
ABSTRACT
Adaptin proteins (APs) play a crucial role in intracellular cell trafficking. The ‘classical’ role of APs is carried out by AP1‒3, which bind to clathrin, cargo, and accessory proteins. Accordingly, AP1–3 are crucial for both vesicle formation and sorting. All APs consist of four subunits that are indispensable for their functions. In fact, based on studies using cells, model organism knockdown/knock-out, and human variants, each subunit plays crucial roles and contributes to the specificity of each AP. These studies also revealed that the sorting and intracellular trafficking function of AP can exert varying effects on pathology by controlling features such as cell development, signal transduction related to the apoptosis and proliferation pathways in cancer cells, organelle integrity, receptor presentation, and viral infection. Although the roles and functions of AP1‒3 are relatively well studied, the functions of the less abundant and more recently identified APs, AP4 and AP5, are still to be investigated. Further studies on these APs may enable a better understanding and targeting of specific diseases.
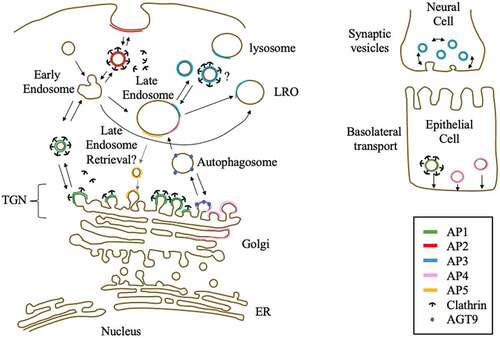
APs known or suggested locations and functions.
GRAPHICAL ABSTRACT
1. Introduction
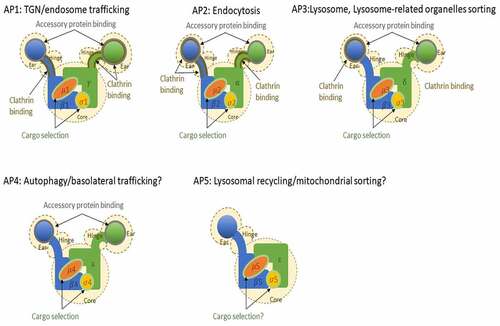
Clathrin adaptor proteins or adaptin proteins (APs) are membrane-bound heterotetrameric complexes localized in cellular buds and vesicles. In addition to clathrin, a structural protein that forms a lattice of hexagons and pentagons around vesicles [Citation1], APs play an important role in intracellular trafficking in the trans-Golgi network (TGN) and beyond. Accordingly, APs are highly conserved and ubiquitously expressed in eukaryotic organisms. Although membrane trafficking from the endoplasmic reticulum to the Golgi is relatively simple and only requires two heptameric coatomer complexes [coat protein type I and II (COPI and COPII)] that form a spherical cage around the membrane to form a vesicle [Citation2], post-TGN trafficking is more complex, as it requires sorting and trafficking to diverse organelles, plasma membranes, and cell specificity. Therefore, post-TGN trafficking requires five APs, several membrane-bound proteins, accessory proteins, and clathrin [Citation3]. The most abundant APs (AP1, AP1, and AP3) and two subunits of the COPI complex share a common ancestral origin [Citation4] and have been identified in various organisms, including model organisms such as D. melanogaster, C. elegans, S. cerevisiae, S. pombe, H. sapiens, M. musculus, and A. thaliana [Citation5]. Shortly after their discovery, a fourth AP, AP4, was identified [Citation6,Citation7] and detected in various organisms, including the model organisms H. sapiens, M. musculus, and A. thaliana [Citation5]. However, unlike AP1‒3, AP4 was not found in D. melanogaster, C. elegans, and S. cerevisiae [Citation8]. AP5, the fifth and last identified complex, has been detected in H. sapiens [Citation9] and putatively in A. thaliana; however, this AP seems to be lost in several organisms [Citation9]. Identification of an ancestral TSET complex in Dictyostelium suggests that eukaryotic AP1‒5s and COPI evolved from a common ancestral complex [Citation10]. All five complexes consist of two large subunits (one common β unit and the other with a specific letter γ, α, δ, ε, or ζ), one middle-size subunit (μ) and one small subunit (σ). AP subunits share an estimated sequence similarity of 20 to 80% and have common domains, which were used for their identification [Citation9]. Based on the structures of AP1 and AP2 complex, these complexes were found to consist of (1) a core structure, that include the N-terminal α-helix domains of the two large subunits and the two smaller subunits, which role is to recognizes cargo and localizes the complex in the membrane; and (2) a hinge plus ear structure consisting of the C-terminal domains of the two large subunits, which recognizes clathrin and accessory proteins [Citation11], except for AP5. AP5 lacks a hinge and ear structure in its large ζ subunit, a deletion that results in the loss of cargo and clathrin binding motif (). AP subunits have isoforms that are encoded by different genes or by alternative splicing, adding more complexity to the AP complexes (). Each of the five APs, as well as some of their isoforms, has specific roles and subcellular localization. AP1 and AP2 are the more ‘classic’ APs that interact with clathrin in the TGN (AP1) or plasma membrane (AP2) to form clathrin-coated vesicles and control intracellular trafficking in the TGN and endocytosis, respectively. The location and function of AP3 are less well defined compared to those of AP1 and AP2. AP3 has been detected in the cytosol, TGN, late endosome, lysosomes, and lysosome-related-organelles (LROs) and can interact with clathrin [Citation12]. However, previous research shows that the function of AP3 can be clathrin independent [Citation13]. AP3 is involved in protein sorting of yeast vacuole and human cell lysosome [Citation14] and iin protein trafficking to the late endosome/lysosome [Citation15]. Further, it has two major isoforms that differ in expression site; AP3A is ubiquitously expressed, while AP3B is brain specific [Citation16,Citation17]. Both AP4 and AP5 are relatively less abundant compared to AP1‒3 (subunit concentration is less than 40-fold in HeLa cells [Citation18]) and are not reported to interact with clathrin. As mentioned above, AP subunits share an estimated similarity of 20–80%. For AP1‒3, the estimated sequence similarity is 60 to 90%, while AP4 subunits similarity to the subunits of other complexes ranges from 17 to 43%. Of note, AP4 lacks clathrin-binding domains and is associated with non clathrin-coated vesicles in TGN [Citation7]. AP4 localizes in the Golgi, TGN, and endosomes [Citation6,Citation7,Citation19] and presumably controls autophagy via intracellular trafficking of AGT9, a key autophagosome protein [Citation20], and its accessory protein, Tepsin [Citation21]. These proteins are localized with cation-independent mannose 6-phosphate receptor (CI-MPR), a transmembrane glycoprotein targeting cargo proteins to the lysosome [Citation19], suggesting that AP4 has a role in lysosome sorting. Furthermore, AP4 was shown to be involved in clathrin-independent basolateral signals of epithelial cells [Citation22]. Although AP5 has the same subunits that share sequence and domain similarities with other APs, it lacks a hinge domain and an ear domain and does not have a clathrin-binding site. While AP5 role is yet not well defined, AP5 was found to be localized in the TGN and late endosome and is speculated to function in late endosome retrieval [Citation9]. This speculation is reinforced by findings in a mouse model, where the deletion of AP5 cause a defect in late endosomal retrieval and in vitro experiments that demonstrated that mutant cells had defective autophagic functions [Citation23].
Table 1. The identified AP complexes
Figure 1. AP complexes with their known or suggested role in cells. Known interaction domains are highlighted
In this paper, we sought to review the findings reported for each AP subunit, including relatively recent discoveries, while focusing on the pathological involvement of AP complexes.
2. AP1 complex
AP1 is an indispensable protein complex in cells as it is known that its main role is in clathrin dependent protein transportation from the TGN to endosome, while evidences show it is also involved in basolateral transport from the TGN in epithelial cells () [Citation24]. Accordingly, its knock-out is embryonically lethal and its missense or nonsense mutations in mouse model organisms result in a serious developmental deficiency [Citation25,Citation26]. In humans, there are two isoforms of the large subunit γ, AP1G1 and AP1G2, and one isoform of the other large subunit β, AP1B1. AP1G1 and AP1G2 are 60% identical. However, AP1G1 is ubiquitously expressed, while AP1G2 is only expressed in plants and vertebrate, with presumably diverse functions and/or stages of expression during development [Citation27]. Based on inhibitor analysis, AP1G1 and AP1G2 might recruit distinct accessory proteins [Citation28] while being indispensable for development [Citation29]. Human AP1G1 variants lead to neurodevelopmental deficiency [Citation26], high risks of pancreatic cancer [Citation30] and cardiac arrest among patients with coronary artery disease [Citation31]. Although required as intracellular trafficking components, AP1G1 and AP1B1 are cofactors that can be hijacked by coronaviruses (SARS-CoV-2, MERS-CoV, and seasonal HCoVs) [Citation32]. Upon HIV infection, AP1G2, but not AP1G1, is used to remove host CD4 receptors [Citation33], which indicates that viruses selectively target the host AP1 complex. In renal cancer, AP1G1 expression is downregulated in patient tumor tissues and suppresses cancer cell proliferation and migration [Citation34]. Upon treatment with cetuximab, an approved anti-cancer drug for head and neck squamous cell carcinoma (HNSCC), membrane-bound epidermal growth factor receptor (EGFR) and alanine-serine-cysteine transporter 2 (ASCT2) were found to interact with AP1G1, resulting in the sensitization of cancer cells to reactive oxygen species (ROS) treatment, which might contribute to a higher survival rate in the patient group showing a higher AP1G1 expression [Citation35]. Taken together this suggest that AP1G1, or AP1 complex might play a role in an intrinsic defense mechanim against cancer. There are two isoforms of AP1 middle-sized subunit μ, AP1M1 that is ubiquitously expressed and AP1M2 that is expressed in polarized epithelial and exocrine cells [Citation36], which specifically function in basolateral transport [Citation37]. Interestingly, as the knock-out of any subunit of AP1 results in AP1 inactivation [Citation38,Citation39] knock-out of one subunit are usually used to study the AP1 complex function [Citation40–42], but two independent studies using forward genetic screening for mutations in cell trafficking detected AP1M1 while, other AP1 subunits were not [Citation41,Citation43]. The ubiquitous AP1M1 subunit is required as it contain the binding domains that recognize proteins andrecruit clathrin [Citation41,Citation44]. However, upregulation of AP1M1 was reported to have pathological consequences; hepatitis B virus, the major cause of liver cancer, was found to cause upregulation of AP1M1 expression, which upregulated cancer cell proliferation [Citation45]. In addition, upregulation of AP1M1 is putatively a biomarker for metastasis to the brain tissue [Citation46]. On the other hand, AP1M2 containing AP1 complex was recently proposed to control β1 integrin transport in the basal membrane of epithelial cells and act as a cell-inherent anticancer mechanism that can inhibit metastasis [Citation47]. There are three AP1 σ subunits: AP1S1, AP1S2, and AP1S3. To date, no reports have been published regarding the cell-specific expression of AP1S1, AP1S2, and AP1S3. Further, each of the three proteins is expressed ubiquitously in human tissues at low levels, with slight enrichment in the brain, epididymis, and a group of tissues (ductus deferens, epididymis, and seminal vesicle) (http://www.proteinatlas.org). However, these subunits also contribute to the complexity of the AP1 complex. For example, during development, AP1S1 and AP1S2 form AP1 complexes with divergent functions in neuronal endosome maturation, that is AP1S1 containing AP1, which binds to the ArfGAP1-Rabex-5 complex, is inhibited by AP1S2 containing AP1 complex, thereby controlling its level [Citation48]. AP1S1 knockdown is embryonically lethal, and in cells, AP1S1 knock-down result in failure of recycling in TGN from endosomes [Citation38] and failure of the targeting low-density lipoprotein-derived cholesterol and glycosphingolipids to late endosome/lysosome [Citation49]. On the other hand, a recent study using bioinformatics have shown that AP1S1 expression is related to glioblastoma multiforme (GBM) pathogenesis [Citation50] and is hypo-methylated and upregulated in acute myeloid leukemia (AML) [Citation51] indicating that its upregulation might have pathological effects in cancer. Similarly, to AP1S1 deficiency, AP1S2 deficiency is linked to failure in the recycling of endosome vesicles and has severe developmental effects [Citation52]. Further, similarly to AP1S1, the main function of AP1S2, which is highly expressed in neurons, is to recognize similar cytosolic domains of proteins for intracellular transport to endosome/lysosome, and exhibit some specificity, for example, sortilin [Citation53]. As AP1S2 is linked to the X-chromosome, its defect is reported to be the cause of X-chromosome-linked mental disease in patients [Citation54]. In addition, AP1S2 downregulation inhibits cancer cell mobility in melanoma cells, suggesting that its regulation might be a target for treatment [Citation55,Citation56]. Mutations in AP1S3 cause pustular psoriasis, an immune disease in skin tissue, via the translocation of Toll-like receptor-3 [Citation57] and disruption of keratinocyte autophagy, resulting in the upregulation of interleukin (IL)-1 signaling [Citation58]. Finally, similarly to that of AP1S2, the upregulation of AP1S3 was found to be linked with cancer cell aggressiveness among breast cancer patients and could be a target for small RNA silencing [Citation59], which highlights its potential as a target for cancer treatment.
3. AP2 complex
AP2 is primarily involved in endocytosis from the plasma membrane, and similarly to AP1, its depletion is embryonic lethal in mouse model, showing it is necessary for proper development [Citation60]. However, AP2 is not necessary for clathrin-dependent endocytosis, as it is partially redundant with other clathrin-associated complexes in the endocytosis pathway ()[Citation61,Citation62]. AP2 complex has relatively fewer isoforms compared to AP1 and AP3, with only two isoforms of its large α subunit (AP2A1 and AP2A2), while the other subunits are unique [the other large β subunit (AP2B1), medium μ subunit (AP2M1), and small σ subunit (AP2S2)]. AP2A1 recognizes and binds to cargo proteins, clathrin, and accessory proteins, and thus plays a key role in the clathrin-dependent endocytosis process [Citation63–65]. AP2A1 was reported to be involved in neural cell development via binding to m-Numb [Citation66], in hematopoietic stem cell differentiation [Citation67], in the synaptic vesicle cycle pathway and related neurodegenerative diseases [Citation68], in cell defense (e.g., in the antifungal defense mechanism by binding to MYO1F) [Citation69] and in inhibition of HIV genomic incorporation [Citation70]. However, this clathrin endocytosis-related subunit can also serve as a target of viral infection, for example, by being hijacked by HAdV-D37 [Citation71] and HEV71 [Citation72] viruses. Interestingly, in A. thaliana, endocytosis was found to involve AP2A1, whereas the uniquely known AP2 middle-sized subunit, AP2M1, was not required, suggesting that other isoforms of AP2 are yet to be discovered [Citation73]. AP2A1 is a candidate biomarker gene among patients with ovarian cancer [Citation74], pediatric medulloblastoma [Citation75], and Alzheimer’s disease [Citation76,Citation77], suggesting that its aberrant expression can be pathological. Both AP2A1 and AP2A2 were reported to confer resistance to erlotinib, an anti-cancer drug, in lung cancer cells [Citation78]; interact with Shc, an SH2-containing proto-oncogene involved in growth factor signaling in mammalian cancer cells [Citation79]; and function in hemopoietic stem cell development [Citation80]. AP2A2 has a partially non-redundant function with AP2A1 owing to its unique peroxisome proliferator-activated receptor (PPAR) α responding domain, which increases fatty acid oxidization in the adipose tissue [Citation81] by interacting with syndecan-2, a mediator of extracellular matrix (ECM) signaling in cardiac tissue [Citation82]. Listeria monocytogenes bacteria-induced cytotoxicity was reportedly inhibited by the binding of bacterial proteins to AP2A2 in a pathway that enable bacterial growth in host cells [Citation83]. Interestingly, unlike AP2A1, no reported correlation was found between AP2A2 and cancer. However, with or without AP2A1, AP2A2 aberrant expression is linked to Alzheimer’s disease [Citation76,Citation77,Citation84–87] and is specifically linked to obesity [Citation81,Citation88], coronary disease [Citation89], chronic bronchitis [Citation90,Citation91] and nicotine withdrawal [Citation92], suggesting that α isoforms may have distinct roles in cells. AP2B1, the other large β subunit of the AP2 subunit, shares common binding proteins with the α subunits. For example, both AP2B1 and AP2A1 bind to full-length IL-33 [Citation93] and both were reported to be involved in common functions, such as synaptic vesicle cycle [Citation9,Citation68,Citation94] and cell development [Citation95,Citation96]. Moreover, the pathological consequences of AP2B1 mutation are similar to those of the AP2α subunit mutation and has been shown to have various effects in neural tissue. In fact, AP2B1 affects dendrite cell morphology by controlling the mTOR pathway [Citation97] through its action as a biosignature upon antidepressant treatment [Citation98], binding to Dynamin-1, and triggering the process of autophagy, which inhibits dementia [Citation99] and is putatively linked to depressive disorder [Citation100]. AP2B1 is also linked to Alzheimer’s disease [Citation101], which explains the importance of the AP2 complex in controlling autophagy and lysosomal protein degradation that regulate this neuronal degenerative disease. AP2B1 was shown to interact with ANO7 in prostate cancer patients, putatively promoting cancer progression [Citation102], increasing chemotherapy resistance in ovarian cancer patients [Citation103], upregulating cancer cell metastasis ability [Citation104], while putatively acting as an anti-cancer agent in triple-negative breast cancer patients [Citation105]. Furthermore, AP2B1 is upregulated as a putative defense mechanism against heavy metals [Citation106] while is identified as a target of influenza A virus promoting virus internalization [Citation107]. AP2M1, the only identified AP2 middle-sized subunit, controls complex-cargo protein affinity via phosphorylation [Citation108,Citation109], a characteristic that is exploited by viruses such as rabies (RABV) [Citation110], dengue (DENV) [Citation111] and hepatitis C [Citation112] viruses. Moreover, a recent report showed that a wide spectrum of viruses, including coronaviruses , that recently garnered increased attention, have a AP2M1 binding motif YxxØ for viral infection [Citation113]. In addition to other subunits, AP2M1 is important in ECM development [Citation114] and its aberrant expression has pathological consequences, such as the development of autism [Citation115] and Parkinson’s disease, in neural tissues [Citation116]. The role of AP2M1 in signal transduction has been reported in several studies. In fact, its endocytosis function is required for maintaining dendrite cell polarity [Citation117]; restraining signals, such as insulin signal responding glucagon-like peptide receptor, by downregulating receptor presentation on the cell surface [Citation118]; or controlling endosomal trafficking of transcription factors, such as EGFR, to the nucleus [Citation119]. These functions have an effect on cancer cells, as AP2M1 regulates insulin-like growth factor-1 receptor (IGF1R) in prostate cancer [Citation120] and EGFR internalization in bladder cancer [Citation121]. AP2M1 is also related to chemoresistance in AML [Citation122], is upregulated in adenoid cystic carcinoma and mucoepidermoid carcinoma [Citation123], and is a putative biomarker for hepatocellular carcinoma [Citation124]. On the other hand, AP2M1 exhibits anti-tumor activity by inhibiting autophagic activity in acute lymphoblastic leukemia [Citation125]. Controlling the expression of AP2M1 was proposed as a potentially effective treatment for cystic fibrosis, a genetic disorder caused by defective cellular trafficking [Citation126], demonstrating its potential as a target gene for diverse diseases. Finally, the small subunit AP2S1 was shown to be critical for cellular calcium-sensing receptor activity [Citation127], and AP2S1 mutations were reported to result in familial hypocalciuric hypercalcemia type 3, which causes high calcium levels in bloodstream [Citation128]. Most studies involving AP2S1 were linked to this genetic disease [Citation129–135], with one exception linking AP2S1 to obesity [Citation88], which suggests that its major function is in calcium-sensing pathway.
4. AP3 complex
AP3 main function is known to be trafficking between endosomes, lysosomes, lysosomes related organelles (LROs) and synaptic vesicles () [Citation136]. It was first identified in rats with two isoforms of the medium μ subunit: one was ubiquitously expressed (μ3A), while the other was specifically expressed in brain tissue (μ3B) [Citation16]. The large β subunit is also expressed in two isoforms, with one ubiquitously expressed (β3A) [Citation137] and the other being neural cell-specific (β-NAP or β3B) [Citation138]. The two identified AP3 small σ subunits are both ubiquitously expressed [Citation139] and interact with the large subunit δ ear domain, leading to a conformational change in the AP3 complex that regulates its recruitment to the membrane [Citation140]. Unlike AP1 and AP2 knock-down [Citation60,Citation141], AP3 knockdown is not embryonically lethal, with the most prominent phenotype being abnormal LRO formation [Citation142]. Although its knockout is not deleterious, AP3 plays an important role in development by affecting Notch signaling [Citation143]. In cells, AP3 was shown to be involved in melanocyte trafficking [Citation144], immune defense by CD1b trafficking [Citation145] and in movement of lytic granules of T cells [Citation146]. Most of AP3 variants in human patients are linked to Hermansky-Pudlak disorder type 2 (HPS2) [Citation147–150], in addition to be involved in other mental disorders such as schizophrenia [Citation151]. The AP3 budding scission system is dynamin 2 independent [Citation152] and is involved in the trafficking of specific proteins such as LAMPI/II [Citation153]. It specifically interacts with the HIV viral Gag protein in endosomes [Citation154]. AP3D1 was first identified from the expressed sequence tag (EST) database through its homology wit the AP1 and AP2 subunits [Citation155]. AP3D1 mutation in its Drosophila ortholog causes defective eye pigmentation phenotype due to dislocation of pigment granules [Citation156]. In humans, AP3D1 has been identified as a candidate gene for chondrogenesis [Citation157]. As the only known δ subunit of AP3, variation in human AP3D1 is usually linked to HPS2, with few examples that have variable phenotypes; for instance, HPS10, which shows symptoms reminiscent of HPS2, in addition to neurological defects [Citation158,Citation159]. The mouse model for HPS, the ‘mocha’ mouse, has a defect in the mouse AP3 δ subunit [Citation160]. This mutation is known to affect retinal cell development [Citation161] and hippocampal LRO sorting [Citation160,Citation162]. In addition, aberrant AP3D1 expression is associated with high risks for several cancers [Citation163,Citation164], heart attack [Citation165] and may be involved in chronic obstructive pulmonary disease through its binding to TGF bta2 [Citation166]. The AP3D1 subunit is also a target for bovine leukemia virus gp51 [Citation167], is involved in HIV viral particles [Citation168] and surface coat proteins [Citation169] release. These findings imply that similar to AP1 and AP2, AP3D1 is also targeted by viruses. AP3D1 has a positive effect in cancer, as it inhibits chemotherapy resistance in colorectal cells by maintaining signal integrity in tumor cells by preventing IFNGR1 lysosomal sorting [Citation170]. Additionally, AP3D1 and all AP3 subunits are downregulated in cervical carcinoma [Citation171], which suggests that AP3 might play a role in the cell-inherent anti-cancer pathway. The ubiquitously expressed AP3B1 [Citation137] is involved in lysosome positioning, as demonstrated by its knockdown [Citation172]. Its deficiency has been reported to cause the deletion or decrease of the whole AP3 complex, leading to HPS2 [Citation148,Citation173–175]. Further, its deletion mouse model, the ‘pearl’ mouse, displays HPS2, night blindness [Citation176], deficient pigmentation in eyes [Citation177] and uteri hypoxia [Citation178]. In addition, pearl mice cells show defects in platelet granules [Citation179]. In human, AP3B1 variants were detected in patients with bleeding diathesis, presumably caused by platelet disorder [Citation180]. AP3B1 also interacts with multidrug resistance protein 4 (MRP4) [Citation181] and is involved in vesicle-associated membrane protein 8 (VAMP8) trafficking and Weibel-Palade body maturation [Citation182] which are crucial processes in platelet function. AP3B1 mutation was reported to cause neutropenia, a deficiency in white blood cells in humans, as well as canine cyclic hematopoiesis in dogs; however, this mutation only resulted in decreased hematopoietic progenitor and decreased granulocyte mobilization upon signaling in pearl mice [Citation183]. Moreover, although hemophagocytic lymphohistiocytosis (HLH) patients, which show excessive immune activation, are reported to have mutations in AP3B1 [Citation184,Citation185], HPS patients or pearl mouse models rarely display HLH symptoms [Citation186,Citation187], indicating that AP3B1 variation might have different effects depending on the mutation and/or species affected. Recently, bioinformatic analysis revealed the putative role of AP3B1 in amyotrophic lateral sclerosis pathogenesis, a neurodegenerative disease that affects muscle movement [Citation188] Alveolar epithelial cells with defective AP3B1 were found to display abnormal mitochondrial formation [Citation189], while lung tissues showed increased matrix metalloproteinase activity [Citation190], suggesting a potential role of AP3B1 in lung tissue. AP3B1 mutation was found to affect natural killer (NK) and NKT cell granule-associated protein release, which presumably explains the susceptibility to infection and lymphoma among HPS2 patients [Citation191]. The cells of HPS patients exhibit less HIV-1 viral particle release [Citation192] presumably due to the interaction of AP3B1 with Kif3A, which is modulated by IP7-mediated pyrophosphorylation [Citation193]. AP3B1 interacts with Nipah, Hendra [Citation194], and SARS-CoV2 virus protein [Citation193], which suggests that its wild-type version is a target of viral infection. AP3B1 expression is controlled by the anti-tumor microRNA-9 in breast cancer [Citation195] and its copy number increases in patients with neurofibromatis type I, an autosomal dominant disorder causing tumor development [Citation196]. Such findings indicate the potential of AP3B1 as a target in cancer treatment. The neuron-specific AP3B2 [Citation139] was first identified as β-NAP [Citation138]. While both ubiquitously expressed AP3B1 and brain specific AP3B2 are expressed in the brain, their roles are non-redundant [Citation197]. AP3B2 is reported to be involved in neural development [Citation198] and proteins sorting of neural lysosomes and of lysosome-related organelles [Citation199]. Further, aberrant AP3B2 was reported to be involved in neural development disorder [Citation200], autoimmune cerebellar ataxia, various extracerebellar symptoms [Citation201] and recently in Alzheimer’s disease [Citation202]. Interestingly, while AP3B2 is reported to be neural specific, it was identified in a screening related to sporadic Hirschsprung disease, a colon developmental disease [Citation203]. Its abberant expression in other tissues was observed as it is a putative biomarker for rectal carcinoma [Citation204]; and possibly involved in skin tropism of melanoma cells [Citation205]. Such findings suggest that AP3B2 might play a developmental role in non-neural tissues, and that its aberrant expression in other tissues might be involved in cancer, putatively in the metastasis mechanism. Only few reports have been published on the two middle-sized AP3 subunits, AP3M1 and AP3M2. In addition to the AP1 and AP2 middle-sized subunits, the ubiquitously expressed AP3M1 was reported to be a target for HIV infection [Citation206]. Neural tissue-specific AP3M2 affects IL-6 levels in astrocytes [Citation207] and clathrin-mediated endocytosis via its phosphatidylinositol (4,5)-bisphosphate binding domain [Citation208]. To our knowledge, no study has reported a significant correlation between the AP3B2 variant and neurological disease. AP3S1 is involved in melanocyte pigmentation [Citation209] and might control insulin receptor substrate-1 subcellular localization in adipocytes [Citation210]. In addition to the fusion mutations detected in colorectal cancer cell lines [Citation211], variants of AP3S2 are associated with diabetes [Citation212,Citation213], a high risk for liver cancer [Citation214] and cardiovascular disorders [Citation215].
5. AP4 complex
Although its exact functions are not well-defined, mutation of AP4 in various subunits leads to mental deficiency, including hereditary spastic paraplegia (HSP), mental disability, and Alzheimer’s disease [Citation216–220], which might be partially due to autophagy serving as a key process in neurons [Citation221] and axonal AGT9 sorting deficiency leading to deficient neuron development and maturation [Citation222]. In addition to its reported role in autophagy, it is also reported that AP4 is involved in basolateral transport of epithelial cells () [Citation22]. AP4E1 mutation has been reported to be involved in neural diseases, including cerebral palsy, HSP, and stuttering [Citation223–229]. In mouse testes, it was shown that along with AP1S3, it increases upon fluoride-induced stress [Citation230], and in Arabidopsis, to be involved in the hypersensitive cell death pathway [Citation231]. Along with the fact that it is ubiquitously detected [Citation6,Citation7], such findings suggest that it might have an additional role in other tissues. AP4B1, the other large subunit of AP4, is known to have a binding domain for Tepsin in its C terminal [Citation21]. However, its deletion is not sufficient to abolish Tepsin binding to AP4, suggesting that an additional binding domain exists in the AP4 complex [Citation232]. Similarly to AP4E1, AP4 mutation was reported to affect neural cells, as variants were identified in HSP patients [Citation233–236], and might cause intellectual disability [Citation237]. Although AP4 lacks clathrin-binding domain, AP4M1 was reported to be localized in TGN with clathrin vesicles [Citation19], which might be partially explained by the presence of a portion of AP4 co-localized with AP1 in the kidney epithelial cell line MDCK [Citation22]. Interestingly, AP4M1 is involved in basolateral vesicle transport and co-localizes with cation-independent mannose 6-phosphate receptor (CI-MPR), but not with the transferrin receptor lysosomal-associated membrane protein-2 (LAMP-2) in MDCK cells [Citation22]. In HeLa cells, AP4M1 reportedly interacts with LAMP-2 during intracellular transport between endosomes and lysosomes independent of the plasma membrane [Citation238], suggesting that AP4 is involved in independent intracellular transport mechanisms based on their partner proteins. AP4M1 is upregulated and redistributed to neuronal axons under oxygen-glucose deprivation stress [Citation239] and variants affect ocular development [Citation240], are involved in Alzheimer’s disease [Citation241,Citation242], neurodegeneration with brain iron accumulation [Citation243], cerebral palsy [Citation244], and congenital spastic tetraplegia [Citation245]. Knockout of the small subunit AP4S1 in zebrafish resulted in symptoms such as spastic paraplegia 52 [Citation246] and affected immune response in frog [Citation247]. Furthermore, the AP4S1 variant caused iron accumulation and neurodegeneration in human patients [Citation248].
6. AP5 complex
AP5 is the most recently identified adaptin complex. Although AP5M1 has a low expression and homology to other subunits, it possess an unique μ homology domain, which led to the discovery of AP5 that is localized in late endosomes and lysosomes and putatively involved in late endosome retrieval ()[Citation9]. Similarly to those in AP3 and AP4, defects in AP5 have also been linked to HSP; two of its accessory proteins, SPG11 and SPG11, were identified in HSP cells [Citation249] and were reported to be involved in localization and/or AP5 stability [Citation250]. Although further analysis is required to define the function and location of AP5, one of its roles is to recycle lysosome proteins to the Golgi complex [Citation251]. AP5Z1 variants were reportedly detected in HSP patients [Citation252–254] and were suggested to be partially the cause of mitochondrial defects in axons [Citation255]. In addition, knockout of AP5Z1 causes defection in theretrieval of CI-MPR, GOLIM4, and GOLM1 proteins from endosomes [Citation256]. Moreover, defects in AP5Z1 cause additional symptoms in brain and skin tissue due to abnormal lysosomal material, suggesting that AP5 dysregulation can result in other diseases related to deficient lysosome recycling [Citation257]. In addition, although the specific mechanisms are yet to be discovered, AP5Z1 and the downregulation of AP5M1 were observed in patients with sporadic spastic paraplegia [Citation254] while AP5B1 variants were identified in patients with allergic disease [Citation258–262]. AP5M1 was reported to be involved in cell apoptosis pathways, which are controlled by mitochondrial protein [Citation263–269], suggesting that lysosome recycling might have a broad effect or the AP5 complex might have additional functions in cells.
7. Conclusion
Understanding and controlling intracellular trafficking can be crucialaspects in disease treatments. For example, intracellular trafficking can be used by viruses to mediate their entry into host cells or promote their replication [Citation270], a deficiency in intracellular transport can cause severe genetic diseases [Citation271] while it is also putatively a target for treating cancer [Citation272]. Moreover, endocytosis is an important feature that should be considered in drug delivery [Citation273]. Endocytosis affects the presentation of crucial receptor proteins at the cell surface [Citation274]. Indeed, an in vitro study using GBM cell lines revealed that the expression of the μ subunit of AP5 controlled GBM cell sensitivity to Tumor Necrosis Factor Related Apoptosis-Inducing Ligand, suggesting that AP5 might be involved in the apoptosis signaling pathway triggered by anti-cancer drugs [Citation263]. As demonstrated in this review, extensive work has already been carried out by several groups in this field. Clathrin dependent protein trafficking in the TGN was not only indispensable during development, as it was shown by AP1 knockout mutant, but it was also involved in cancer cell proliferation, while its precise mechanism has yet to be investigated. Deficient AP1 subunits were also linked to defection in neuron cells and pigment misallocation in skin cells, presumably because of aberrant protein sorting in TGN. Impairment of the also developmental necessary AP2, which major role is to control endocytosis from plasma membrane, was reported in diseases with a broad spectrum by its involvement in mechanisms such as endocytic cargo transportion, ECM communication and displaying of receptors at the membrane surface. Further studies will be needed to elucidate involved mechanisms as it was shown to be controlling yet uncurable diseases, such Alzheimer’s disease. Patients case studies has also shown that AP2 was also hijacked by pathogen for infection or cancer cells for controlling receptor display, suggesting that its study can potentially improve further treatments. Interestingly, reports show that some of its tissue specific subunits are expressed in unexpected tissues and/or aberrantly expressed in pathological tissue, suggesting that yet additional roles for AP2 and consequences in diseases are yet to be identified. AP3, which function is to sort proteins in lysosomes, LRO and synaptic vesicles recycling in neural cells is notably linked to HPS2, while also being reported to affect various diseases such as Alzheimer’s disease, autoimmune diseases and melanoma, reflecting the variable impact of aberrant sorting in organelles. AP4 and AP5 precise mechanisms are yet to be defined and therefore pathways involved in known diseases linked to their deficiency such as HSP or Alzheimer’s disease is yet to be identified. Adaptin-related intracellular trafficking pathways are complex and diverse, which can be interfered with based on the few known binding domains and corresponding partner proteins listed in . Although all known partner proteins are not listed herein, more partner proteins and mechanism are yet to be identified, particularly in the less known AP4 and AP5 complexes. It is notable that while diseases or genetic disorders linked to APs deficiency are various, reflecting the multifaceted role of selective vesicular sorting and transport have in cell function, most of the case studies results are from screening for candidate genes from a list of genes known to be involved to vesicle intracellular trafficking. Hence, most APs variants were detected in patient with diseases such as neuronal disorders, melanosomes missorting or in cohorts with high virus infection susceptibility. Admittedly, proper vesicular trafficking and protein sorting might be specifically critical in those conditions. For example, as neuron cell major function is to receive and transmit signals, mislocation of proteins in synaptic vesicles might result in severe phenotypes, which can be reflected on the fact that all APs variants are shown to be detected in case studies related to neuron defect disease or disorder. However, as mentioned above, all isoforms expressed and binding partners are yet to be discovered, especially for the less well studied AP4 and 5. Moreover, while APs link to diseases and disorders has been identified, their mechanisms have only been partially explained, especially for its role in cancer cell. This suggest more APs linked diseases/disorders are to yet be discovered and show their potential as biomarkers and/or putative target for treatments.
Table 2. Motifs and binding partners of AP subunits
Author contributions
Conceptualization, J.S. and J.-W.O; methodology, J.S. and A.N.; validation, J.S., J.‐W.O.; formal analysis, J.S. and A. N.; investigation, J.S. and A. N.; resources, J.S. and A. N.; data curation, J.S.; writing—original draft preparation, J.S.; writing—review and editing, J.S., J.‐W.O.; supervision, J.‐W.O.; project administration, J.S. and A. N.; funding acquisition, J.S., and J.‐W.O. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
The author(s) declare no conflict of interest.
Additional information
Funding
References
- Pearse BM. Coated vesicles from pig brain: purification and biochemical characterization. J Mol Biol. 1975;97(1):93–98.
- Lee C, Goldberg J. Structure of coatomer cage proteins and the relationship among COPI, COPII, and clathrin vesicle coats. Cell. 2010;142(1):123–132.
- Jarousse N, Kelly RB. Selective inhibition of adaptor complex-mediated vesiculation. Traffic. 2000;1(5):378–384.
- Schledzewski K, Brinkmann H, Mendel RR. Phylogenetic analysis of components of the eukaryotic vesicle transport system reveals a common origin of adaptor protein complexes 1, 2, and 3 and the F subcomplex of the coatomer COPI. J Mol Evol. 1999;48(6):770–778.
- Boehm M, Bonifacino JS. Adaptins: the final recount. Mol Biol Cell. 2001;12(10):2907–2920.
- Dell’Angelica EC, Mullins C, Bonifacino JS. AP-4, a novel protein complex related to clathrin adaptors. J Biol Chem. 1999;274(11):7278–7285.
- Hirst J, Bright NA, Rous B, et al. Characterization of a fourth adaptor-related protein complex. Mol Biol Cell. 1999;10(8):2787–2802.
- Robinson MS, Bonifacino JS. Adaptor-related proteins. Curr Opin Cell Biol. 2001;13(4):444–453.
- Hirst J, Barlow LD, Francisco GC, et al. The fifth adaptor protein complex. PLoS Biol. 2011;9(10):e1001170.
- Hirst J, Schlacht A, Norcott JP, et al. Characterization of TSET, an ancient and widespread membrane trafficking complex. Elife. 2014;3:e02866.
- Edeling MA, Smith C, Owen D. Life of a clathrin coat: insights from clathrin and AP structures. Nat Rev Mol Cell Biol. 2006;7(1):32–44.
- Dell’Angelica EC, Klumperman J, Stoorvogel W, et al. Association of the AP-3 adaptor complex with clathrin. Science. 1998;280(5362):431–434.
- Simpson F, Bright NA, West MA, et al. A novel adaptor-related protein complex. J Cell Biol. 1996;133(4):749–760.
- Llinares E, Barry AO, Andre B. The AP-3 adaptor complex mediates sorting of yeast and mammalian PQ-loop-family basic amino acid transporters to the vacuolar/lysosomal membrane. Sci Rep. 2015;5(1):16665.
- Peden AA, Oorschot V, Hesser BA, et al. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol. 2004;164(7):1065–1076.
- Pevsner J, Volknandt W, Wong BR, et al. Two rat homologs of clathrin-associated adaptor proteins. Gene. 1994;146(2):279–283.
- Shi G, Faundez V, Roos J, et al. Neuroendocrine synaptic vesicles are formed in vitro by both clathrin-dependent and clathrin-independent pathways. J Cell Biol. 1998;143(4):947–955.
- Itzhak DN, Tyanova S, Cox J, et al. Global, quantitative and dynamic mapping of protein subcellular localization. Elife. 2016;5. DOI:10.7554/eLife.16950.
- Barois N, Bakke O. The adaptor protein AP-4 as a component of the clathrin coat machinery: a morphological study. Biochem J. 2005;385(2):503–510.
- Davies AK, Itzhak DN, Edgar JR, et al. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat Commun. 2018;9(1):3958.
- Borner GH, Antrobus R, Hirst J, et al. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol. 2012;197(1):141–160.
- Simmen T, Honing S, Icking A, et al. AP-4 binds basolateral signals and participates in basolateral sorting in epithelial MDCK cells. Nat Cell Biol. 2002;4(2):154–159.
- Khundadze M, Ribaudo F, Hussain A, et al. A mouse model for SPG48 reveals a block of autophagic flux upon disruption of adaptor protein complex five. Neurobiol Dis. 2019;127:419–431.
- Nakatsu F, Hase K, Ohno H. The role of the clathrin adaptor AP-1: polarized sorting and beyond. Membranes (Basel). 2014;4(4):747–763.
- Johnson KR, Gagnon LH, Chang B. A hypomorphic mutation of the gamma-1 adaptin gene (Ap1g1) causes inner ear, retina, thyroid, and testes abnormalities in mice. Mamm Genome. 2016;27(5–6):200–212.
- Usmani MA, Ahmed ZM, Magini P, et al. De novo and bi-allelic variants in AP1G1 cause neurodevelopmental disorder with developmental delay, intellectual disability, and epilepsy. Am J Hum Genet. 2021;108(7):1330–1341.
- Zizioli D, Forlanelli E, Guarienti M, et al. Characterization of the AP-1 mu1A and mu1B adaptins in zebrafish (Danio rerio). Dev Dyn. 2010;239(9):2404–2412.
- Lewin DA, Sheff D, Ooi CE, et al. Cloning, expression, and localization of a novel gamma-adaptin-like molecule. FEBS Lett. 1998;435(2–3):263–268.
- Zizioli D, Geumann C, Kratzke M, et al. gamma2 and gamma1AP-1 complexes: different essential functions and regulatory mechanisms in clathrin-dependent protein sorting. Eur J Cell Biol. 2017;96(4):356–368.
- Grant RC, Denroche RE, Borgida A, et al. Exome-wide association study of pancreatic cancer risk. Gastroenterology. 2018;154(3):719–722 e713.
- Aouizerat BE, Vittinghoff E, Musone SL, et al. GWAS for discovery and replication of genetic loci associated with sudden cardiac arrest in patients with coronary artery disease. BMC Cardiovasc Disord. 2011;11(1):29.
- Goujon C, Rebendenne A, Roy P, et al. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal HCoVs. Res Sq. 2021. DOI:10.21203/rs.3.rs-555275/v1.
- Tavares LA, Da Silva EM, Da Silva-januario ME, et al. CD4 downregulation by the HIV-1 protein Nef reveals distinct roles for the gamma1 and gamma2 subunits of the AP-1 complex in protein trafficking. J Cell Sci. 2017;130:429–443.
- Yun WK, Hu YM, Zhao CB, et al. HCP5 promotes colon cancer development by activating AP1G1 via PI3K/AKT pathway. Eur Rev Med Pharmacol Sci. 2019;23:2786–2793.
- Tao X, Lu Y, Qiu S, et al. AP1G1 is involved in cetuximab-mediated downregulation of ASCT2-EGFR complex and sensitization of human head and neck squamous cell carcinoma cells to ROS-induced apoptosis. Cancer Lett. 2017;408:33–42.
- Ohno H, Tomemori T, Nakatsu F, et al. μ1B, a novel adaptor medium chain expressed in polarized epithelial cells. FEBS Lett. 1999;449(2–3):215–220.
- Mostov K, ter Beest MB, Chapin SJ. Catch the mu1B train to the basolateral surface. Cell. 1999;99(2):121–122.
- Meyer C, Zizioli D, Lausmann S, et al. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 2000;19(10):2193–2203.
- Peden AA, Rudge RE, Lui WW, et al. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol. 2002;156(2):327–336.
- Moreira C, Batista CM, Fernandes JC, et al. Knockout of the gamma subunit of the AP-1 adaptor complex in the human parasite Trypanosoma cruzi impairs infectivity and differentiation and prevents the maturation and targeting of the major protease cruzipain. PLoS One. 2017;12(7):e0179615.
- Navarro Negredo P, Edgar JR, Wrobel AG, et al. Contribution of the clathrin adaptor AP-1 subunit micro1 to acidic cluster protein sorting. J Cell Biol. 2017;216(9):2927–2943.
- Teh OK, Shimono Y, Shirakawa M, et al. The AP-1 mu adaptin is required for KNOLLE localization at the cell plate to mediate cytokinesis in Arabidopsis. Plant Cell Physiol. 2013;54(6):838–847.
- Tafesse FG, Guimaraes CP, Maruyama T, et al. GPR107, a G-protein-coupled receptor essential for intoxication by Pseudomonas aeruginosa exotoxin A, localizes to the Golgi and is cleaved by furin. J Biol Chem. 2014;289(35):24005–24018.
- Ohno H, Fournier MC, Poy G, et al. Structural determinants of interaction of tyrosine-based sorting signals with the adaptor medium chains. J Biol Chem. 1996;271(46):29009–29015.
- Kou Y, Yan X, Liu Q, et al. HBV upregulates AP-1 complex subunit mu-1 expression via the JNK pathway to promote proliferation of liver cancer cells. Oncol Lett. 2019;18:456–464.
- Rojas LK, Trilla-Fuertes L, Gamez-Pozo A, et al. Proteomics characterisation of central nervous system metastasis biomarkers in triple negative breast cancer. Ecancermedicalscience. 2019;13:891.
- Kell MJ, Ang SF, Pigati L, et al. Novel function for AP-1B during cell migration. Mol Biol Cell. 2020;31(22):2475–2493.
- Candiello E, Kratzke M, Wenzel D, et al. AP-1/sigma1A and AP-1/sigma1B adaptor-proteins differentially regulate neuronal early endosome maturation via the Rab5/Vps34-pathway. Sci Rep. 2016;6(1):29950.
- Poirier S, Mayer G, Murphy SR, et al. The cytosolic adaptor AP-1A is essential for the trafficking and function of Niemann-pick type C proteins. Traffic. 2013;14(4):458–469.
- Alshabi AM, Vastrad B, Shaikh IA, et al. Identification of crucial candidate genes and pathways in glioblastoma multiform by bioinformatics analysis. Biomolecules. 2019;9(5):201.
- Mallik S, Zhao Z. Towards integrated oncogenic marker recognition through mutual information-based statistically significant feature extraction: an association rule mining based study on cancer expression and methylation profiles. Quant Biol. 2017;5(4):302–327.
- Glyvuk N, Tsytsyura Y, Geumann C, et al. AP-1/sigma1B-adaptin mediates endosomal synaptic vesicle recycling, learning and memory. EMBO J. 2010;29(8):1318–1330.
- Baltes J, Larsen JV, Radhakrishnan K, et al. sigma1B adaptin regulates adipogenesis by mediating the sorting of sortilin in adipose tissue. J Cell Sci. 2014;127:3477–3487.
- Borck G, Molla-Herman A, Boddaert N, et al. Clinical, cellular, and neuropathological consequences of AP1S2 mutations: further delineation of a recognizable X-linked mental retardation syndrome. Hum Mutat. 2008;29(7):966–974.
- Vitiello M, Tuccoli A, D’Aurizio R, et al. Context-dependent miR-204 and miR-211 affect the biological properties of amelanotic and melanotic melanoma cells. Oncotarget. 2017;8(15):25395–25417.
- Luan W, Ding Y, Ma S, et al. Long noncoding RNA LINC00518 acts as a competing endogenous RNA to promote the metastasis of malignant melanoma via miR-204-5p/AP1S2 axis. Cell Death Dis. 2019;10(11):855.
- Setta-Kaffetzi N, Simpson MA, Navarini AA, et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am J Hum Genet. 2014;94(5):790–797.
- Mahil SK, Twelves S, Farkas K, et al. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J Invest Dermatol. 2016;136(11):2251–2259.
- Toda H, Kurozumi S, Kijima Y, et al. Molecular pathogenesis of triple-negative breast cancer based on microRNA expression signatures: antitumor miR-204-5p targets AP1S3. J Hum Genet. 2018;63(12):1197–1210.
- Mitsunari T, Nakatsu F, Shioda N, et al. Clathrin adaptor AP-2 is essential for early embryonal development. Mol Cell Biol. 2005;25(21):9318–9323.
- Motley A, Bright NA, Seaman MN, et al. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162(5):909–918.
- Rappoport JZ, Benmerah A, Simon SM. Analysis of the AP-2 adaptor complex and cargo during clathrin-mediated endocytosis. Traffic. 2005;6(7):539–547.
- Di Rubbo S, Irani NG, Kim SY, et al. The clathrin adaptor complex AP-2 mediates endocytosis of BRASSINOSTEROID INSENSITIVE1 in arabidopsis. Plant Cell. 2013;25(8):2986–2997.
- Kadlecova Z, Spielman SJ, Loerke D, et al. Regulation of clathrin-mediated endocytosis by hierarchical allosteric activation of AP2. J Cell Biol. 2017;216(1):167–179.
- Beattie EC, Howe CL, Wilde A, et al. NGF signals through TrkA to increase clathrin at the plasma membrane and enhance clathrin-mediated membrane trafficking. J Neurosci. 2000;20(19):7325–7333.
- Corallini S, Fera S, Grisanti L, et al. Expression of the adaptor protein m-Numb in mouse male germ cells. Reproduction. 2006;132(6):887–897.
- Han T, Yang CS, Chang KY, et al. Identification of novel genes and networks governing hematopoietic stem cell development. EMBO Rep. 2016;17(12):1814–1828.
- Wang C, Zhao D, Shah SZA, et al. Proteome analysis of potential synaptic vesicle cycle biomarkers in the cerebrospinal fluid of patients with sporadic Creutzfeldt-Jakob disease. Mol Neurobiol. 2017;54(7):5177–5191.
- Sun W, Ma X, Wang H, et al. MYO1F regulates antifungal immunity by regulating acetylation of microtubules. Proc Natl Acad Sci U S A. 2021;118(30):e2100230118.
- Sapsutthipas S, Kitagawa Y, Tokunaga K, et al. Viral factors involved in adapter-related protein complex 2 alpha 1 subunit-mediated regulation of human immunodeficiency virus type 1 replication. Southeast Asian J Trop Med Public Health. 2011;42:311–319.
- Lee JS, Mukherjee S, Lee JY, et al. Entry of epidemic keratoconjunctivitis-associated human adenovirus type 37 in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2020;61(10):50.
- Hussain KM, Leong KL, Ng MM, et al. The essential role of clathrin-mediated endocytosis in the infectious entry of human enterovirus 71. J Biol Chem. 2011;286(1):309–321.
- Johnson A, Vert G. Single event resolution of plant plasma membrane protein endocytosis by TIRF microscopy. Front Plant Sci. 2017;8:612.
- Jiao J, Jiang L, Luo Y. N6-methyladenosine-related RNA signature predicting the prognosis of ovarian cancer. Recent Pat Anticancer Drug Discov. 2021;16. DOI:10.2174/1574892816666210615164645.
- Huang P, Guo YD, Zhang HW. Identification of hub genes in pediatric medulloblastoma by multiple-microarray analysis. J Mol Neurosci. 2020;70(4):522–531.
- Raj T, Li YI, Wong G, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet. 2018;50(11):1584–1592.
- Tian Y, Chang JC, Fan EY, et al. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A. 2013;110(42):17071–17076.
- Saafan H, Foerster S, Parra-Guillen ZP, et al. Utilising the EGFR interactome to identify mechanisms of drug resistance in non-small cell lung cancer - Proof of concept towards a systems pharmacology approach. Eur J Pharm Sci. 2016;94:20–32.
- Okabayashi Y, Sugimoto Y, Totty NF, et al. Interaction of Shc with adaptor protein adaptins. J Biol Chem. 1996;271(9):5265–5269.
- Ting SB, Deneault E, Hope K, et al. Asymmetric segregation and self-renewal of hematopoietic stem and progenitor cells with endocytic Ap2a2. Blood. 2012;119(11):2510–2522.
- Montgomery MK, Bayliss J, Keenan S, et al. The role of Ap2a2 in PPARalpha-mediated regulation of lipolysis in adipose tissue. FASEB J. 2019;33(12):13267–13279.
- Mathiesen SB, Lunde M, Stensland M, et al. The cardiac syndecan-2 interactome. Front Cell Dev Biol. 2020;8:792.
- Chen C, Nguyen BN, Mitchell G, et al. The listeriolysin O PEST-like sequence Co-opts AP-2-mediated endocytosis to prevent plasma membrane damage during listeria infection. Cell Host Microbe. 2018;23(6):786–795 e785.
- Nelson PT, Fardo DW, Katsumata Y. The MUC6/AP2A2 Locus and Its Relevance to Alzheimer’s Disease: a Review. J Neuropathol Exp Neurol. 2020;79(6):568–584.
- Katsumata Y, Fardo DW, Bachstetter AD, et al. Alzheimer disease pathology-associated polymorphism in a complex variable number of tandem repeat region within the MUC6 gene, near the AP2A2 gene. J Neuropathol Exp Neurol. 2020;79(1):3–21.
- Espinosa A, Hernandez-Olasagarre B, Moreno-Grau S, et al. Exploring genetic associations of Alzheimer’s disease Loci with mild cognitive impairment neurocognitive endophenotypes. Front Aging Neurosci. 2018;10:340.
- Kawalia SB, Raschka T, Naz M, et al. Analytical strategy to prioritize Alzheimer’s disease candidate genes in gene regulatory networks using public expression data. J Alzheimers Dis. 2017;59(4):1237–1254.
- Platek T, Polus A, Goralska J, et al. DNA methylation microarrays identify epigenetically regulated lipid related genes in obese patients with hypercholesterolemia. Mol Med. 2020;26(1):93.
- Wang S, Ma Z, Zhang Y, et al. A genetic variant near adaptor-related protein complex 2 alpha 2 subunit gene is associated with coronary artery disease in a Chinese population. BMC Cardiovasc Disord. 2018;18(1):161.
- Lee JH, Cho MH, Hersh CP, et al. Genetic susceptibility for chronic bronchitis in chronic obstructive pulmonary disease. Respir Res. 2014;15(1):113.
- Arathimos R, Suderman M, Sharp GC, et al. Epigenome-wide association study of asthma and wheeze in childhood and adolescence. Clin Epigenetics. 2017;9(1):112.
- Hallfors J, Palviainen T, Surakka I, et al. Genome-wide association study in Finnish twins highlights the connection between nicotine addiction and neurotrophin signaling pathway. Addict Biol. 2019;24(3):549–561.
- Luzina IG, Fishelevich R, Hampton BS, et al. Full-length IL-33 regulates Smad3 phosphorylation and gene transcription in a distinctive AP2-dependent manner. Cell Immunol. 2020;357:104203.
- Balasubramanian D, Pearson JF, Kennedy MA. Gene expression effects of lithium and valproic acid in a serotonergic cell line. Physiol Genomics. 2019;51(2):43–50.
- Li B, Ma L, Zhang C, et al. Associations of genetic variants in endocytic trafficking of epidermal growth factor receptor super pathway with risk of nonsyndromic cleft lip with or without cleft palate. Mol Genet Genomic Med. 2018;6(6):1157–1167.
- Katikireddy KR, Schmedt T, Price MO, et al. Existence of neural crest-derived progenitor cells in normal and fuchs endothelial dystrophy corneal endothelium. Am J Pathol. 2016;186(10):2736–2750.
- Koscielny A, Malik AR, Liszewska E, et al. Adaptor complex 2 controls dendrite morphology via mTOR-dependent expression of GluA2. Mol Neurobiol. 2018;55(2):1590–1606.
- Mendez-David I, Boursier C, Domergue V, et al. Differential peripheral proteomic biosignature of fluoxetine response in a mouse model of anxiety/depression. Front Cell Neurosci. 2017;11:237.
- Diling C, Yinrui G, Longkai Q, et al. Circular RNA NF1-419 enhances autophagy to ameliorate senile dementia by binding Dynamin-1 and Adaptor protein 2 B1 in AD-like mice. Aging (Albany NY). 2019;11(24):12002–12031.
- Feng J, Zhou Q, Gao W, et al. Seeking for potential pathogenic genes of major depressive disorder in the gene expression Omnibus database. Asia Pac Psychiatry. 2020;12(1):e12379.
- Sjodin S, Brinkmalm G, Ohrfelt A, et al. Endo-lysosomal proteins and ubiquitin CSF concentrations in Alzheimer’s and Parkinson’s disease. Alzheimers Res Ther. 2019;11(1):82.
- Kaikkonen E, Takala A, Pursiheimo JP, et al. The interactome of the prostate-specific protein Anoctamin 7. Cancer Biomark. 2020;28(1):91–100.
- Cheng L, Lu W, Kulkarni B, et al. Analysis of chemotherapy response programs in ovarian cancers by the next-generation sequencing technologies. Gynecol Oncol. 2010;117(2):159–169.
- Pignatelli J, Jones MC, LaLonde DP, et al. Beta2-adaptin binds actopaxin and regulates cell spreading, migration and matrix degradation. PLoS One. 2012;7(10):e46228.
- Rangel R, Guzman-Rojas L, Kodama T, et al. Identification of new tumor suppressor genes in triple-negative breast cancer. Cancer Res. 2017;77(15):4089–4101.
- Lee JY, Tokumoto M, Fujiwara Y, et al. Gene expression analysis using DNA microarray in HK-2 human proximal tubular cells treated with cadmium. J Toxicol Sci. 2013;38(6):959–962.
- Wang G, Jiang L, Wang J, et al. The G protein-coupled receptor FFAR2 promotes internalization during influenza A virus entry. J Virol. 2020;94. DOI:10.1128/JVI.01707-19.
- Fingerhut A, von Figura K, Honing S. Binding of AP2 to sorting signals is modulated by AP2 phosphorylation. J Biol Chem. 2001;276(8):5476–5482.
- Ricotta D, Conner SD, Schmid SL, et al. Phosphorylation of the AP2 mu subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J Cell Biol. 2002;156(5):791–795.
- Wang C, Wang J, Shuai L, et al. The serine/threonine kinase AP2-associated kinase 1 plays an important role in rabies virus entry. Viruses. 2019;12(1):45.
- Pu S, Schor S, Karim M, et al. BIKE regulates dengue virus infection and is a cellular target for broad-spectrum antivirals. Antiviral Res. 2020;184:104966.
- Neveu G, Ziv-Av A, Barouch-Bentov R, et al. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J Virol. 2015;89(8):4387–4404.
- Yuan S, Chu H, Huang J, et al. Viruses harness YxxO motif to interact with host AP2M1 for replication a vulnerable broad-spectrum antiviral target. Sci Adv. 2020;6(35):eaba7910.
- Lee S, Lim GE, Kim YN, et al. AP2M1 supports TGF-beta signals to promote collagen expression by inhibiting caveolin expression. Int J Mol Sci. 2021;22. DOI:10.3390/ijms22041639.
- Kanduri C, Kantojarvi K, Salo PM, et al. The landscape of copy number variations in finnish families with autism spectrum disorders. Autism Res. 2016;9(1):9–16.
- Liu Q, Bautista-Gomez J, Higgins DA, et al. Dysregulation of the AP2M1 phosphorylation cycle by LRRK2 impairs endocytosis and leads to dopaminergic neurodegeneration. Sci Signal. 2021;14(693):eabg3555.
- Lee KH, Ho WK, Lee SH. Endocytosis of somatodendritic NCKX2 is regulated by Src family kinase-dependent tyrosine phosphorylation. Front Cell Neurosci. 2013;7:14.
- Huang X, Dai FF, Gaisano G, et al. The identification of novel proteins that interact with the GLP-1 receptor and restrain its activity. Mol Endocrinol. 2013;27(9):1550–1563.
- Mikula M, Skrzypczak M, Goryca K, et al. Genome-wide co-localization of active EGFR and downstream ERK pathway kinases mirrors mitogen-inducible RNA polymerase 2 genomic occupancy. Nucleic Acids Res. 2016;44:10150–10164.
- Meisel Sharon S, Pozniak Y, Geiger T, et al. TMPRSS2-ERG fusion protein regulates insulin-like growth factor-1 receptor (IGF1R) gene expression in prostate cancer: involvement of transcription factor Sp1. Oncotarget. 2016;7(32):51375–51392.
- Song X, Li M, Wu W, et al. Regulation of BMP2K in AP2M1-mediated EGFR internalization during the development of gallbladder cancer. Signal Transduct Target Ther. 2020;5(1):154.
- Yu DH, Chen C, Liu XP, et al. Dysregulation of miR-138-5p/RPS6KA1-AP2M1 is associated with poor prognosis in AML. Front Cell Dev Biol. 2021;9:641629.
- Wu CC, Li H, Xiao Y, et al. Expression levels of SIX1, ME2, and AP2M1 in adenoid cystic carcinoma and mucoepidermoid carcinoma. Oral Dis. 2020;26(8):1687–1695.
- Cho SH, Pak K, Jeong DC, et al. The AP2M1 gene expression is a promising biomarker for predicting survival of patients with hepatocellular carcinoma. J Cell Biochem. 2019;120(3):4140–4146.
- Shi C, Lan W, Wang Z, et al. Alantolactone inhibits cell autophagy and promotes apoptosis via AP2M1 in acute lymphoblastic leukemia. Cancer Cell Int. 2020;20(1):442.
- Trzcinska-Daneluti AM, Ly D, Huynh L, et al. High-content functional screen to identify proteins that correct F508del-CFTR function. Mol Cell Proteomics. 2009;8(4):780–790.
- Hendy GN, Cole DE. Ruling in a suspect: the role of AP2S1 mutations in familial hypocalciuric hypercalcemia type 3. J Clin Endocrinol Metab. 2013;98(12):4666–4669.
- Hannan FM, Howles SA, Rogers A, et al. Adaptor protein-2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype-phenotype correlations, codon bias and dominant-negative effects. Hum Mol Genet. 2015;24(18):5079–5092.
- Vahe C, Benomar K, Espiard S, et al. Diseases associated with calcium-sensing receptor. Orphanet J Rare Dis. 2017;12(1):19.
- Kerut S, Kovvuru KR, Yanes-Cardozo L, et al. Familial hypocalciuric hypercalcaemia type 3: AP2S1 missense mutation. BMJ Case Rep. 2020;13(11):e236631.
- Wong FCK, Wong WS, Kwok JSS, et al. Chinese kindred with familial hypocalciuric hypercalcaemia caused by AP2S1 mutation. F1000Res. 2019;8:1612.
- Aashiq M, Malallah AJ, Khan F, et al. Clinical and biochemical features in a case of familial hypocalciuric hypercalcemia type 3 with AP2S1 gene mutation in Codon Arg15His. Case Rep Pediatr. 2020;2020:7312894.
- Hannan FM, Stevenson M, Bayliss AL, et al. Ap2s1 mutation causes hypercalcaemia in mice and impairs interaction between calcium-sensing receptor and adaptor protein-2. Hum Mol Genet. 2021;30(10):880–892.
- Scheers I, Sokal E, Limaye N, et al. Cinacalcet sustainedly prevents pancreatitis in a child with a compound heterozygous SPINK1/AP2S1 mutation. Pancreatology. 2019;19(6):801–804.
- Krupinova JA, Almaskhanova AA, Eremkina AK, et al. A series of clinical cases of familial hypocalciuric hypercalcemia syndrome. Probl Endokrinol (Mosk). 2020;66(5):61–69.
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell. 1998;93(3):423–432.
- Dell’Angelica EC, Ooi CE, Bonifacino JS. Beta3A-adaptin, a subunit of the adaptor-like complex AP-3. J Biol Chem. 1997;272(24):15078–15084.
- Newman LS, McKeever MO, Okano HJ, et al. Beta-NAP, a cerebellar degeneration antigen, is a neuron-specific vesicle coat protein. Cell. 1995;82(5):773–783.
- Dell’Angelica EC, Ohno H, Ooi CE, et al. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO J. 1997;16(5):917–928.
- Lefrancois S, Janvier K, Boehm M, et al. An ear-core interaction regulates the recruitment of the AP-3 complex to membranes. Dev Cell. 2004;7(4):619–625.
- Zizioli D, Meyer C, Guhde G, et al. Early embryonic death of mice deficient in gamma-adaptin. J Biol Chem. 1999;274(9):5385–5390.
- Odorizzi G, Cowles CR, Emr SD. The AP-3 complex: a coat of many colours. Trends Cell Biol. 1998;8(7):282–288.
- Wilkin M, Tongngok P, Gensch N, et al. Drosophila HOPS and AP-3 complex genes are required for a Deltex-regulated activation of notch in the endosomal trafficking pathway. Dev Cell. 2008;15(5):762–772.
- Huizing M, Sarangarajan R, Strovel E, et al. AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol Biol Cell. 2001;12(7):2075–2085.
- Sugita M, Cao X, Watts GF, et al. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity. 2002;16(5):697–706.
- Del Val M, Yewdell JW. The latest killer AP. Nat Immunol. 2003;4(11):1049–1050.
- Clark RH, Stinchcombe JC, Day A, et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol. 2003;4(11):1111–1120.
- Dell’Angelica EC, Shotelersuk V, Aguilar RC, et al. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3(1):11–21.
- Fontana S, Parolini S, Vermi W, et al. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood. 2006;107(12):4857–4864.
- Huizing M, Malicdan MCV, Wang JA, et al. Hermansky-Pudlak syndrome: mutation update. Hum Mutat. 2020;41(3):543–580.
- Hashimoto R, Ohi K, Okada T, et al. Association analysis between schizophrenia and the AP-3 complex genes. Neurosci Res. 2009;65(1):113–115.
- Kural C, Tacheva-Grigorova SK, Boulant S, et al. Dynamics of intracellular clathrin/AP1- and clathrin/AP3-containing carriers. Cell Rep. 2012;2(5):1111–1119.
- Le Borgne R, Alconada A, Bauer U, et al. The mammalian AP-3 adaptor-like complex mediates the intracellular transport of lysosomal membrane glycoproteins. J Biol Chem. 1998;273(45):29451–29461.
- Van Damme N, Guatelli J. HIV-1 Vpu inhibits accumulation of the envelope glycoprotein within clathrin-coated, Gag-containing endosomes. Cell Microbiol. 2008;10(5):1040–1057.
- Simpson F, Peden AA, Christopoulou L, et al. Characterization of the adaptor-related protein complex, AP-3. J Cell Biol. 1997;137(4):835–845.
- Ooi CE, Moreira JE, Dell’Angelica EC, et al. Altered expression of a novel adaptin leads to defective pigment granule biogenesis in the Drosophila eye color mutant garnet. EMBO J. 1997;16(15):4508–4518.
- Cappato S, Giacopelli F, Tonachini L, et al. Identification of reference genes for quantitative PCR during C3H10T1/2 chondrogenic differentiation. Mol Biol Rep. 2019;46(3):3477–3485.
- Ammann S, Schulz A, Krageloh-Mann I, et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. 2016;127(8):997–1006.
- Mohammed M, Al-Hashmi N, Al-Rashdi S, et al. Biallelic mutations in AP3D1 cause Hermansky-Pudlak syndrome type 10 associated with immunodeficiency and seizure disorder. Eur J Med Genet. 2019;62(11):103583.
- Kantheti P, Qiao X, Diaz ME, et al. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21(1):111–122.
- Baguma-Nibasheka M, Kablar B. Altered retinal cell differentiation in the AP-3 delta mutant (Mocha) mouse. Int J Dev Neurosci. 2009;27(7):701–708.
- Miller CL, Burmeister M, Stevens KE. Hippocampal auditory gating in the hyperactive mocha mouse. Neurosci Lett. 1999;276(1):57–60.
- Li SY, Yoshida Y, Kobayashi E, et al. Serum anti-AP3D1 antibodies are risk factors for acute ischemic stroke related with atherosclerosis. Sci Rep. 2021;11(1):13450.
- Li H, Li M, Tang C, et al. Screening and prognostic value of potential biomarkers for ovarian cancer. Ann Transl Med. 2021;9(12):1007.
- Hirokawa M, Morita H, Tajima T, et al. A genome-wide association study identifies PLCL2 and AP3D1-DOT1L-SF3A2 as new susceptibility loci for myocardial infarction in Japanese. Eur J Hum Genet. 2015;23(3):374–380.
- Gong L, Bates S, Li J, et al. Connecting COPD GWAS genes: FAM13A controls TGFbeta2 secretion by modulating AP-3 transport. Am J Respir Cell Mol Biol. 2021. DOI:10.1165/rcmb.2021-0016OC
- Corredor AP, Gonzalez J, Baquero LA, et al. In silico and in vitro analysis of boAP3d1 protein interaction with bovine leukaemia virus gp51. PLoS One. 2018;13(6):e0199397.
- Garcia E, Nikolic DS, Piguet V. HIV-1 replication in dendritic cells occurs through a tetraspanin-containing compartment enriched in AP-3. Traffic. 2008;9(2):200–214.
- Nishimura N, Plutner H, Hahn K, et al. The delta subunit of AP-3 is required for efficient transport of VSV-G from the trans-Golgi network to the cell surface. Proc Natl Acad Sci U S A. 2002;99(10):6755–6760.
- Du W, Hua F, Li X, et al. Loss of optineurin drives cancer immune evasion via palmitoylation-dependent IFNGR1 lysosomal sorting and degradation. Cancer Discov. 2021;11(7):1826–1843.
- Petrenko AA, Pavlova LS, Karseladze AI, et al. Downregulation of genes encoding for subunits of adaptor complex-3 in cervical carcinomas. Biochemistry (Mosc). 2006;71(10):1153–1160.
- Ivan V, Martinez-Sanchez E, Sima LE, et al. AP-3 and Rabip4ʹ coordinately regulate spatial distribution of lysosomes. PLoS One. 2012;7(10):e48142.
- Meric R, Ercan-Sencicek AG, Uludag Alkaya D, et al. A patient with mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, keratodermia syndrome caused by AP1B1 gene variant. Clin Dysmorphol. 2021;30(1):54–57.
- Jones ML, Murden SL, Brooks C, et al. Disruption of AP3B1 by a chromosome 5 inversion: a new disease mechanism in Hermansky-Pudlak syndrome type 2. BMC Med Genet. 2013;14(1):42.
- De Boer M, van Leeuwen K, Geissler J, et al. Hermansky-Pudlak syndrome type 2: aberrant pre-mRNA splicing and mislocalization of granule proteins in neutrophils. Hum Mutat. 2017;38(10):1402–1411.
- Feng L, Seymour AB, Jiang S, et al. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet. 1999;8(2):323–330.
- Jing R, Dong X, Li K, et al. The Ap3b1 gene regulates the ocular melanosome biogenesis and tyrosinase distribution differently from the Hps1 gene. Exp Eye Res. 2014;128:57–66.
- Jing R, Kong Y, Han G, et al. The mutation of the Ap3b1 gene causes uterine hypoplasia in pearl mice. Reprod Sci. 2020;27(1):182–191.
- Zhen L, Jiang S, Feng L, et al. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood. 1999;94(1):146–155.
- Andres O, Konig EM, Althaus K, et al. Use of targeted high-throughput sequencing for genetic classification of patients with bleeding diathesis and suspected platelet disorder. TH Open. 2018;2(4):e445–e454.
- Schaletzki Y, Kromrey ML, Broderdorf S, et al. Several adaptor proteins promote intracellular localisation of the transporter MRP4/ABCC4 in platelets and haematopoietic cells. Thromb Haemost. 2017;117(1):105–115.
- Karampini E, Schillemans M, Hofman M, et al. Defective AP-3-dependent VAMP8 trafficking impairs Weibel-Palade body exocytosis in Hermansky-Pudlak syndrome type 2 blood outgrowth endothelial cells. Haematologica. 2019;104(10):2091–2099.
- Vallejo MO, Niemeyer GP, Vaglenov A, et al. Decreased hematopoietic progenitor cell mobilization in pearl mice. Exp Hematol. 2013;41(10):848–857.
- Miao Y, Zhu HY, Qiao C, et al. Pathogenic gene mutations or variants identified by targeted gene sequencing in adults with hemophagocytic lymphohistiocytosis. Front Immunol. 2019;10:395.
- Gao L, Zhu L, Huang L, et al. Synergistic defects of UNC13D and AP3B1 leading to adult hemophagocytic lymphohistiocytosis. Int J Hematol. 2015;102(4):488–492.
- Jessen B, Bode SF, Ammann S, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type 2. Blood. 2013;121(15):2943–2951.
- Nishikawa T, Okamura K, Moriyama M, et al. Novel AP3B1 compound heterozygous mutations in a Japanese patient with Hermansky–Pudlak syndrome type 2. J Dermatol. 2020;47(2):185–189.
- Liu D, Zuo X, Zhang P, et al. The novel regulatory role of lncRNA-miRNA-mRNA axis in amyotrophic lateral sclerosis: an integrated bioinformatics analysis. Comput Math Methods Med. 2021;2021:5526179.
- Cuevas-Mora K, Roque W, Shaghaghi H, et al. Hermansky-Pudlak syndrome-2 alters mitochondrial homeostasis in the alveolar epithelium of the lung. Respir Res. 2021;22(1):49.
- Summer R, Krishna R, Schriner D, et al. Matrix metalloproteinase activity in the lung is increased in Hermansky-Pudlak syndrome. Orphanet J Rare Dis. 2019;14(1):162.
- Lorenzi L, Tabellini G, Vermi W, et al. Occurrence of nodular lymphocyte-predominant hodgkin lymphoma in hermansky-pudlak type 2 syndrome is associated to natural killer and natural killer T cell defects. PLoS One. 2013;8(11):e80131.
- Liu L, Sutton J, Woodruff E, et al. Defective HIV-1 particle assembly in AP-3-deficient cells derived from patients with Hermansky-Pudlak syndrome type 2. J Virol. 2012;86(20):11242–11253.
- Azevedo C, Burton A, Ruiz-Mateos E, et al. Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 Gag release. Proc Natl Acad Sci U S A. 2009;106(50):21161–21166.
- Sun W, McCrory TS, Khaw WY, et al. Matrix proteins of Nipah and Hendra viruses interact with beta subunits of AP-3 complexes. J Virol. 2014;88(22):13099–13110.
- Selcuklu SD, Donoghue MT, Rehmet K, et al. MicroRNA-9 inhibition of cell proliferation and identification of novel miR-9 targets by transcriptome profiling in breast cancer cells. J Biol Chem. 2012;287(35):29516–29528.
- Mantripragada KK, Diaz de Stahl T, Patridge C, et al. Genome-wide high-resolution analysis of DNA copy number alterations in NF1-associated malignant peripheral nerve sheath tumors using 32K BAC array. Genes Chromosomes Cancer. 2009;48(10):897–907.
- Seong E, Wainer BH, Hughes ED, et al. Genetic analysis of the neuronal and ubiquitous AP-3 adaptor complexes reveals divergent functions in brain. Mol Biol Cell. 2005;16(1):128–140.
- Sanuki R, Watanabe S, Sugita Y, et al. Protein-4.1G-mediated membrane trafficking is essential for correct rod synaptic location in the retina and for normal visual function. Cell Rep. 2015;10(5):796–808.
- Newell-Litwa K, Salazar G, Smith Y, et al. Roles of BLOC-1 and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell. 2009;20(5):1441–1453.
- Ueda K, Ogawa S, Matsuda K, et al. Blended phenotype of combination of HERC2 and AP3B2 deficiency and Angelman syndrome caused by paternal isodisomy of chromosome 15. Am J Med Genet A. 2021;185(10):3092–3098.
- Mange L, Haitao R, Lixin Z, et al. Cerebellar ataxia and myeloradiculopathy associated with AP3B2 antibody: a case report and literature review. J Neurol. 2021. DOI:10.1007/s00415-021-10496-8
- Zhu M, Jia L, Li F, et al. Identification of KIAA0513 and other hub genes associated with Alzheimer disease using weighted gene coexpression network analysis. Front Genet. 2020;11:981.
- Zhang Z, Li Q, Diao M, et al. Sporadic hirschsprung disease: mutational spectrum and novel candidate genes revealed by next-generation sequencing. Sci Rep. 2017;7(1):14796.
- Sun M, Sun T, He Z, et al. Identification of two novel biomarkers of rectal carcinoma progression and prognosis via co-expression network analysis. Oncotarget. 2017;8(41):69594–69609.
- Koh Y, Kim D, Jung WJ, et al. Revealing genomic profile that underlies tropism of myeloma cells using whole exome sequencing. Int J Genomics. 2015;2015:675379.
- Ohno H, Aguilar RC, Fournier MC, et al. Interaction of endocytic signals from the HIV-1 envelope glycoprotein complex with members of the adaptor medium chain family. Virology. 1997;238(2):305–315.
- Sullivan SE, Liao M, Smith RV, et al. Candidate-based screening via gene modulation in human neurons and astrocytes implicates FERMT2 in Aβ and TAU proteostasis. Hum Mol Genet. 2019;28(5):718–735.
- Rohde G, Wenzel D, Haucke V. A phosphatidylinositol (4,5)-bisphosphate binding site within mu2-adaptin regulates clathrin-mediated endocytosis. J Cell Biol. 2002;158(2):209–214.
- Ishizaki H, Spitzer M, Wildenhain J, et al. Combined zebrafish-yeast chemical-genetic screens reveal gene-copper-nutrition interactions that modulate melanocyte pigmentation. Dis Model Mech. 2010;3(9–10):639–651.
- VanRenterghem B, Morin M, Czech MP, et al. Interaction of insulin receptor substrate-1 with the sigma3A subunit of the adaptor protein complex-3 in cultured adipocytes. J Biol Chem. 1998;273(45):29942–29949.
- Nome T, Thomassen GO, Bruun J, et al. Common fusion transcripts identified in colorectal cancer cell lines by high-throughput RNA sequencing. Transl Oncol. 2013;6(5):546–553.
- Kazakova EV, Zghuang T, Li T, et al. The Gas6 gene rs8191974 and Ap3s2 gene rs2028299 are associated with type 2 diabetes in the northern Chinese Han population. Acta Biochim Pol. 2017;64(2):227–231.
- Kanthimathi S, Chidambaram M, Bodhini D, et al. Association of recently identified type 2 diabetes gene variants with gestational diabetes in Asian Indian population. Mol Genet Genomics. 2017;292(3):585–591.
- Zhang YQ, Peng LJ, Cao YR, et al. Risk factors for hepatocellular carcinoma in cirrhotic patients with chronic hepatitis B. Genet Test Mol Biomarkers. 2016;20(9):535–543.
- Dong C, Beecham A, Slifer S, et al. Genomewide linkage and peakwide association analyses of carotid plaque in caribbean hispanics. Stroke. 2010;41(12):2750–2756.
- Ebrahimi-Fakhari D, Behne R, Davies AK, et al. AP-4-associated hereditary spastic paraplegia. In: Adam MP, Ardinger HH, Pagon RA, et al, editors GeneReviews((R)). Seattle (WA): University of Washington: 1993 Dec 13.
- Abou Jamra R, Philippe O, Raas-Rothschild A, et al. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet. 2011;88(6):788–795.
- Ebrahimi-Fakhari D, Teinert J, Behne R, et al. Defining the clinical, molecular and imaging spectrum of adaptor protein complex 4-associated hereditary spastic paraplegia. Brain. 2020;143:2929–2944.
- Tuysuz B, Bilguvar K, Kocer N, et al. Autosomal recessive spastic tetraplegia caused by AP4M1 and AP4B1 gene mutation: expansion of the facial and neuroimaging features. Am J Med Genet A. 2014;164A:1677–1685.
- Novikova G, Kapoor M, Tcw J, et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat Commun. 2021;12(1):1610.
- Maday S. Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res. 2016;1649:143–150.
- Ivankovic D, Drew J, Lesept F, et al. Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy. 2020;16(3):391–407.
- Moreno-De-Luca A, Helmers SL, Mao H, et al. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J Med Genet. 2011;48(2):141–144.
- Murakami H, Uehara T, Tsurusaki Y, et al. Blended phenotype of AP4E1 deficiency and Angelman syndrome caused by paternal isodisomy of chromosome 15. Brain Dev. 2020;42(3):289–292.
- Frigerio-Domingues CE, Gkalitsiou Z, Zezinka A, et al. Genetic factors and therapy outcomes in persistent developmental stuttering. J Commun Disord. 2019;80:11–17.
- Sager G, Turkyilmaz A, Ates EA, et al. Correction to: HACE1, GLRX5, and ELP2 gene variant cause spastic paraplegies. Acta Neurol Belg. 2021. DOI:10.1007/s13760-021-01685-3
- Winkler I, Miotla P, Lejman M, et al. A new family with spastic paraplegia type 51 and novel mutations in AP4E1. BMC Med Genomics. 2021;14(1):131.
- Kong XF, Bousfiha A, Rouissi A, et al. A novel homozygous p.R1105X mutation of the AP4E1 gene in twins with hereditary spastic paraplegia and mycobacterial disease. PLoS One. 2013;8(3):e58286.
- Raza MH, Mattera R, Morell R, et al. Association between rare variants in AP4E1, a component of intracellular trafficking, and persistent stuttering. Am J Hum Genet. 2015;97(5):715–725.
- Li Y, Min C, Zhao Y, et al. Effects of fluoride on PIWI-interacting RNA expression profiling in testis of mice. Chemosphere. 2021;269:128727.
- Hatsugai N, Nakatsuji A, Unten O, et al. Involvement of adapter protein complex 4 in hypersensitive cell death induced by avirulent bacteria. Plant Physiol. 2018;176(2):1824–1834.
- Frazier MN, Davies AK, Voehler M, et al. Molecular basis for the interaction between AP4 β4 and its accessory protein, Tepsin. Traffic. 2016;17(4):400–415.
- Ebrahimi-Fakhari D, Cheng C, Dies K, et al. Clinical and genetic characterization of AP4B1-associated SPG47. Am J Med Genet A. 2018;176(2):311–318.
- Teinert J, Behne R, D’Amore A, et al. Generation and characterization of six human induced pluripotent stem cell lines (iPSC) from three families with AP4B1-associated hereditary spastic paraplegia (SPG47). Stem Cell Res. 2019;40:101575.
- Abdollahpour H, Alawi M, Kortum F, et al. An AP4B1 frameshift mutation in siblings with intellectual disability and spastic tetraplegia further delineates the AP-4 deficiency syndrome. Eur J Hum Genet. 2015;23(2):256–259.
- Szczaluba K, Mierzewska H, Smigiel R, et al. AP4B1-associated hereditary spastic paraplegia: expansion of phenotypic spectrum related to homozygous p.Thr387fs variant. J Appl Genet. 2020;61(2):213–218.
- Lamichhane DM. New AP4B1 mutation in an African-American child associated with intellectual disability. J Pediatr Genet. 2013;2:191–195.
- Aguilar RC, Boehm M, Gorshkova I, et al. Signal-binding specificity of the mu4 subunit of the adaptor protein complex AP-4. J Biol Chem. 2001;276(16):13145–13152.
- Zhang J, Cheng XY, Sheng GY. AP4M1 is abnormally expressed in oxygen-glucose deprived hippocampal neurons. Neurosci Lett. 2014;563:85–89.
- Accogli A, Hamdan FF, Poulin C, et al. A novel homozygous AP4B1 mutation in two brothers with AP-4 deficiency syndrome and ocular anomalies. Am J Med Genet A. 2018;176(4):985–991.
- Burgos PV, Mardones GA, Rojas AL, et al. Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev Cell. 2010;18(3):425–436.
- Toh WH, Tan JZ, Zulkefli KL, et al. Amyloid precursor protein traffics from the Golgi directly to early endosomes in an Arl5b- and AP4-dependent pathway. Traffic. 2017;18(3):159–175.
- Roubertie A, Hieu N, Roux CJ, et al. AP4 deficiency: a novel form of neurodegeneration with brain iron accumulation? Neurol Genet. 2018;4(1):e217.
- Jameel M, Klar J, Tariq M, et al. A novel AP4M1 mutation in autosomal recessive cerebral palsy syndrome and clinical expansion of AP-4 deficiency. BMC Med Genet. 2014;15(1):133.
- Verkerk AJ, Schot R, Dumee B, et al. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. 2009;85(1):40–52.
- D’Amore A, Tessa A, Naef V, et al. Loss of ap4s1 in zebrafish leads to neurodevelopmental defects resembling spastic paraplegia 52. Ann Clin Transl Neurol. 2020;7(4):584–589.
- Price SJ, Garner TW, Balloux F, et al. A de novo assembly of the common frog (Rana temporaria) transcriptome and comparison of transcription following exposure to ranavirus and batrachochytrium dendrobatidis. PLoS One. 2015;10(6):e0130500.
- Vill K, Muller-Felber W, Alhaddad B, et al. A homozygous splice variant in AP4S1 mimicking neurodegeneration with brain iron accumulation. Mov Disord. 2017;32(5):797–799.
- Slabicki M, Theis M, Krastev DB, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010;8(6):e1000408.
- Hirst J, Borner GH, Edgar J, et al. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell. 2013;24(16):2558–2569.
- Seaman MNJ. Back from the brink: retrieval of membrane proteins from terminal compartments: unexpected pathways for membrane protein retrieval from vacuoles and endolysosomes. Bioessays. 2019;41(3):e1800146.
- Breza M, Hirst J, Chelban V, et al. Expanding the spectrum of AP5Z1- related hereditary spastic paraplegia (HSP-SPG48): a multicenter study on a rare disease. Mov Disord. 2021;36(4):1034–1038.
- Maruta K, Ando M, Otomo T, et al. A case of spastic paraplegia 48 with a novel mutation in the AP5Z1 gene. Rinsho Shinkeigaku. 2020;60:543–548.
- Schlipf NA, Schule R, Klimpe S, et al. AP5Z1/SPG4 8 frequency in autosomal recessive and sporadic spastic paraplegia. Mol Genet Genomic Med. 2014;2(5):379–382.
- Denton K, Mou Y, Xu CC, et al. Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum Mol Genet. 2018;27(14):2517–2530.
- Hirst J, Itzhak DN, Antrobus R, et al. Role of the AP-5 adaptor protein complex in late endosome-to-Golgi retrieval. PLoS Biol. 2018;16(1):e2004411.
- Hirst J, Madeo M, Smets K, et al. Complicated spastic paraplegia in patients with AP5Z1 mutations (SPG48). Neurol Genet. 2016;2(5):e98.
- Marenholz I, Esparza-Gordillo J, Ruschendorf F, et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun. 2015;6(1):8804.
- Peng C, Van Meel ER, Cardenas A, et al. Epigenome-wide association study reveals methylation pathways associated with childhood allergic sensitization. Epigenetics. 2019;14(5):445–466.
- Calender A, Rollat Farnier PA, Buisson A, et al. Whole exome sequencing in three families segregating a pediatric case of sarcoidosis. BMC Med Genomics. 2018;11(1):23.
- Zuo X, Sun L, Yin X, et al. Whole-exome SNP array identifies 15 new susceptibility loci for psoriasis. Nat Commun. 2015;6(1):6793.
- Christou MA, Ntritsos G, Markozannes G, et al. A genome-wide scan for pleiotropy between bone mineral density and nonbone phenotypes. Bone Res. 2020;8(1):26.
- Choi JH, Lim JB, Wickramanayake DD, et al. Characterization of MUDENG, a novel anti-apoptotic protein. Oncogenesis. 2016;5(5):e221.
- Won M, Luo Y, Lee DH, et al. BAX is an essential key mediator of AP5M1-induced apoptosis in cervical carcinoma cells. Biochem Biophys Res Commun. 2019;518(2):368–373.
- Muthu M, Chun S, Gopal J, et al. The MUDENG augmentation: a genesis in anti-cancer therapy? Int J Mol Sci. 2020;21. DOI:10.3390/ijms21155583.
- Shin JN, Han JH, Kim JY, et al. MUDENG is cleaved by caspase-3 during TRAIL-induced cell death. Biochem Biophys Res Commun. 2013;435(2):234–238.
- Iacobas S, Iacobas DA. A personalized genomics approach of the prostate cancer. Cells. 2021;10(7):1644.
- Jung S, Shin J, Oh J, et al. Cytotoxic and apoptotic potential of Phyllodium elegans extracts on human cancer cell lines. Bioengineered. 2019;10(1):501–512.
- Lee MR, Shin JN, Moon AR, et al. A novel protein, MUDENG, induces cell death in cytotoxic T cells. Biochem Biophys Res Commun. 2008;370(3):504–508.
- Strazic Geljic I, Kucan Brlic P, Musak L, et al. Viral interactions with adaptor-protein complexes: a Ubiquitous trait among viral species. Int J Mol Sci. 2021;22(10):5274.
- Amara JF, Cheng SH, Smith AE. Intracellular protein trafficking defects in human disease. Trends Cell Biol. 1992;2(5):145–149.
- Goldenring JR. A central role for vesicle trafficking in epithelial neoplasia: intracellular highways to carcinogenesis. Nat Rev Cancer. 2013;13(11):813–820.
- Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–758.
- Irannejad R, Tsvetanova NG, Lobingier BT, et al. Effects of endocytosis on receptor-mediated signaling. Curr Opin Cell Biol. 2015;35:137–143.
- Mattera R, Ritter B, Sidhu SS, et al. Definition of the consensus motif recognized by gamma-adaptin ear domains. J Biol Chem. 2004;279(9):8018–8028.
- Doray B, Kornfeld S. Gamma subunit of the AP-1 adaptor complex binds clathrin: implications for cooperative binding in coated vesicle assembly. Mol Biol Cell. 2001;12(7):1925–1935.
- Shih W, Gallusser A, Kirchhausen T. A clathrin-binding site in the hinge of the beta 2 chain of mammalian AP-2 complexes. J Biol Chem. 1995;270(52):31083–31090.
- Craig HM, Reddy TR, Riggs NL, et al. Interactions of HIV-1 nef with the mu subunits of adaptor protein complexes 1, 2, and 3: role of the dileucine-based sorting motif. Virology. 2000;271(1):9–17.
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72(1):395–447.
- Benmerah A, Begue B, Dautry-Varsat A, et al. The ear of alpha-adaptin interacts with the COOH-terminal domain of the Eps 15 protein. J Biol Chem. 1996;271(20):12111–12116.
- Brett TJ, Traub LM, Fremont DH. Accessory protein recruitment motifs in clathrin-mediated endocytosis. Structure. 2002;10(6):797–809.
- Jha A, Agostinelli NR, Mishra SK, et al. A novel AP-2 adaptor interaction motif initially identified in the long-splice isoform of synaptojanin 1, SJ170. J Biol Chem. 2004;279(3):2281–2290.
- Walther K, Diril MK, Jung N, et al. Functional dissection of the interactions of stonin 2 with the adaptor complex AP-2 and synaptotagmin. Proc Natl Acad Sci U S A. 2004;101(4):964–969.