also contributed to morphine tolerance [Citation4–6]. Stimulation of activated microglia by morphine can promote the release of pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin-6 (IL-6) and IL-1β, which in turn causes severe neuroinflammatory reaction and increases the hyperactivity of dorsal horn neurons, leading to central sensitization and a decrease of analgesic efficacy of morphine [Citation4,Citation5].
Artemisinins comprise a group of sesquiterpene trioxane lactones extracted from the Chinese herb Artemisia annua L, which has been a WHO-recommended mainstay in treating malaria [Citation7]. Dihydroartemisinin (DHA), a semisynthetic derivative that derives from artemisinin, has also been widely considered as the most optimal antimalarial drug. Recently, DHA has been found to exhibit other effects, such as anti-neurodegeneration, anti-inflammatory, anti-cancer, anti-oxidant, and anti-organizational fibrosis effects, thus protecting against systemic lupus erythematosus, multiple cancers, colitis, pulmonary fibrosis, etc.[Citation8–12] Of note, DHA has been also found to ameliorate lipopolysaccharide (LPS)-induced neuroinflammation by inhibiting the phosphatidylinositol 3-kinase (PI3K)/serine/threonine kinase (AKT) pathway in vivo [Citation13]; however, more precise molecular mechanisms need to be explored. It is worth mentioning that LPS has been widely recognized as a classic toll-like receptor 4 (TLR4) receptor agonist and is used to induce glial cell activation to simulate neuroinflammation. Meanwhile, morphine has been shown to directly activate TLR4 receptor and promote inflammatory response [Citation14]. Considering these pharmacological activities of DHA, we hypothesized that DHA might have an inhibitory effect on morphine-induced neuroinflammation.
With the aim of exploring the effects of DHA on morphine-induced microglial activation and inflammation, as well as discovering its more precise mechanism of actions, we conducted this study.
Materials and methods
Cell culture and treatment
Mouse BV-2 cell lines that were purchased from BeNa Culture Collection (Beijing, China) were incubated with Dulbecco’s modified Eagle medium (DMEM; Hyclone) containing with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM glutamine (Gibco BRL, Grand Island, NY, USA) at 37°C in humidified 5% CO2 and 95% air.
For morphine induction and treatment, BV-2 cells were inoculated in 6-well plates (1 × 105 cells/well) overnight. Then, cells were administrated with morphine (200 μM) with or without DHA for 6 h [Citation15,Citation16]. Before morphine induction, the cells were treated with DHA (1, 5, 10, and 20 μM) for 30 min. Minocycline (10 μM), an inhibitor of microglia activation, was adopted as the positive control.
To introduce miR-16 inhibitor or miR-16 mimic into BV-2 cells to regulate miR-16 expression, cell transfection was operated. Briefly, miR-16 inhibitor, miR-16 mimic and their negative controls (inhibitor-NC, mimic-NC) that provided by Shanghai GenePharma Co., Ltd. (Shanghai, China) were transfected into BV-2 cells with the application of Lipofectamine 3000 (Invitrogen).
Cell viability assay
BV-2 cells were inoculated in 96-well plates (5 × 103 cells/well) and cultured at 37°C overnight. After indicated treatment, each well was added with 10 μl of Cell Counting Kit-8 (CCK-8) agent (Dojingdo Molecular Technologies, Inc) and the cells were incubated with mixture for another 2 h. Eventually, a microplate reader (Bio-Rad Laboratories, France) was employed to determine the absorbance at 450 nm.
Western blot
Total protein was extracted from cells using RIPA lysis buffer containing phenylmethylsulfonyl fluoride (PMSF) (Boster Biological Technology, Wuhan, China). After measuring protein concentration using a Bicinchoninic Acid (BCA) kit (Boster Biological Technology), the proteins were subjected to 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel for separation and then transferred onto a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). Impeded with 5% nonfat milk for 1 h at room temperature, the membranes were incubated with primary antibodies against Ki67 (cat. no. 16,667, Abcam), IBa-1 (cat. no. 178,846, Abcam), TLR4 (cat. no. 13,556, Abcam), and GAPDH (cat. no. 9485, Abcam) at 4°C overnight, after which horseradish peroxidase-labeled Rabbit anti-mouse IgG secondary antibody (cat. no. ab6728, Abcam) was utilized to incubate membranes for 2 h. At last, the visualization and quantification of protein bands were performed by an enhanced ECL substrate (Tanon, Shanghai, China) and Image J software (version 2.0.0, USA), respectively.
Quantitative real-time PCR (qRT-PCR)
The isolation of total RNA from samples was performed by Trizol reagent (Invitrogen) strictly in line with manufacture’s specification. Subsequently, 2 μg of RNA was synthesized to cDNA using the PrimeScript RT reagent Kit (Takara Bio Inc, Japan). The operation of SYBR Green quantitative PCR of IL-1β, IL-6, TNF-α, and miR-16 was performed with the adoption of Applied Bio systems 7500 Fast Real-Time PCR System. The mRNA level of IL-1β, IL-6, and TNF-α was calculated by 2−ΔΔCt and normalized to β-actin, and the level of miR-16 was normalized to U6.
Cytokine determination
Mouse TNF-α, IL-1β, and IL-6 in the supernatant from cell culture were determined using their corresponding ELISA kits (Shanghai Enzyme Link Biotechnology Co., Ltd., Shanghai, China).
Luciferase reporter assay
Starbase website predicted the potential-binding sites between miR-16 and TLR4. To verify this connection, the binding sequences of miR-16 on TLR4 was cloned into pSI-check Dual-luciferase vectors. The co-transfection of BV-2 cells with miR-16 mimic and the wild type (WT) or mutated type (MUT) of TLR4 was operated by a lipofectamine 3000 (Invitrogen). At last, the luciferase activity was assessed by Dual-luciferase reporter assay system (Solaribio Life Sciences, Shanghai, China).
Immunofluorescence
After being inoculated into the glass bottom cell culture dishes, the BV-2 cells were administrated with morphine with or without DHA. After 6 h, 4% paraformaldehyde and 0.3% Triton X-100 were employed to fix and permeate BV-2 cells. Blocked with 1% bovine serum albumin (BSA), cells were then incubated with primary antibody against nuclear transcription factor (NF)-κB p65 (cat. no. ab32536, Abcam) at 4°C overnight. On the following day, the fluorescent secondary antibody (cat. no. ab150113, Abcam) was used to incubate cells for 1 h in dark. After the procedure of staining cells with DAPI for 3 min, the cells were observed under a laser confocal microscope (Olympus, Japan).
Statistical analysis
All data collected from experiments were displayed as mean ± standard deviation (SD). GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA) was employed to analyze the statistics. A one-way analysis of variance (ANOVA) followed by post hoc Tukey’s tests was employed for difference comparison. The difference was considered significant at p < 0.05.
Results
DHA reduced morphine-induced cell viability in BV-2 cells
With the aim of investigating the role of DHA in BV-2 that induced by morphine, the appropriate concentrations of DHA for application was evaluated. As demonstrated, after the treatment with different concentrations (1, 5, 10, 20, and 40 μM) of DHA, a slight reduction of the cell viability was observed upon DHA treatment from 1~20 μM, whereas a significant reduction of cell viability was observed upon DHA treatment at 40 μM, indicating that DHA (40 μM) caused cell injury. In view of this, DHA with concentrations of 1, 5, 10, and 20 μM were applied for the subsequent experiments.
Figure 1. DHA reduced morphine-induced cell viability in BV-2 cells
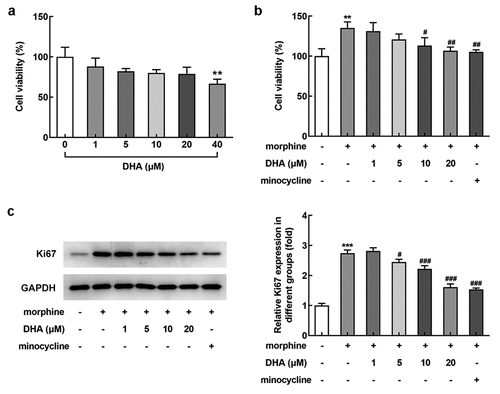
Subsequently, the effect of DHA on cell viability in morphine-induced BV-2 cells was evaluated. Results obtained from CCK-8 assay exhibited that morphine greatly stimulated the viability of BV-2 cells, whereas this elevated viability was reduced by DHA treatment, especially at 10 and 20 μM (). Additionally, the elevated protein expression of Ki67 was remarkably reduced by DHA treatment (5, 10, and 20 μM) (), indirectly reflecting that DHA could partly inhibit morphine-induced BV-2 cell viability, and the inhibitory effect of DHA at 20 μM was similar with that of minocycline treatment, the positive drug control.
DHA inhibited morphine-induced microglial activation in BV-2 cells
Next, the effect of DHA on microglial activation in morphine-induced BV-2 cells was evaluated. Ionized calcium-binding adaptor molecule 1 (IBa-1) is a classical microglial activation hallmark in the spinal cord, and its expression level reflects the degree of microglial activation. As displayed, morphine induction hugely promoted the protein expression of IBa-1, suggesting that the BV-2 cells were highly activated upon morphine induction. Minocycline, an inhibitor of microglia activation, was also expected to abolish microglia activation by inhibiting the protein expression of IBa-1 in morphine-induced BV-2 cells. Surprisingly, an inhibitory effect on IBa-1 expression was also observed by DHA treatment. These results suggested that DHA exerted inhibitory effects on morphine-induced microglial activation.
Figure 2. DHA inhibited morphine-induced microglial activation in BV-2 cells
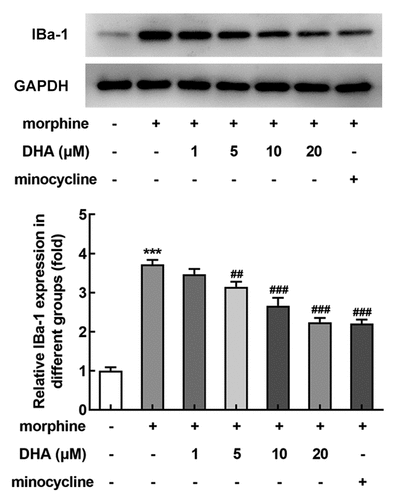
DHA alleviated morphine-induced inflammatory response in BV-2 cells
Then, we also assessed the effect of DHA on inflammatory response in morphine-induced BV-2 cells. A series of inflammatory cytokines were detected by ELISA and qRT-PCR assays. According to , the release of IL-1β, IL-6, and TNF-α in BV-2 cells gained a huge growth upon morphine induction, which were then reversed by DHA treatment in a dose-dependent manner. Minocycline suppressed the release of IL-1β, IL-6, and TNF-α as well. At the same time, the mRNA level of IL-1β, IL-6, and TNF-α in different treatment detected by qRT-PCR was in the same trend with that detected by ELISA ().
Figure 3. DHA alleviated morphine-induced inflammatory response in BV-2 cells
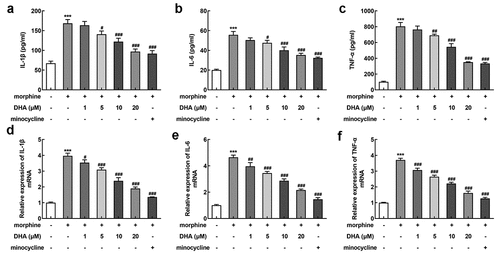
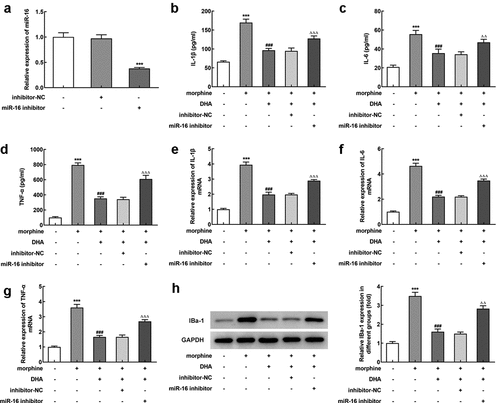
miR-16 was regulated by DHA and miR-16 inhibitor partly abolished the protective effects of DHA in morphine-induced BV-2 cells
Next, we further tried to clarify the specific mechanism of DHA underlying its role in morphine-induced BV-2 cells. Interestingly, we found that miR-16 was greatly downregulated by morphine, but was upregulated by DHA concentration-dependently or minocycline treatment (), indicating that miR-16 might be an important gene that regulated the gene network response to morphine induction or these treatments in BV-2 cells, resulting in multiple cellular biological activities.
Figure 4. miR-16 was regulated by morphine and DHA
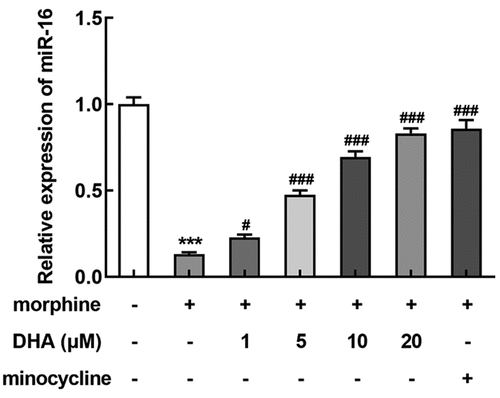
To verify this hypothesis, miR-16 inhibitor was introduced to BV-2 cells to inhibit the expression level of miR-16 (). BV-2 cells were pretreated with DHA with or without transfection with inhibitor-NC or miR-16 inhibitor, prior to morphine induction. Subsequently, the detection results of inflammatory cytokines production by ELISA and qRT-PCR assays both revealed that the suppressive effect of DHA on the production of IL-1β, IL-6, and TNF-α in morphine-induced BV-2 cells was partly abolished by miR-16 inhibitor (). In addition, the suppressive effect of DHA on the protein expression of IBa-1 in morphine-induced BV-2 cells was greatly hindered by miR-16 inhibitor (). These results suggested the protective effects of DHA on morphine-induced microglial activation and inflammatory response were partly abolished by miR-16.
Figure 5. miR-16 inhibitor partly abolished the protective effects of DHA in morphine-induced BV-2 cells
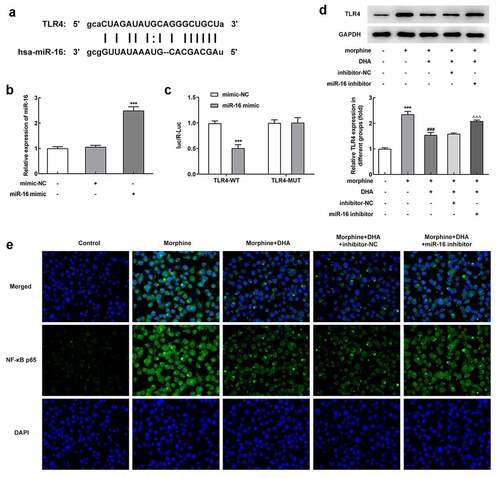
miR-16 was a direct target of TLR4 and regulated TLR4/NF-κB signaling
To understand the relationship between miR-16 and TLR4, a potential binding site between miR-16 and TLR4 was found by searching Starbase database (). To verify this interaction, miR-16 mimic was introduced to overexpress miR-16, and the binding relationship was further verified by luciferase reporter assay (). The increased protein expression of TLR4 in morphine-induced BV-2 cells was decreased by DHA treatment; however, miR-16 inhibition partly reversed the changes made by DHA (). In addition, results in exhibited that the number of NF-κB p65-positive cells was increased upon morphine induction, but decreased upon DHA treatment, which was further partly abolished by miR-16 inhibitor (). These results suggested that DHA treatment could repress morphine-mediated TLR4/NF-κB signaling partly by upregulating miR-16.
Figure 6. miR-16 was a direct target of TLR4 and regulated NF-κB signaling
Discussion
In this study, we discovered that DHA, a semisynthetic derivative of artemisinin, exhibited inhibitory effects on morphine-induced microglial activation and inflammatory response. TLR4, an important inducer of neuroinflammation, could be bound to miR-16. DHA-induced upregulation of miR-16 could block TLR4/NF-κB signaling pathway, contributing to suppression of microglial activation and inflammatory response in morphine-induced BV-2 cells. Given that microglial activation and neuroinflammation has been widely recognized as important contributors to morphine tolerance, this study may provide a new solution to improve clinical analgesic efficacy of morphine.
In order to make out the mechanism of DHA inhibiting LPS-induced neuroinflammation, we assessed its effects on TLR4/NF-κB signaling pathway. TLR4 is an innate immune receptor in the body, mainly distributed in microglia, and is considered to be one of the key mediators of morphine-induced microglia activation and neuro-inflammatory response. Morphine-evoked neuroinflammation mediated by TLR4 has been evidenced to be important for morphine tolerance, and suppression of TLR4-mediated neuroinflammation attenuated morphine tolerance [Citation15]. NF-κB is a crucial transcription factor in the induction of inflammatory injury, the activation of which was found to contribute to chronic morphine-induced analgesic tolerance [Citation17,Citation18]. TLR4 has been considered as an inducer of NF-κB by TLR4-MyD88 signaling, followed by the degradation of IκBα, an inhibitor of NF-κB, and translocation of NF-κB subunit p65 from the cytoplasm to nucleus, thus leading to the release of multiple cytokines, and contributing to inflammatory response [Citation19,Citation20]. The previous reports also revealed that silence of TLR4 gene expression or blocking TLR4/NF-κB contributed to an increased intensity and prolonged duration of opioid analgesia [Citation21,Citation22]. What’s more, DHA, especially its derivative with alkynyl as the end functional group, was considered to be a TLR4 antagonist [Citation23]. As expected, our study revealed that DHA significantly reduced morphine-induced TLR4 expression, thus blocking TLR4/NF-κB signaling pathway. DHA exerted its protective effects in morphine-induced neuroinflammation through suppressing TLR4/NF-κB signaling pathway.
Moreover, a further investigation was carried out to illuminate more precise molecular mechanism underlying the regulatory effect of DHA on TLR4. To our surprise, there is a potential binding relationship between miR-16 and TLR4 according to Starbase database, which was then further verified by luciferase reporter assay, indicating that TLR4 was a direct target of miR-16, and miR-16 might exert effect through regulating TLR4. Consistent with this finding, the existing evidence also revealed that the expressions of TLR4 and NF-κB were reduced by miR-16 overexpression, and miR-16 could relieve inflammatory response of acute lung injury via targeting TLR4/NF-κB pathway [Citation24]. Additionally, miR-16 was reported to participate in the release of inflammatory response, evidenced by the decreased levels of IL-6 and TNF-α as well as the increased level of IL-10, indicating that miR-16 might act as an anti-inflammatory gene and attenuate inflammatory response [Citation25,Citation26]. Therefore, considering the important role of TLR4/NF-κB signaling and the binding relationship between miR-16 and TLR4 as aforementioned, the inhibition of miR-16 might downregulate TLR4 expression, and block the degradation of IκBα and p65 translocation, thus reducing NF-κB signaling-mediated inflammatory response. Coincidentally, DHA was demonstrated to increase miR-16 expression in gastric cancer cells, disclosing a positively regulator relationship between DHA and miR-16 [Citation27]. In addition, DHA-induced miRNA-mRNA regulatory network has been demonstrated to be a potential pathway that partly explains the multiple therapeutic effects of DHA in various diseases, including cancers, diabetic vascular complications, and asthma [Citation28–30]. Here, DHA-induced increase in miR-16 and down-regulated TLR4 expression, likely contributing to the ability of DHA to inhibit microglial activation and inflammatory response in morphine-induced BV-2 cells. Therefore, miR-16-mediated TLR4/NF-κB pathway might be a novel mechanism by which DHA alleviated morphine-induced neuroinflammation.
Conclusion
In conclusion, our findings demonstrated that DHA remarkably attenuated the development of chronic morphine tolerance in an efficient manner by suppressing microglial activation and neuroinflammation. In addition, the possible mechanism by which DHA exerted its protective effects might partly depend on the upregulation of miR-16, which directly inhibited TLR4 expression and then blocked TLR4/NF-κB signaling pathway. However, this study mainly focuses on in vitro experiments, an in vivo mice experiment is more convincing to verify the protective effects of DHA on chronic morphine tolerance, as well as its potential mechanism, which will be further explored in our future work. Anyway, our study may potentially offer a new clinical application of DHA in attenuating morphine tolerance.
Highlight
1. Dihydroartemisinin could suppress microglial activation and neuroinflammation.
2. miR-16 was upregulated by Dihydroartemisinin in morphine-induced BV-2 cells, and directly targeted to TLR4.
3. Dihydroartemisinin exerted its protective effects through regulating miR-16-mediated TLR4/NF-κB signaling.
Authors’ contributions
Yongxin Liang conducted the concept and designed the experiments. Sen Guan, Tingting Jin, Shuai Han, and Wenjie Fan performed the experiments and obtained the data. Sen Guan, Tingting Jin, and Haichen Chu analyzed and interpreted the results. Sen Guan drafted the manuscript and Yongxin Liang revised the manuscript. All authors read and approved the final version of the manuscript.
Disclosure statement
The authors declare that there is no potential interests.
Additional information
Funding
References
- Gretton SK, Ross JR, Rutter D, et al. Plasma morphine and metabolite concentrations are associated with clinical effects of morphine in cancer patients. J Pain Symptom Manage. 2013;45(4):670–680.
- Williams JT, Ingram SL, Henderson G, et al. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65(1):223–254.
- Corder G, Tawfik VL, Wang D, et al. Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med. 2017;23(2):164–173.
- Eidson LN, Murphy AZ. Persistent peripheral inflammation attenuates morphine-induced periaqueductal gray glial cell activation and analgesic tolerance in the male rat. J Pain. 2013;14(4):393–404.
- Ferrini F, Trang T, Mattioli TA, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 2013;16(2):183–192.
- Cai Y, Kong H, Pan YB, et al. Procyanidins alleviates morphine tolerance by inhibiting activation of NLRP3 inflammasome in microglia. J Neuroinflammation. 2016;13(1):53.
- Tu Y. Artemisinin-A gift from traditional Chinese medicine to the world (Nobel lecture). Angew Chem Int Ed Engl. 2016;55(35):10210–10226.
- Huang X, Xie Z, Liu F, et al. Dihydroartemisinin inhibits activation of the Toll-like receptor 4 signaling pathway and production of type I interferon in spleen cells from lupus-prone MRL/lpr mice. Int Immunopharmacol. 2014;22(1):266–272.
- Hui HY, Wu N, Wu M, et al. Dihydroartemisinin suppresses growth of squamous cell carcinoma A431 cells by targeting the Wnt/beta-catenin pathway. Anticancer Drugs. 2016;27(2):99–105.
- Yi R, Wang H, Deng C, et al. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci Rep. 2020;40(6). DOI:10.1042/BSR20193314.
- Li N, Sun W, Zhou X, et al. Dihydroartemisinin protects against Dextran Sulfate Sodium-Induced colitis in mice through inhibiting the PI3K/AKT and NF-kappaB signaling pathways. Biomed Res Int. 2019;2019:1415809.
- Yang DX, Qiu J, Zhou HH, et al. Dihydroartemisinin alleviates oxidative stress in bleomycin-induced pulmonary fibrosis. Life Sci. 2018;205:176–183.
- Gao Y, Cui M, Zhong S, et al. Dihydroartemisinin ameliorates LPS-induced neuroinflammation by inhibiting the PI3K/AKT pathway. Metab Brain Dis. 2020;35(4):661–672.
- Wang X, Loram LC, Ramos K, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A. 2012;109(16):6325–6330.
- Qu J, Tao XY, Teng P, et al. Blocking ATP-sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70-TLR4-NLRP3-mediated neuroinflammation. J Neuroinflammation. 2017;14(1):228.
- Han Y, Jiang C, Tang J, et al. Resveratrol reduces morphine tolerance by inhibiting microglial activation via AMPK signalling. Eur J Pain. 2014;18(10):1458–1470.
- Bai L, Zhai C, Han K, et al. Toll-like receptor 4-mediated nuclear factor-kappaB activation in spinal cord contributes to chronic morphine-induced analgesic tolerance and hyperalgesia in rats. Neurosci Bull. 2014;30(6):936–948.
- Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034.
- Cao C, Yin C, Shou S, et al. Ulinastatin protects against LPS-Induced acute lung injury by attenuating TLR4/NF-kappaB pathway activation and reducing inflammatory mediators. Shock. 2018;50(5):595–605.
- Ju M, Liu B, He H, et al. MicroRNA-27a alleviates LPS-induced acute lung injury in mice via inhibiting in fl ammation and apoptosis through modulating TLR4/MyD88/NF-kappaB pathway. Cell Cycle. 2018;17(16):2001–2018.
- Shafie A, Moradi F, Izadpanah E, et al. Neuroprotection of donepezil against morphine-induced apoptosis is mediated through toll-like receptors. Eur J Pharmacol. 2015;764:292–297.
- Hutchinson MR, Zhang Y, Shridhar M, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2010;24(1):83–95.
- Wang S, Wang H, Lin C, et al. Structure-activity relationship study of dihydroartemisinin C-10 hemiacetal derivatives as toll-like receptor 4 antagonists. Bioorg Chem. 2021;114:105107.
- Yang Y, Yang F, Yu X, et al. miR-16 inhibits NLRP3 inflammasome activation by directly targeting TLR4 in acute lung injury. Biomed Pharmacother. 2019;112:108664.
- Liang X, Xu Z, Yuan M, et al. MicroRNA-16 suppresses the activation of inflammatory macrophages in atherosclerosis by targeting PDCD4. Int J Mol Med. 2016;37(4):967–975.
- Talari M, Kapadia B, Kain V, et al. MicroRNA-16 modulates macrophage polarization leading to improved insulin sensitivity in myoblasts. Biochimie. 2015;119:16–26.
- Sun H, Meng X, Han J, et al. Anti-cancer activity of DHA on gastric cancer–an in vitro and in vivo study. Tumour Biol. 2013;34(6):3791–3800.
- Li Y, Wang Y, Kong R, et al. Dihydroartemisinin suppresses pancreatic cancer cells via a microRNA-mRNA regulatory network. Oncotarget. 2016;7(38):62460–62473.
- Yang B, Gao X, Sun Y, et al. Dihydroartemisinin alleviates high glucose-induced vascular smooth muscle cells proliferation and inflammation by depressing the miR-376b-3p/KLF15 pathway. Biochem Biophys Res Commun. 2020;530(3):574–580.
- Zhu H, Ji W. Dihydroartemisinin ameliorated ovalbumin-Induced asthma in mice via regulation of miR-183C. Med Sci Monit. 2019;25:3804–3814.